
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuo vadis niobium? Divergent coordination behavior of early-transition metals towards MOF-5†
Maciej D.
Korzyński
 a,
Luca
Braglia
b,
Elisa
Borfecchia
cd,
Kirill A.
Lomachenko
e,
Amgalanbaatar
Baldansuren
f,
Christopher H.
Hendon
g,
Carlo
Lamberti‡
hi and
Mircea
Dincă
*a
a,
Luca
Braglia
b,
Elisa
Borfecchia
cd,
Kirill A.
Lomachenko
e,
Amgalanbaatar
Baldansuren
f,
Christopher H.
Hendon
g,
Carlo
Lamberti‡
hi and
Mircea
Dincă
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: mdinca@mit.edu
bCNR-Istituto Officina dei Materiali, TASC Laboratory in Area Science Park - Basovizza, Strada Statale 14 km 163.5, 34149 Trieste, Italy
cDepartment of Chemistry, NIS, CrisDi, INSTM Centre of Reference, University of Turin, Via Quarello 15, I-10135 Torino, Italy
dCenter for Materials Science and Nanotechnology (SMN), Department of Chemistry, University of Oslo, 1033 Blindern, 0315 Oslo, Norway
eEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France
fEPSRC National EPR Facility, School of Chemistry, The University of Manchester, Oxford Road, Manchester M13 9PL, UK
gMaterials Science Institute, Department of Chemistry and Biochemistry, University of Oregon, Eugene, Oregon 97403, USA
hDepartment of Physics, NIS, CrisDi, Interdepartmental Centers, INSTM Centre of Reference, University of Turin, Via Giuria 1, I-10125 Torino, Italy
iThe Smart Materials Research Institute, Southern Federal University, 178/24 Sladkova Street, Rostov-on-Don, 344090, Russia
First published on 9th May 2019
Abstract
Treatment of MOF-5 with NbCl4(THF)2 in acetonitrile leads to incorporation of Nb(IV) centers in a fashion that diverges from the established cation metathesis reactivity of this iconic material. A combination of X-ray absorption spectroscopy analysis and reactivity studies altogether supported by density functional theory computational studies document an unprecedented binding mode for the Zn4O(O2C–)6 secondary building units (SBUs), which in Nb(IV)-MOF-5 function as κ2-chelating ligands for NbCl4 moieties, with no exchange of Zn2+ observed. This unusual reactivity expands the portfolio of post-synthetic modification techniques available for MOFs, exemplified here by MOF-5, and underscores the diverse coordination environments offered by this and potentially other MOFs towards heterometal species.
Introduction
Nominally, the compositional variety of metal–organic frameworks (MOFs) is unlimited, with virtually any stable metal in the periodic table potentially serving as the basis for the inorganic building blocks, or the secondary building units (SBUs).1–9 Yet, the vast majority of SBUs are made from a very limited set of metal species, nearly all MOFs featuring first-row transition metals, with heavy representation among the late first row metals. Although early transition metal (ETM)-based MOFs have distinguished themselves with remarkable stability, especially under aqueous conditions,10 MOFs made from ETMs are still rare outside of Zr and Ti. One of the major challenges associated with de novo synthesis of ETM-based MOFs is the tendency of metal precursors to undergo deleterious oxygen-scavenging reactivity under solvothermal conditions encountered in typical MOF syntheses.11With an eye towards enriching the compositional space of MOFs with other ETMs and to explore the fundamentals of bonding for ETMs within this set of materials, we turned our attention to niobium, which remains essentially unexplored in this sense. Indeed, to our knowledge, there are only two reported Nb-containing frameworks in the literature.12,13 With its flexible coordination sphere, large covalent radius, and extreme oxophilicity, niobium is the epitome of a difficult substrate for MOF formation and should therefore provide insight into the behavior of other early transition metals for this purpose.14
To circumvent the expected challenges associated with the direct synthesis of Nb MOFs (vide supra), we decided to focus on post-synthetic modification (PSM), a method that has previously proven successful in altering the chemical structure of pre-formed MOFs.15–17 Because PSM allows for mild reaction conditions, contrasting with the harsh solvothermal synthetic routes encountered in de novo MOF preparation, it can lead to the isolation of materials that are difficult to form otherwise.18 In particular, within the context of post-synthetic cation exchange19 our group and others have shown, initially through the iconic Zn4O(BDC)3 (MOF-5; BDC = terephthalate) (Fig. 1a), that the SBUs within MOFs can be thought of as supramolecular chelating ligands for incoming metal ions. The exchanged SBUs behave spectroscopically and chemically as isolated molecules with unusual coordination environments that are often unknown from traditional solution chemistry.20 These earlier studies showed that the [Zn4O]6+ SBU (Fig. 1b) is particularly prone to exchanging Zn2+ for the majority of the first row transition metal cations in various oxidation states (Fig. 1c).21–23 MOF was therefore identified as a promising platform for exploring Nb chemistry. Herein, we document our quest in understanding niobium immobilization in MOF-5, and we unveil an unprecedented coordination mode within its SBU.
 | ||
| Fig. 1 (a) Structure of MOF-5, (b) its SBU, and (c) idealized cation exchange process. Hydrogen atoms were omitted for clarity. | ||
Results and discussion
Appropriate starting materials are available for Nb in several oxidation states, with the Nb(IV) d1 system offering an additional characterization handle in the form of electron paramagnetic resonance (EPR) spectroscopy, when compared to the more common Nb(V) oxidation state. To this end, the soluble niobium(IV) chloride tetrahydrofuran complex (NbCl4(THF)2) serves as a good entry point to Nb(IV) chemistry.24 The routine method to introduce a foreign metal into a framework involves soaking the MOF crystals in a solution of the desired precursor. Here, treatment of pre-activated, colorless MOF-5 crystals25 with a yellow-green solution of NbCl4(THF)2 in acetonitrile (MeCN) led to an almost instantaneous change of color for the crystals to dark violet (Fig. S1†). The observed fast kinetics of this process were surprising given that reported first row cation exchange processes proceed rather slowly. After 48 h, the isolated dark violet crystals of Nb(IV)-treated MOF-5 were washed extensively with MeCN then dichloromethane (DCM) until no characteristic features of Nb(IV) could be observed in the UV spectrum of the effluent solutions. Residual solvent molecules within the pores and/or weakly-bound to the metal centers, were removed by heating to 150 °C under high vacuum (0.1 mTorr).Powder X-ray diffraction (PXRD) analysis confirmed that treatment of MOF-5 with the niobium solution and subsequent thermal treatment proceed with preservation of crystallinity (Fig. S2†). The R1 (9.7°-to-6.8°) and R2 (13.8°-to-6.8°) peak ratios26,27 were low indicating no pore obstruction by the guest molecules and lack of framework interpenetration. Inspection of the attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectrum of the activated material showed predominantly typical MOF-5 features (Fig. S3†). Additional FTIR bands in the spectrum of Nb(IV)-treated MOF-5, which are absent in the spectrum of MOF-5 itself, are also present in the vacuum-dried residue obtained from a solution of NbCl4(THF)2 in acetonitrile (Fig. S4†). In particular, a prominent feature at 2282 cm−1 in this latter residue suggests that excess acetonitrile partially replaces THF in the primary coordination sphere of Nb(IV), in agreement with the literature data on MeCN adducts of Nb(IV),28 and is likely carried over upon reaction with MOF-5. Importantly, this band does not persist in fully activated Nb(IV)-MOF-5, indicating full removal of MeCN from this material.
The documented tendency of Nb(IV) compounds to disproportionate thermally29 required confirmation that niobium immobilization within MOF-5 occurs with preservation of the +IV oxidation state, which came from EPR spectroscopy and X-ray absorption spectroscopy (XAS). Although the typical hyperfine splitting pattern for 93Nb (I = 9/2) is observed in the EPR spectrum of Nb(IV)-MOF-5 at X-band, the peaks are significantly broadened, with a first-order hyperfine splitting (ANb) of approximately 557 MHz (Fig. 3, Table S1†). In the simulated spectrum (Fig. S5 and S6†), the hyperfine interactions of both isotopes 35Cl and 37Cl (both I = 3/2 nuclei) are included, and although they do not affect the alignment of the electron spins, these contribute to inhomogeneous line broadening. Notably, EPR signals obtained for Nb(IV)-MOF-5 and a toluene-MeCN frozen glass of NbCl4(THF)2 are similar, the aforementioned line broadening in the MOF notwithstanding (Fig. S7†). Finally, analysis of the XAS data in the Nb K-edge X-ray absorption near edge structure region (XANES) for Nb(IV)-treated MOF-5 and a series of Nb(IV) standards30 confirmed that the edge energy, ≈19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 001 eV, was in line with that of niobium(IV) oxide (Fig. S8†). These results offer confidence that Nb(IV) does not disproportionate and remains in its original oxidation state when immobilized within MOF-5.
001 eV, was in line with that of niobium(IV) oxide (Fig. S8†). These results offer confidence that Nb(IV) does not disproportionate and remains in its original oxidation state when immobilized within MOF-5.
 | ||
| Fig. 2 (a) PXRD patterns and (b) N2 adsorption isotherms for Nb(IV)-treated MOF-5 samples with increasing niobium content. | ||
 | ||
| Fig. 3 EPR spectra of Nb(IV)-MOF-5 and of Nb(V)-MOF-5 treated with (TMS)2pyr. | ||
The impact of immobilized Nb content on the properties of the MOF was probed by preparing samples containing different Nb-to-SBU molar ratios (0.16, 0.20, 0.52, and 1.12; determined by inductively coupled plasma – atomic emission spectroscopy, ICP-AES). Even when using less than one equivalent of Nb precursor with respect to the [Zn4O]6+ SBU, Nb uptake into the MOF progressed rapidly and in a qualitatively similar manner, with retention of crystallinity (Fig. 2a). This stands in contrast with cation exchange, which occurs typically only in an excess of incoming metal species and often leads to loss of crystallinity when exchange is rapid.
The porosity of the Nb(IV)-treated samples was further confirmed by N2 adsorption isotherms at 77 K (Fig. 2b). Fitting these to the Brunauer–Emmett–Teller (BET) equation gave apparent surface areas ranging from 2943 m2 g−1 to 2010 m2 g−1 for Nb(IV)-MOF-5 samples with 0.16 mol Nb per SBU and 1.12 mol Nb per SBU, respectively. Although these are lower than anticipated for cation-exchanged materials, the decreased porosity was not caused by surface deposition of certain species that would block access to the pores. Indeed, scanning electron microscopy – energy dispersive X-ray spectroscopy mapping revealed that niobium deposition occurs not only on the surface, but also homogeneously throughout the crystal (Fig. S9 and S10†).
With solid evidence that Nb is immobilized inside the pores of MOF-5, but with conflicting evidence regarding its substitution of Zn2+ ions inside the SBU, the primary coordination environment of Nb(IV) within MOF-5 was probed by XAS.31 These studies were aided by density functional theory (DFT) calculations that modelled three Nb(IV) bonding scenarios. The first model assumed that typical cation exchange occurred, with a [NbCl2]2+ unit replacing a Zn2+ ion to produce a neutral Cl2NbZn3O(O2C–)6 cluster (Fig. 4a). As shown with Ni2+-exchanged MOF-5,21 the SBU in this material is able to support octahedral metal ions, as would be required for the Nb center in this model. A second model assumed that the [Zn4O]6+ constitution of the node is preserved and that instead of exchanging any Zn2+ ions, a NbCl4 moiety binds to the SBU in an exogenous κ2-fashion utilizing two carboxylate oxygen atoms as anchor points (Fig. 4b). Finally, to account for the possibility of adventitious oxygen or water scavenging during the deposition, we also considered a niobyl unit (NbOCl2) bound to the SBU exogenously in a κ2-fashion similar to the NbCl4 model (Fig. 4c).
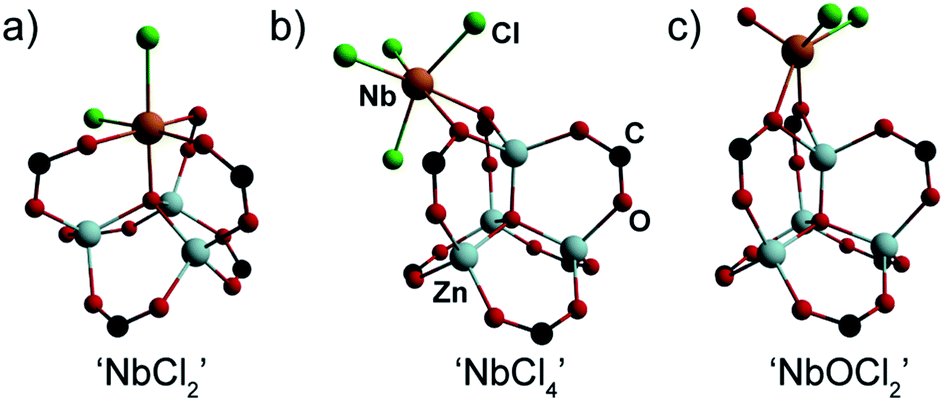 | ||
| Fig. 4 DFT-optimized models of plausible Nb coordination modes in MOF-5. | ||
The XAS data coupled with these DFT models was sufficient to discriminate the local Nb environment within MOF-5. Extended X-ray absorption fine structure (EXAFS) analysis of the XAS data for a wide range of Nb30 allowed adoption of a two-step refinement strategy to analyze the experimental EXAFS spectrum of Nb(IV)-MOF-5: (i) in a first step, all the parameters for the three DFT-structures were optimized (Table S2†) apart from the S02, which was fixed to 1 and the Debye–Waller factor of O, σO2, and of Cl, σCl2, which were fixed from the EXAFS results obtained on the reference samples; (ii) the second step involved refinement of S02 (Table S3†). Unexpectedly, the first step revealed that the fit for ‘NbCl4’ matched the experimental data much better than other models. Confirming that no hydrolysis at the Nb center occurs, the fit for the ‘NbOCl2’ model yielded a meaningless value of ΔE (15 ± 31) eV and an unphysically high σ2 (0.14 ± 0.12) Å2 for the scattering paths involving C and O atoms at distances larger than 3.2 Å from the Nb absorber. Although the parameters obtained for the ‘NbCl2’ cation exchange model were also reasonable, the fit for this model did not match the experimental curves as well as the ‘NbCl4’ fit. Indeed, the R-factor for the ‘NbCl2’ model was seven times larger than that for ‘NbCl4’ (Table S2†).
The second step in the EXAFS fitting routine (Table S3†) definitely disqualified the cation exchanged (‘NbCl2’) model as a likely candidate for Nb(IV)-MOF-5. Indeed, optimizing the S02 for all three models excluded ‘NbCl2’ both on the basis of an unreasonably high value for the energy shift (ΔE = 10 ± 2 eV) and a value that was too low for the S02 parameter (0.51 ± 0.04). Lastly, the best qualitative agreement between the fit and experimental curves was obtained for the ‘NbCl4’ model (Fig. 5, red curves), which we therefore favor as the dominant coordination environment of Nb in Nb(IV)-MOF-5. The final fit parameters for this model (Fig. S11†) gave Nb–O distances of 2.49(5) Å and two independent sets of Cl atoms situated at 2.23(4) Å and 2.41(2) Å from the niobium center.
 | ||
| Fig. 5 Comparison of EXAFS fits for various models of Nb moieties within MOF-5. | ||
Immobilization of hetero-metal species in MOF-5 by means other than cation exchange has received little attention in the literature. In fact, the vast majority of examples that employ decoration of the SBUs with exogenous metals, rather than cation exchange, are reported for Zr-based materials.16,32–35 One recent example with transition metals in MOF-5 involved the installation of Fe(OH)3(H2O)2 moieties bridging SBUs on two interpenetrated lattices,36 with limited additional examples focusing on alkali metal decoration of MOF-5.37–42 These findings point to a previously unrecognized divergence in the reactivity of early and late transition metals with oxygen-based SBUs: whereas the late transition metals favor cation exchange reactivity (the Zr-based MOFs notwithstanding43), we show here that the high-valent early transition metals prefer to adhere to the SBU rather than exchange within the SBU, presumably because of their extreme oxophilicity. Notably, MOF-5 is a rare instance that allows both processes to occur.§ Further proof of exogenous SBU binding of Nb(IV), rather than exchange within the SBU, came from a clean reaction between Nb(IV)-MOF-5 and XeF2 in MeCN, which caused a swift color change of the violet crystals to off-white and emergence of a yellow solution. Inspection of the off-white solid by ICP-AES showed that niobium was almost quantitatively removed from the material, with preservation of crystallinity (Fig. S12†). This would be unlikely if niobium served a structural role as part of the SBU.
Immobilization of early transition metal ions by decoration of the MOF-5 SBU can be extended to Nb(V), which furthermore engages in molecule-like, well behaved redox chemistry within the MOF, reminiscent of the behavior of late transition metals in the same material. Thus, soaking MOF-5 in a solution of NbCl5 in MeCN yielded Nb(V)-MOF-5 as an off-white crystalline powder. Despite the higher Lewis acidity44 and even higher oxophilicity of Nb(V) relative to Nb(IV), Nb(V)-decorated MOF-5 also retains crystallinity (Fig. S14†) and, upon activation in a similar manner to the Nb(IV) MOF, shows porosity with a BET surface area of 1437 m2 g−1 (Fig. S16†). This value is lower than that of Nb(IV)-MOF-5, as may be expected for a material with an additional chloride protruding into the pore. A testament to the molecule-like, site-isolated environment of Nb(V) within this material was its clean reduction with 2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene ((TMS)2pyr), a practical reagent for the reduction of high-valent early transition metal halides.45 Treatment of Nb(V)-MOF-5 with an excess of (TMS)2pyr in THF led to rapid change of color of the solids from off-white to dark blue, a transformation that once again occurred with preservation of crystallinity (Fig. S17†). The reduced MOF exhibited an X-band EPR spectrum in THF that was qualitatively similar to that of independently-synthesized Nb(IV)-MOF-5 obtained from NbCl4(THF)2 (Fig. 3, blue trace). Although a simple inner-sphere electron transfer reaction, this transformation demonstrates that metal centers decorating the MOF-5 SBU are amenable for further synthetic elaboration in a manner similar to those exchanged into the SBU.
Conclusions
In our endeavors to advance the niobium chemistry in MOFs, we discovered a novel coordination mode of foreign metal species to the SBU of MOF-5. Our results show that it is possible both to introduce and to remove niobium ions with the preservation of the parent framework's crystallinity. The chemical competence of these extra framework Nb sites is exemplified by simple reduction of Nb(V) to Nb(IV) within the framework. Excitingly, literally 20 years after its initial discovery, MOF-5, arguably one of the most iconic materials in its class, continues to surprise. This bodes well for the richness of fundamental chemistry yet to be discovered with the other thousands of MOFs out there.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through a CAREER grant to M. D. from the National Science Foundation (DMR-1452612). Computational work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. C. L. acknowledges the Mega-grant of the Russian Federation Government, No. 14.Y26.31.0001. The authors would like to acknowledge Dr Wesley J. Transue for aid with computational work, Dr Lei Sun for assistance in SEM-EDX measurements, Dr Giovanni Agostini for the assistance during the experiment at BM23 beamline46 of the ESRF and Prof. Dr Nicholas Chilton for helpful discussions regarding the EPR data.Notes and references
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- F.-Y. Yi, D. Chen, M.-K. Wu, L. Han and H.-L. Jiang, Chempluschem, 2016, 81, 675–690 CrossRef CAS.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- C. A. Downes and S. C. Marinescu, ChemSusChem, 2017, 10, 4374–4392 CrossRef CAS PubMed.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- J.-S. Qin, S. Yuan, C. Lollar, J. Pang, A. Alsalme and H.-C. Zhou, Chem. Commun., 2018, 54, 4231–4249 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- S. Ahn, N. E. Thornburg, Z. Li, T. C. Wang, L. C. Gallington, K. W. Chapman, J. M. Notestein, J. T. Hupp and O. K. Farha, Inorg. Chem., 2016, 55, 11954–11961 CrossRef CAS PubMed.
- L. G. Hubert-Pfalzgraf, Niobium & Tantalum: Inorganic & Coordination Chemistry in Encyclopedia of Inorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, 2006 Search PubMed.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- R. J. Marshall and R. S. Forgan, Eur. J. Inorg. Chem., 2016, 2016, 4310–4331 CrossRef CAS.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M.-H. Zeng, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- C. K. Brozek and M. Dincă, Chem. Sci., 2012, 3, 2110 RSC.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS.
- C. K. Brozek, V. K. Michaelis, T.-C. Ong, L. Bellarosa, N. López, R. G. Griffin and M. Dincă, ACS Cent. Sci., 2015, 1, 252–260 CrossRef CAS PubMed.
- J. W. Herndon, Niobium(IV) Chloride in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS.
- B. Chen, X. Wang, Q. Zhang, X. Xi, J. Cai, H. Qi, S. Shi, J. Wang, D. Yuan and M. Fang, J. Mater. Chem., 2010, 20, 3758 RSC.
- J. Hafizovic, M. Bjørgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- T. A. Dougherty, PhD thesis, Iowa State University, 1967.
- R. E. McCarley and B. A. Torp, Inorg. Chem., 1963, 2, 540–546 CrossRef CAS.
- M. D. Korzyński, L. Braglia, E. Borfecchia, C. Lamberti and M. Dincă, Inorg. Chem., 2018, 57, 13998–14004 CrossRef PubMed.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736–1850 CrossRef CAS PubMed.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS PubMed.
- S. Yuan, L. Zou, H. Li, Y.-P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CrossRef CAS PubMed.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuño, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS PubMed.
- M. D. Korzyński, D. F. Consoli, S. Zhang, Y. Román-Leshkov and M. Dincă, J. Am. Chem. Soc., 2018, 140, 6956–6960 CrossRef PubMed.
- R. J. Holmberg, T. Burns, S. M. Greer, L. Kobera, S. A. Stoian, I. Korobkov, S. Hill, D. L. Bryce, T. K. Woo and M. Murugesu, Chem.–Eur. J., 2016, 22, 7711–7715 CrossRef CAS PubMed.
- A. Blomqvist, C. M. Araujo, P. Srepusharawoot and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20173–20176 CrossRef CAS PubMed.
- S. S. Han and W. A. Goddard, J. Am. Chem. Soc., 2007, 129, 8422–8423 CrossRef CAS PubMed.
- S. J. Kolmann, B. Chan and M. J. T. Jordan, Chem. Phys. Lett., 2008, 467, 126–130 CrossRef CAS.
- M. Dixit, T. A. Maark and S. Pal, Int. J. Hydrogen Energy, 2011, 36, 10816–10827 CrossRef CAS.
- N. T. T. Ha, O. V. Lefedova and N. N. Ha, Russ. J. Phys. Chem. A, 2016, 90, 220–225 CrossRef CAS.
- S. Chaemchuem, Z. Kui and F. Verpoort, CrystEngComm, 2016, 18, 7614–7619 RSC.
- M. S. Denny, L. R. Parent, J. P. Patterson, S. K. Meena, H. Pham, P. Abellan, Q. M. Ramasse, F. Paesani, N. C. Gianneschi and S. M. Cohen, J. Am. Chem. Soc., 2018, 140, 1348–1357 CrossRef CAS PubMed.
- C. Z. Andrade, Curr. Org. Synth., 2004, 1, 333–353 CrossRef CAS.
- T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161–5170 CrossRef CAS PubMed.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, J. Synchrotron Radiat., 2015, 22, 1548–1554 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01553a |
| ‡ Deceased February 1, 2019. |
| § Even though it has been reported that SBUs undergo cation exchange in the UiO-66 framework, it was later shown that deposition of the foreign metal oxide was instead occurring, see ref. 43. |
| This journal is © The Royal Society of Chemistry 2019 |