
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExclusive occurrence of photoinduced energy transfer and switching of its direction by rectangular π-extension of nanographenes†
Tomokazu
Umeyama
 *a,
Takuma
Hanaoka
a,
Hiroki
Yamada
a,
Yuki
Namura
a,
Satoshi
Mizuno
a,
Tomoya
Ohara
a,
Jinseok
Baek
a,
JaeHong
Park
a,
Yuta
Takano
b,
Kati
Stranius
c,
Nikolai V.
Tkachenko
*c and
Hiroshi
Imahori
*ab
*a,
Takuma
Hanaoka
a,
Hiroki
Yamada
a,
Yuki
Namura
a,
Satoshi
Mizuno
a,
Tomoya
Ohara
a,
Jinseok
Baek
a,
JaeHong
Park
a,
Yuta
Takano
b,
Kati
Stranius
c,
Nikolai V.
Tkachenko
*c and
Hiroshi
Imahori
*ab
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: umeyama@scl.kyoto-u.ac.jp; imahori@scl.kyoto-u.ac.jp
bInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
cFaculty of Engineering and Natural Sciences, Tampere University, Korkeakoulunkatu 8, 33720 Tampere, Finland. E-mail: nikolai.tkachenko@tuni.fi
First published on 5th June 2019
Abstract
As structure defined cutouts of the graphene lattice, nanographene molecules have gained plenty of attention because of their high potential for versatile applications in organic electronics and energy conversion devices and as ideal model systems for the better understanding of intrinsic structure–property correlations of graphenes. In this study, well-defined nanographenes with sp2 carbon networks of different sizes, hexa-peri-hexabenzocoronene (HBC) and its rectangularly π-extended version, a short graphene nanoribbon (GNR), have been covalently functionalized with photoactive porphyrin molecules. On the basis of their spectroscopic studies, the photodynamics of the porphyrin-linked nanographenes was found to be influenced substantially by the size of the nanographenes. Photoexcitation of the porphyrin–HBC linked system led to exclusive energy transfer (EnT) from the first singlet excited state (S1) of the nanographene to the porphyrin, whereas opposite selective EnT occurred from the first and second singlet excited states (S1 and S2) of the porphyrin to the nanographene in the porphyrin–GNR linked system. In particular, ultrafast efficient EnTs from both the S2 and S1 states of the porphyrin to GNR mimic the corresponding ultrafast EnTs from the S2 and S1 states of carotenoids to chlorophylls in light-harvesting systems of natural photosynthesis. Such unique photophysical properties will be useful for the rational design of carbon-based photofunctional nanomaterials for optoelectronics and solar energy conversion devices.
Introduction
Nanographenes, characterized by well-defined planar structures of sp2 carbon networks with sizes over 1 nm, have attracted tremendous attention,1 as their extended π-conjugation offers intriguing characteristics in optical and electronic properties, such as large extinction coefficients, high photoluminescence quantum yields, and excellent charge transport abilities.2–4 These properties suggest their potential utility as basic components in various electronic devices including light-emitting diodes, solar cells, and field-effect transistors.5–8 Along this line, nanographenes with different sizes and edge configurations have been prepared, demonstrating the possibility of fine-tuning their electronic and optoelectronic properties through structural modulation.1 Among them, hexa-peri-hexabenzocoronenes (HBCs) consisting of 42 sp2 carbons in the π-conjugated core can be regarded as the minimum unit of nanographenes.1,9 In contrast to nanographene molecules, other two-dimensional (2D) nanocarbons such as graphenes, graphene oxides (GOs), chemically converted graphenes (CCGs), and graphene quantum dots (GQDs) are structurally inhomogeneous at the molecular level, containing different π-conjugation sizes, edge structures, and defect sites.10 Nanographenes can be regarded as part of larger 2D nanocarbons, and therefore, π-extended HBC derivatives with well-defined structures are ideal model units to understand the intrinsic structure–property correlations of the 2D nanocarbons.1Covalent linking of nanographenes and photoactive molecules is an effective strategy to endow them with new functions, as well as to examine their optical and photophysical properties, without changing the nanographene core structures.11–14 Various HBC linked systems with photoactive molecules including porphyrins,11 perylene diimides,12 and fullerenes13 were prepared and their basic optical properties such as steady-state absorption and fluorescence spectra were investigated. Nevertheless, so far such covalent functionalization of nanographenes has been limited to HBCs solely, and, to the best of our knowledge, covalently linked systems of photoactive molecule–nanographenes larger than HBC have never been reported. Therefore, the size and shape effects of nanographenes on the optical and photophysical properties of nanographene–photoactive molecule linked systems have yet to be studied systematically.
To this end, we employed two nanographenes as models: the symmetrical HBC and its π-extended rectangular strip-shaped nanographene consisting of 114 sp2 carbons,15 which is a short graphene nanoribbon (GNR). They were linked with two zincporphyrin (ZnP) units through a p-phenylene bridge at the periphery positions (Scheme 1) for the better understanding of their intrinsic properties. The short, rigid phenylene spacer ensures a well-defined geometry between the nanographene and the attached porphyrins. Hereafter, the HBC and GNR reference molecules are denoted as HBC-ref and GNR-ref, and the porphyrin linked systems with HBC-ref and GNR-ref are referred to as ZnP–HBC and ZnP–GNR (Scheme 1). To improve their solubility in organic solvents, long alkyl chains were introduced into the HBC and GNR moieties. The HBC core was selected as a typical, basic example of nanographenes.1,9 Although various linked systems of HBC with photoactive molecules including porphyrins have already been reported,11–14 ZnP–HBC linked systems with a p-phenylene spacer have yet to be synthesized. Meanwhile the GNR core was chosen because of its closely related structure to HBC. GNR possesses the same size on the short axis, but a large size on the long axis compared to HBC. Furthermore, GNR and HBC have the same edge structures, which provides an opportunity to focus on the size effect of nanographenes.15a The symmetric extension of the π-conjugation network of HBC in a regular hexagon shape would be another option, but even the synthesis is a great challenge.1,4c,16 Both HBC-ref and GNR-ref possess two reactive C–H groups at less sterically hindered positions,17 which are useful for introducing the porphyrin units regioselectively (Scheme 1).
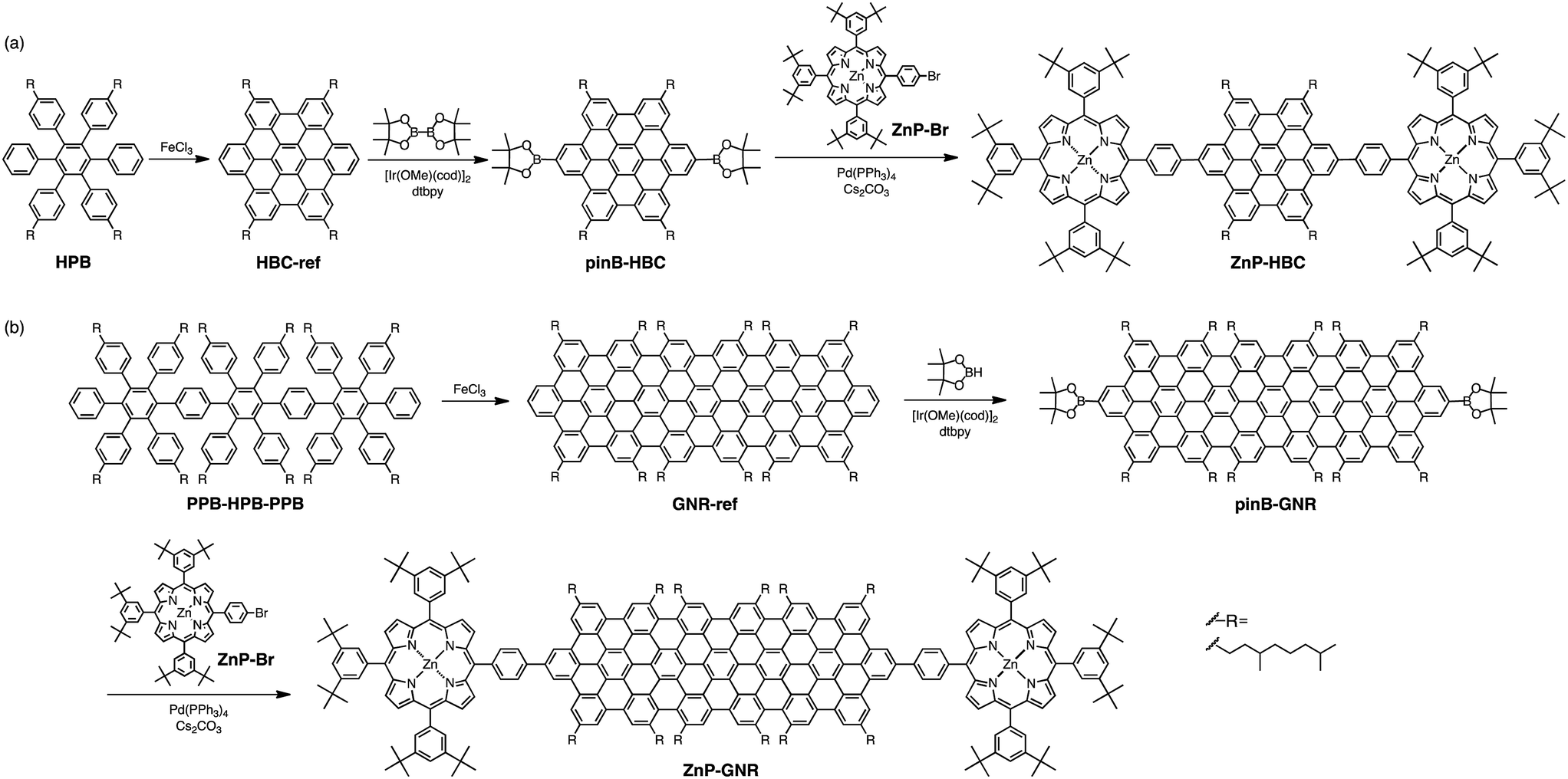 | ||
| Scheme 1 Synthetic procedures for (a) ZnP–HBC and (b) ZnP–GNR. | ||
Nanocomposites of porphyrins with 2D nanocarbons such as graphenes, GOs, CCGs, and GQDs have been synthesized because of their potential applications in photoelectrochemical devices, photocatalysts, photodynamic and photothermal therapies, and biosensors.18–21 To achieve synergistic effects of the combinations of porphyrins and 2D nanocarbons on their performances in the applications, the occurrence of efficient photoinduced energy transfer (EnT) or electron transfer (ET) from the attached porphyrins to the 2D nanocarbons or vice versa is of extreme importance. Systematic investigations on porphyrin–2D nanocarbon covalently linked systems revealed that their photodynamics is profoundly affected by the spacer structures that govern separation distances and mutual orientations between porphyrins and 2D nanocarbons.22–24 However, 2D nanocarbons in the porphyrin-linked systems reported so far consist of complicated mixtures with a range of sizes, shapes, and defects of the sp2 carbon network. In addition, the attachment positions of the porphyrins on the 2D nanocarbons, i.e., the center or the edge of the sp2 carbon network, are non-uniform. These factors may also exert a significant influence on the interaction between porphyrins and 2D nanocarbons in the excited states. In contrast to the porphyrin–2D nanocarbon linked systems, ZnP–HBC and ZnP–GNR in the present study have well-defined molecular structures. Therefore, they are excellent model systems to investigate the intrinsic photophysical properties of porphyrin–2D nanocarbon nanocomposite materials. Furthermore, the porphyrin–nanographene linked molecules themselves are excellent candidates as photoactive materials in the above practical applications. Nevertheless, the detailed photophysical properties of previously synthesized porphyrin-linked HBC molecules have not been scrutinized using time-resolved spectroscopy.11 Herein, we report the photophysical properties of ZnP–HBC and ZnP–GNR. The size increase in the nanographenes was found to switch the EnT direction together with the exclusive occurrence of photoinduced EnT.
Experimental
Instruments
NMR spectra were measured with a JEOL JNM-EX400 NMR spectrometer or JEOL JNM-AL300 NMR spectrometer. Attenuated total reflectance (ATR) FT-IR spectra were recorded on a Thermo Fisher Scientific Nicolet 6700 FT-IR. High-resolution mass spectra (HRMS) were obtained with a Thermo Fisher Scientific EXACTIVE. UV-visible absorption spectra were measured with a PerkinElmer Lambda 900 UV/vis/NIR spectrometer. Steady-state fluorescence spectra were recorded on a HORIBA SPEX Fluoromax-3 spectrofluorometer. Phosphorescence spectra were measured on a HORIBA NanoLOG-TCSPC at 77 K. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements were performed using an ALS 630A electrochemical analyzer in THF containing 0.1 M tetra-n-butylammonium hexafluorophosphate (Bu4PF6) as a supporting electrolyte. Time-correlated single photon counting (TCSPC) measurements using a HORIBA NanoLOG-TCSPC were conducted for fluorescence lifetime measurements on the nanosecond and subnanosecond time-scale. The time resolution was ca. 200 ps. Ultrafast fluorescence decays were measured using a photon up-conversion method described in detail elsewhere,25 with a temporal resolution of ca. 150 fs. In brief, fundamental pulses of 810 nm produced by a Ti:sapphire laser (TiF50, CDP-Avesta) at an 80 MHz repetition rate were split into two beams. One portion underwent second harmonic generation to excite the sample at 405 or 425 nm, generating emission. Pump–probe measurements were performed using Libra-F (Coherent) and Topas-C (Light Conversion) laser systems to produce pump and probe pulses and an ExciPro (CDP) measurement system to record the time-resolved absorption spectra.26 Typically, the time resolution of the instrument was 150 fs. Transient absorption measurements for nanoseconds to microseconds were carried out with a laser system provided by Unisoku according to the following procedure: a sample solution was excited using a Panther optical parametric oscillator pumped by a Nd:YAG laser (Continuum, Surelite SLI-10; 4–6 ns fwhm) at λ = 410 nm. The photodynamics was monitored by continuous exposure to a Xe lamp as a probe light and a photomultiplier tube (Unisoku, PMT for 440–780 nm) and a photodiode (Unisoku, NIR-PD1 for 780–1100 nm) as detectors. The output from the photodiode and photomultiplier tube was recorded with a digitizing oscilloscope (Iwatsu, Digital Oscilloscope DS-5532).Theoretical calculations
Geometry optimization and electronic structure calculations were performed using density functional theory (DFT) at the spin-restricted B3LYP/6-31G level. Calculations were carried out using the Gaussian 09 program.27 All structures were fully optimized without any symmetry restriction.Synthesis
All solvents and chemicals were of reagent grade quality, purchased and used without further purification. Synthetic details of oligo(p-phenylene)s HPB and PPB-HPB-PPB (Scheme 1) are described in the ESI.† The abbreviations of compounds are defined in Scheme 1 and the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:2) and gel permeation chromatography (toluene), yielding ZnP–HBC as a deep purple solid (2.4 mg, 7.7 × 10−4 mmol, 18%). 1H NMR (400 MHz, CDCl3, 333 K) δ 9.50 (s, 4H), 9.30–9.27 (m, 4H), 9.12–9.01 (m, 12H), 8.89 (s, 4H), 8.64 (t, J = 16.6 Hz 4H), 8.57 (s, 4H), 8.16–8.12 (m, 12H), 7.81 (s, 6H), 7.35 (d, J = 3.44 Hz 4H), 3.29 (m, 8H), 2.34–0.74 (m, 130H). IR (ATR): 2950, 2876, 1714, 1442, 1362, 1261, 1182, 1092, 1039, 921, 805 cm−1. HRMS (MALDI-TOFMS, m/z) = 3103.8039 (M˙+); calcd for C218H246N8Zn2: 3103.8078.
:hexane = 1:2) and gel permeation chromatography (toluene), yielding ZnP–GNR as a deep red solid (2.2 mg, 4.3 × 10−4 mmol, 22%). 1H NMR (400 MHz, C2D2Cl4 393 K) δ 10.01–9.62 (m, 12H), 9.21–8.95 (m, 16H), 8.62 (s, 4H), 8.08 (d, J = 15.2 Hz 12H), 7.77 (s, 10H), 7.17–7.05 (m, 8H), 3.57–3.35 (m, 24H), 2.25–0.88 (m, 336H). IR (ATR): 2950, 2932, 2919, 2859, 1717, 1617, 1554, 1456, 1363, 1260, 1086, 803 cm−1. HRMS (MALDI-TOFMS, m/z) = 5105.1846 (M˙+); calcd for C370H446N8Zn2: 5105.1850.
:hexane = 1:2) and gel permeation chromatography (toluene), yielding ZnP–HBC as a deep purple solid (2.4 mg, 7.7 × 10−4 mmol, 18%). 1H NMR (400 MHz, CDCl3, 333 K) δ 9.50 (s, 4H), 9.30–9.27 (m, 4H), 9.12–9.01 (m, 12H), 8.89 (s, 4H), 8.64 (t, J = 16.6 Hz 4H), 8.57 (s, 4H), 8.16–8.12 (m, 12H), 7.81 (s, 6H), 7.35 (d, J = 3.44 Hz 4H), 3.29 (m, 8H), 2.34–0.74 (m, 130H). IR (ATR): 2950, 2876, 1714, 1442, 1362, 1261, 1182, 1092, 1039, 921, 805 cm−1. HRMS (MALDI-TOFMS, m/z) = 3103.8039 (M˙+); calcd for C218H246N8Zn2: 3103.8078.
:hexane = 1:2) and gel permeation chromatography (toluene), yielding ZnP–GNR as a deep red solid (2.2 mg, 4.3 × 10−4 mmol, 22%). 1H NMR (400 MHz, C2D2Cl4 393 K) δ 10.01–9.62 (m, 12H), 9.21–8.95 (m, 16H), 8.62 (s, 4H), 8.08 (d, J = 15.2 Hz 12H), 7.77 (s, 10H), 7.17–7.05 (m, 8H), 3.57–3.35 (m, 24H), 2.25–0.88 (m, 336H). IR (ATR): 2950, 2932, 2919, 2859, 1717, 1617, 1554, 1456, 1363, 1260, 1086, 803 cm−1. HRMS (MALDI-TOFMS, m/z) = 5105.1846 (M˙+); calcd for C370H446N8Zn2: 5105.1850.
Results and discussion
Synthetic procedures are depicted in Scheme 1. HBC-ref and GNR-ref were prepared by a conventional intramolecular oxidation reaction of the corresponding oligo(p-phenylene)s (HPB and PPB-HPB-PPB) with FeCl3 as an oxidant. The iridium-catalyzed borylation reaction was applied to the less hindered C–H groups of HBC-ref and GNR-ref,17 and then the Suzuki coupling reactions between the bis-borylated nanographenes (pinB–HBC and pinB–GNR) and zinc-porphyrins with a p-bromophenyl group at a meso-position (ZnP–Br) were conducted to yield ZnP–HBC and ZnP–GNR, respectively. The UV-visible absorption spectra of ZnP–HBC and ZnP–GNR are largely the sum of the spectra of HBC-ref/GNR-ref and a porphyrin reference (5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)-porphyrinatozinc(II), ZnP-ref) (Fig. 1). The Soret bands of ZnP–HBC and ZnP–GNR are slightly red-shifted by ca. 2 nm compared to that of ZnP-ref. Bearing in mind that two ZnP units are present in ZnP–HBC and ZnP–GNR, the extinction coefficients (ε) at the maxima of the Soret bands are decreased by 30% and 44% with respect to ZnP-ref (Fig. S1†), indicating that the interaction between the ZnP and nanographene moieties in the ground state is moderate and more pronounced in ZnP–GNR than in ZnP–HBC due to the larger size of the GNR unit than the HBC unit.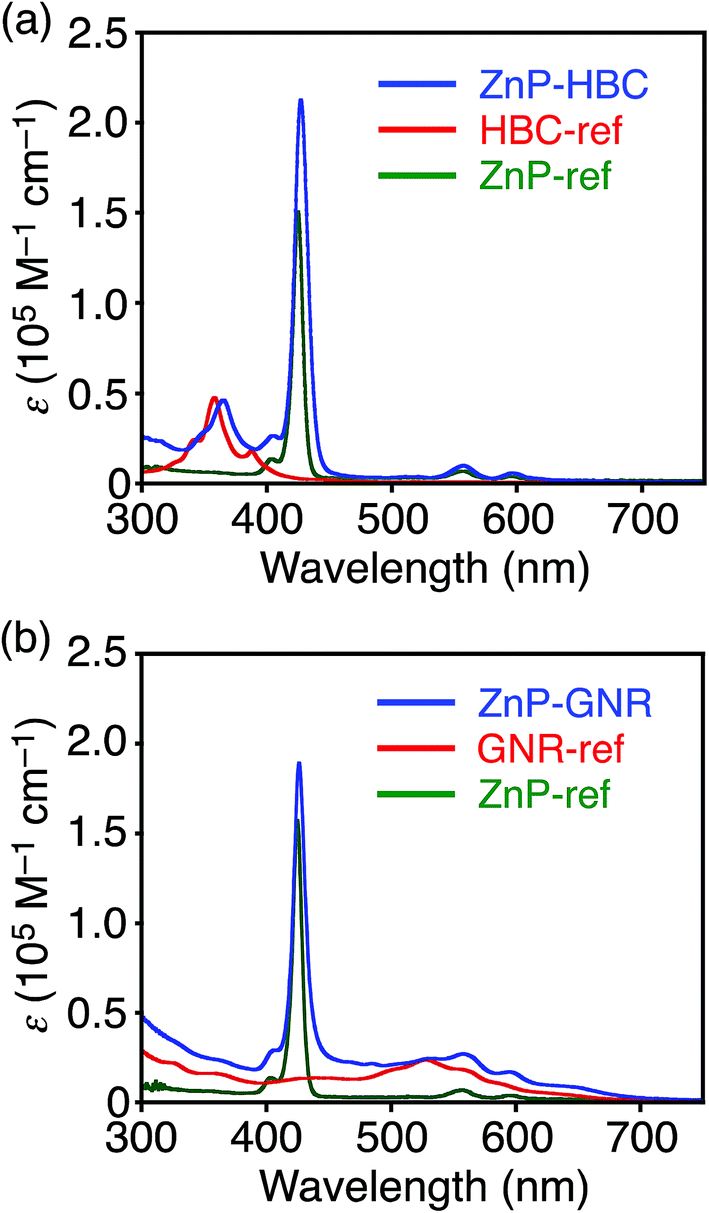 | ||
| Fig. 1 UV-visible absorption spectra of (a) ZnP–HBC (blue), HBC-ref (red), ZnP-ref (green), (b) ZnP–GNR (blue), GNR-ref (red), and ZnP-ref (green) in THF. | ||
To evaluate the interactions between ZnP and HBC units in the excited states of ZnP–HBC, we measured steady-state fluorescence spectra of ZnP–HBC and the references (Fig. 2 and S2†). When ZnP–HBC and ZnP-ref were excited at λex = 426 nm, where the absorbances were adjusted to be identical, the emission spectra showed comparable shapes and intensities (Fig. S2†). Since the HBC moiety has almost no absorbance at 426 nm (ε < 3.0 × 103 M−1 cm−1), the ZnP moieties (ε = 2.1 × 105 M−1 cm−1) in ZnP–HBC predominantly absorb the excitation light and emit with negligible interaction with the HBC moiety. In contrast, HBC-ref showed a strong emission in the region of 460–600 nm with multiple peaks (λex = 360 nm, Fig. 2). However, the fluorescence spectrum of ZnP–HBC, where the absorption ratio of ZnP and HBC is 1:4 (λex = 360 nm), exhibited two broad peaks at 604 and 656 nm (Fig. 2) with efficient quenching of the emission from the HBC unit. This spectrum matches with that of ZnP-ref. In addition, the nanosecond fluorescence lifetime (τ) of the ZnP moiety in ZnP–HBC (2.0 ns (100%)), excited mainly at the HBC moiety, was similar to that of ZnP-ref (2.1 ns (100%)), but differed significantly from those of HBC-ref (4.6 ns (8%) and 43 ns (92%))28 (Fig. S3†). These results demonstrate the occurrence of EnT from the first singlet excited state (S1) of HBC (1HBC*) to ZnP in ZnP–HBC, as reported in other porphyrin–HBC linked systems.11 The occurrence of EnT is reasonable because the optical bandgap of the HBC moiety (2.73 eV) is larger than that of the ZnP moiety (2.03 eV). Furthermore, the fluorescence intensities of ZnP–HBC and ZnP-ref with the same absorbance at λex = 360 nm are virtually the same (Fig. 2), indicating the highly efficient, exclusive EnT. In the picosecond fluorescence of the ZnP moiety in ZnP–HBC, a rise component (τ = 45 ps) was observed (Fig. S4†). This corresponds to an EnT rate constant (kEnT) of 2.2 × 1010 s−1. Given the average lifetime of 1HBC* in HBC-ref (40 ns), the quantum efficiency (φEnT) of EnT from 1HBC* to ZnP in ZnP–HBC was estimated to be unity (100%).
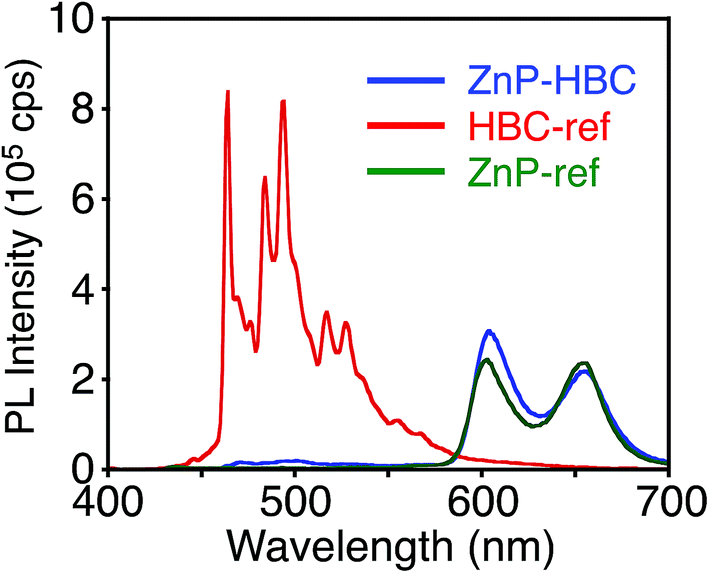 | ||
| Fig. 2 Fluorescence spectra of ZnP–HBC (blue), HBC-ref (red), and ZnP-ref (green) in THF. The λex values are 360 nm, where the absorbances are identical. | ||
DFT calculations also disclosed the larger bandgap of the HBC moiety than that of the ZnP moieties in ZnP–HBC (Fig. S5†). In addition, both the HOMO and LUMO of ZnP–HBC are distributed solely on the ZnP moieties (Fig. S5†). Considering the HOMO and LUMO energy levels experimentally estimated by electrochemical measurements and the optical bandgaps of HBC and ZnP, ET from 1HBC* to ZnP is also energetically favorable with a driving force (−ΔGET) of 0.54 eV in THF (Fig. S6†). To get a thorough insight into the interaction between HBC and ZnP in the excited states, femto- to picosecond time-resolved transient absorption (TA) spectra measurements for ZnP–HBC and HBC-ref were conducted at λex = 360 nm (Fig. 3). The TA spectrum of HBC-ref revealed a positive absorption signal with a peak around 550 nm corresponding to 1HBC* (Fig. 3b),3c which has a lifetime longer than the limit of the detection system (6 ns). Meanwhile, even though the HBC moiety in ZnP–HBC was mainly excited, the positive peak at 550 nm was not evident even at 0.2 ps delay (Fig. 3a). The TA spectra of ZnP–HBC at 0.2 and 5 ps delays are similar to the sum of the TA spectra of HBC-ref (Fig. 3b) and ZnP-ref (Fig. S7†). The TA spectral shapes at 100 ps and 2.1 ns coincide well with those of ZnP-ref, corresponding to the singlet (1ZnP*) and triplet (3ZnP*) excited states of ZnP. The characteristic TA signal for the porphyrin radical anion (ZnP˙−) at 700–750 nm (ref. 29) was absent in the TA spectrum of ZnP–HBC, implying no occurrence of ET. This result agrees with the high φEnT value of EnT from 1HBC* to ZnP as estimated by the fluorescence analysis. Four decay components were obtained from the global fitting of the TA data for ZnP–HBC (Fig. 4): the transition from the second (S2) to first (S1) singlet excited state of ZnP (τ1 = 1.3 ps), EnT from 1HBC* to ZnP (τ2 = 49 ps), the decay of 1ZnP* (τ3 = 1.7 ns), and that of 3ZnP* (τ4 > 10 ns). The kEnT value estimated by TA (2.0 × 1010 s−1) also matches well with that obtained from the fluorescence rise component (2.2 × 1010 s−1, Fig. S4†). This rapid EnT may prevail over ET from 1HBC* to ZnP to generate the HBC radical cation and ZnP˙−, although ET is also energetically favorable.
 | ||
| Fig. 3 Femto- to picosecond time-resolved TA spectra of (a) ZnP–HBC and (b) HBC-ref in THF (λex = 360 nm). The delay times of 0.2 ps to 6 ns are shown. | ||
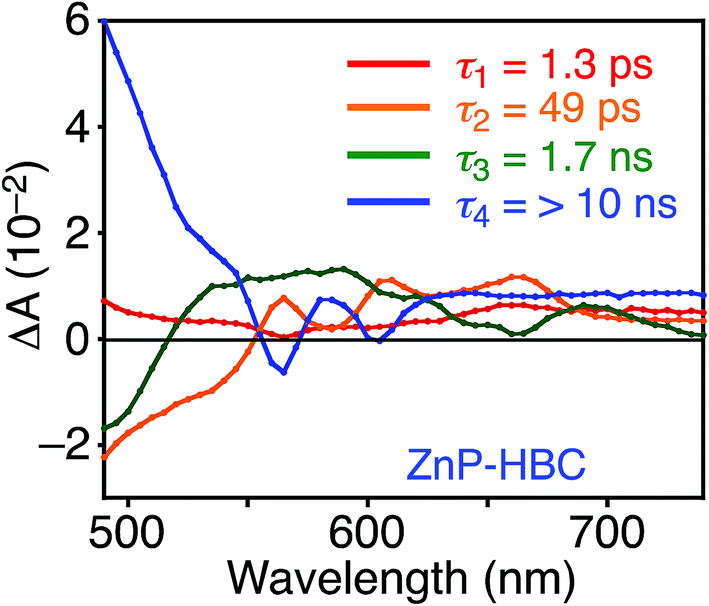 | ||
| Fig. 4 Femto- to picosecond transient absorption decay component spectra of ZnP–HBC in THF obtained with global four component fit of the data. The excitation wavelength is 360 nm. Lifetimes of respective components are given in the figures. | ||
The nano- to microsecond time-resolved TA spectrum of ZnP–HBC at 10 μs after the photoexcitation of the HBC moiety corresponded to that of 3ZnP* with a peak at 840 nm (Fig. S8†). The monoexponential decay of the TA signal without forming any subsequent TA signifies a direct relaxation of 3ZnP* to the ground state. The energy levels of 3HBC* and 3ZnP* were determined using the phosphorescence spectra in frozen 2-methyltetrahydrofuran glass at 77 K (ref. 30 and 31), and the relaxation pathways of ZnP–HBC are summarized in Fig. S9.†
Unlike ZnP–HBC, the fluorescence spectrum of ZnP–GNR with the Soret band excitation bears a remarkable resemblance to that of GNR-ref (Fig. 5). Consistently, the nanosecond fluorescence lifetimes and the component ratio of ZnP–GNR (2.7 ns (60%) and 25 ns (40%)) at λex = 416 nm (absorption ratio, ZnP:GNR = 5:1) and a detection wavelength (λobs) of 650 nm were virtually identical with those of GNR-ref (3.0 ns (64%) and 27 ns (36%)) (Fig. S10†), but were totally different from that of ZnP-ref (2.0 ns (100%)). These results indicate that the efficient EnT from 1ZnP* to GNR also takes place in ZnP–GNR, which is in the opposite direction from that observed in ZnP–HBC and other porphyrin-linked HBCs.11 To the best of our knowledge, this is the first example of well-defined nanographene as an energy acceptor in covalently linked donor–acceptor systems. The reverse EnT results from the narrowed optical bandgap of the GNR moiety (1.84 eV) arising from the rectangularly extended π-conjugation from HBC. A clear rise component with τ = 0.2 ps was detected in the picosecond fluorescence profile of ZnP–GNR at λex = 425 nm and λobs = 650 nm, where the absorption ratio of ZnP and GNR is 6:1 (Fig. S11†). This corresponds to a kEnT of 5 × 1012 s−1, which is much larger than the rate of EnT from 1HBC* to ZnP (kEnT of 2.2 × 1010 s−1, Fig. S4†). This ultrafast EnT indicates the violation of the Kasha's rule; EnT takes place mainly from the S2 state of 1ZnP* to GNR, taking into account the internal conversion rate constant of S2 to S1 of 1ZnP* (6.7 × 1011 s−1, Fig. S7b†). Although ultrafast ET from the S2 state of 1ZnP* to an acceptor in donor–acceptor linked systems has been frequently observed,22a,32 the corresponding ultrafast EnT from the S2 state of 1ZnP* to an acceptor is rare.33 Meanwhile, in natural light-harvesting systems ultrafast EnT starting from carotenoids to chlorophylls is essential for efficient light-harvesting and subsequent charge separation.34
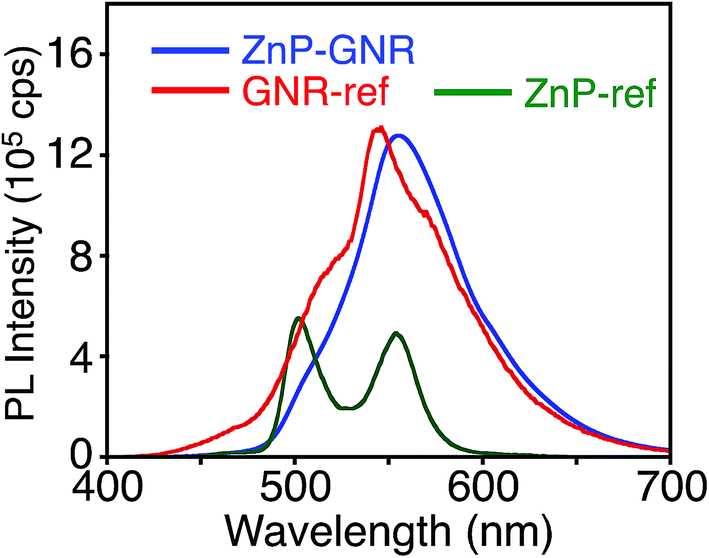 | ||
| Fig. 5 Fluorescence spectra of ZnP–GNR (blue), GNR-ref (red), and ZnP-ref (green) in THF. The λex values are 422 nm, where the absorbances are identical, respectively. | ||
A DFT calculation provided an insight into the electronic state of ZnP–GNR, which revealed that the HOMO and LUMO are localized on the GNR moiety (Fig. S5†). This is in sharp contrast to ZnP–HBC, where the HOMO and LUMO are localized on the ZnP moieties (Fig. S5†). Considering the experimentally estimated HOMO and LUMO energy levels and optical bandgaps of the GNR and ZnP moieties, ETs from the singlet excited state of GNR (1GNR*) to ZnP (−ΔGET = 0.04 eV in THF) and from GNR to 1ZnP* (−ΔGET = 0.23 eV in THF) as well as EnT from 1ZnP* to GNR are energetically possible by the excitation of ZnP–GNR (Fig. S12†). To shed light on the possible ET and EnT processes, the TA spectra of ZnP–GNR and GNR-ref were measured at λex = 425 nm (Fig. 6). In contrast to HBC-ref, the TA spectra of GNR-ref did not show clear positive signals in the visible region, but exhibited a negative peak at 530 nm derived from the ground state absorption (Fig. 6b). Meanwhile, the TA spectra of ZnP–GNR largely resembled those of GNR-ref with a few exceptions (Fig. 6a). The TA spectrum of ZnP–GNR at 0.2 ps has features similar to that of ZnP-ref and GNR-ref at 0.2 ps (Fig. S7†), implying the formation of the S2 state of 1ZnP* and 1GNR*. The TA spectra at 5 ps and 100 ps exhibited a negative peak at 655 nm, which is absent in the TA spectra of GNR-ref. Four components with τ1 = 0.2 ps, τ2 = 7.0 ps, τ3 = 3.2 ns, and τ4 > 10 ns were extracted from the TA spectra of ZnP–GNR (Fig. 7). No characteristic signals of ZnP˙− (ref. 29) were observed, supporting no occurrence of ET from GNR to ZnP.
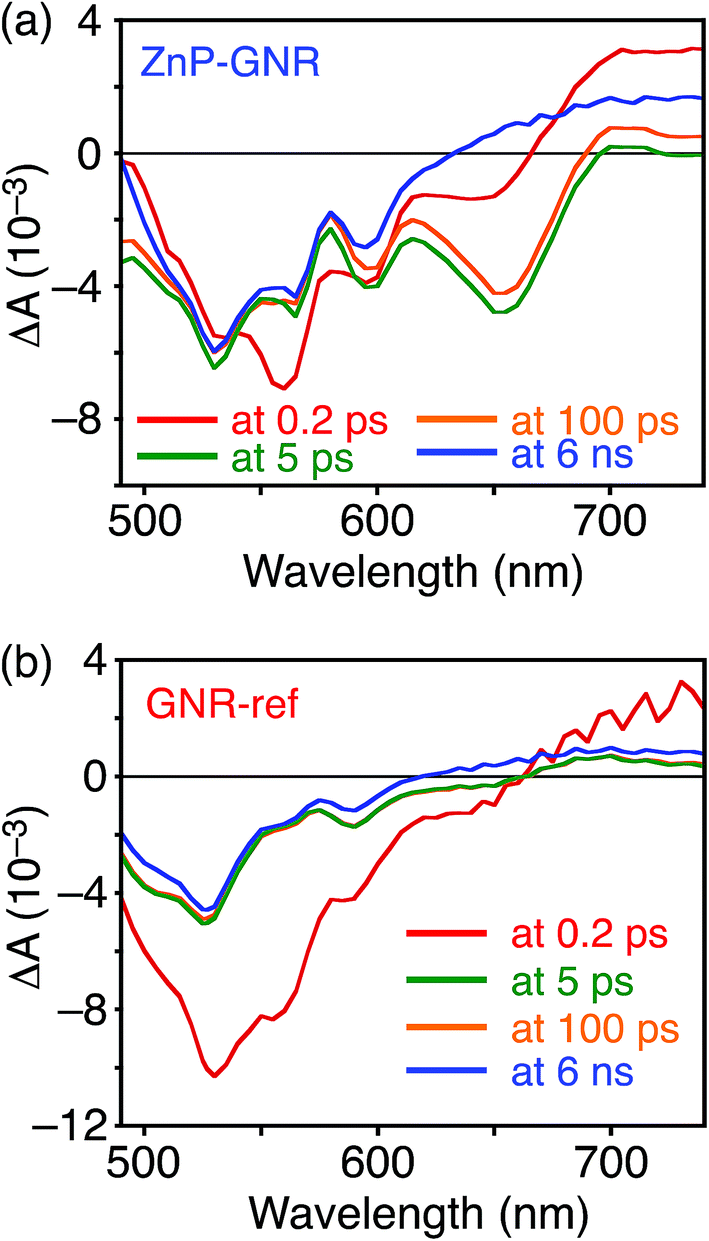 | ||
| Fig. 6 Femto- to picosecond time-resolved TA spectra of (a) ZnP–GNR and (b) GNR-ref in THF (λex = 425 nm). The delay times of 0.2 ps to 6 ns are shown. | ||
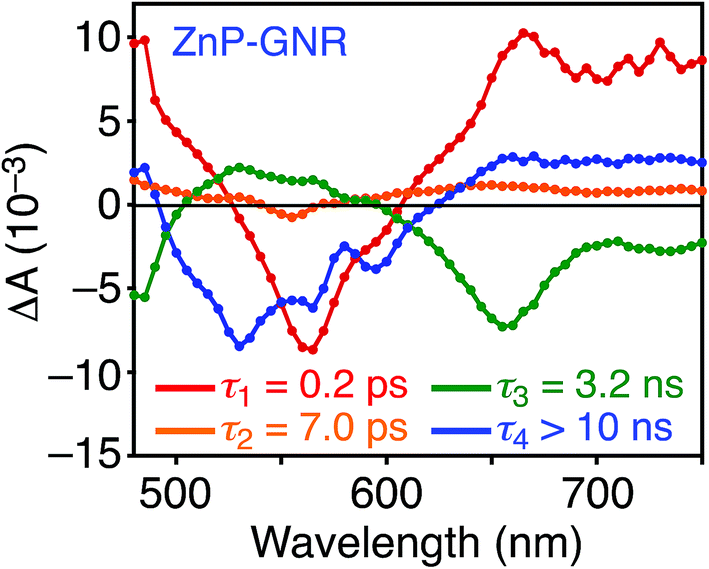 | ||
| Fig. 7 Transient absorption decay component spectra of ZnP–GNR in THF obtained with global four component fit of the data. The excitation wavelength is 425 nm. Lifetimes of respective components are given in the figures. | ||
The first TA decay component (τ1 = 0.2 ps) corresponds to the rise component (τ = 0.2 ps) in the picosecond fluorescence decay, which reflects the ultrafast EnT from the S2 state of 1ZnP* to GNR. Given the good agreement between the second components of the fluorescence lifetime (τ2 = 6.5 ps, Fig. S11†) and TA (τ2 = 7.0 ps, Fig. 7), EnT from the S1 state of 1ZnP* to GNR also occurs with kEnT = 1.5 × 1011 s−1. The overall φEnT of EnT from 1ZnP* (S2 and S1) to GNR in ZnP–GNR is unity considering the ultrafast EnTs, exceeding the energetically favorable ET from 1ZnP* to GNR (−ΔGET = 0.23 eV). ET from 1GNR* to ZnP was also absent probably due to the insufficient driving force (−ΔGET = 0.04 eV). The negative peak at 655 nm in the second decay component spectrum of ZnP–GNR is presumably ascribed to the bleaching of the ground state absorption of the GNR moiety in ZnP–GNR, thereby supporting the formation of 1GNR*. Indeed, the corresponding ground-state absorption at 655 nm is enhanced in ZnP–GNR relative to that in GNR-ref (Fig. S13†). The peripheral functionalization of the GNR unit with ZnP through the p-phenylene bridge may influence the electronic state of the GNR edge structure, causing the difference in the absorption behaviors of ZnP–GNR and GNR-ref. Nevertheless, the TA spectrum of ZnP–GNR at 6 ns is almost identical with that of GNR-ref at 6 ns (Fig. 6), suggesting the exclusive formation of the excited state of GNR by the excitation of ZnP–GNR on this time scale. The third and fourth components of transient absorption decay of ZnP–GNR (Fig. 7) correspond to the decays of 1GNR*. Consistently, the first components (τ = 2.7–3.0 ns) of the nanosecond fluorescence decays of ZnP–GNR and GNR-ref (Fig. S10†) match well with the third decay component of TA of ZnP–GNR (Fig. 7). Nano- to microsecond resolved TA spectra of ZnP–GNR and GNR-ref revealed the direct relaxation of the triplet excited state of GNR (3GNR*) to the ground state (Fig. S14†). Although the energy level of 3GNR* remains obscure owing to the absence of the phosphorescence signal of GNR-ref at 77 K, the relaxation pathways of ZnP–GNR are illustrated in Fig. S15.†
It is worthwhile to compare the photodynamics of porphyrin-linked graphenes and nanographenes. We previously reported ZnP-linked CCG with a p-phenylene bridge (ZnP–CCG),24 which showed entirely different photodynamic behavior from ZnP–HBC and ZnP–GNR. The photoexcitation of the porphyrin moiety in ZnP–CCG yielded an exciplex state, i.e., a mixed state of the locally excited states (1ZnP* and 1CCG*) and charge-separated state (ZnP˙+–CCG˙−), which rapidly decayed to the ground state. EnT from 1ZnP* to CCG as well as complete ET did not occur in ZnP–CCG.24 A stronger electronic coupling between ZnP and large graphenes than between ZnP and small nanographenes may facilitate the formation of the exciplex state in ZnP–CCG.22,24 It is noteworthy that the ZnP groups stand on a graphene plane with a large tilt angle (ca. 49°) in ZnP–CCG, whereas the ZnP moieties are attached to the nanographene at the periphery positions in ZnP–HBC and ZnP–GNR. The difference in the mutual orientation of ZnP and graphenes or nanographenes may have an impact on the electronic coupling and dipole moment interaction and thereby the photodynamics.
Conclusion
In summary, porphyrins were covalently linked with well-defined nanographenes as graphene models at the periphery through a short, rigid p-phenylene spacer. Photoexcitation of the ZnP–HBC linked system led to exclusive EnT from the HBC first excited singlet state to ZnP, whereas opposite selective EnT occurred from the ZnP second and first excited singlet states (S2 and S1) to GNR in the ZnP–GNR linked system. In particular, the ultrafast EnTs took place from both the S2 and S1 states of ZnP, mimicking efficient light-harvesting and EnT from the S2 and S1 states of carotenoids to chlorophylls in natural photosynthesis. The size of the π-conjugation system in nanographenes influences the EnT direction with maintaining exclusive occurrence of EnT, although the reason for selective occurrence of EnT is unclear at this stage. In contrast to the covalently linked systems of ZnP and large, ununiform graphenes with a p-phenylene bridge, charge-separated and exciplex states were not formed in the present systems probably owing to the relatively weak electronic coupling between ZnP and nanographenes. The results demonstrate that the photophysical properties of photoactive molecule–graphene composites can be tuned by altering the size of the sp2 carbon network in graphenes and the attachment orientation of the photoactive molecule to the graphene plane. Such unique photophysical properties will be useful for their future practical applications in optoelectronic devices, photocatalysts, and biosensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant Number 18H03898).Notes and references
- (a) L. Chen, Y. Hernandez, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2012, 51, 7640 CrossRef CAS PubMed; (b) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef CAS PubMed; (c) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (d) Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS; (e) X.-Y. Wang, A. Narita and K. Müllen, Nat. Rev. Chem., 2017, 2, 0100 CrossRef.
- (a) X. Yan, X. Cui, B. Li and L.-s. Li, Nano Lett., 2010, 10, 1869 CrossRef CAS PubMed; (b) X. Yan, B. Li, X. Cui, Q. Wei, K. Tajima and L.-s. Li, J. Phys. Chem. Lett., 2011, 2, 1119 CrossRef CAS PubMed; (c) X. Yan, B. Li and L.-s. Li, Acc. Chem. Res., 2013, 46, 2254 CrossRef CAS PubMed.
- (a) M. L. Mueller, X. Yan, J. A. McGuire and L.-s. Li, Nano Lett., 2010, 10, 2679 CrossRef CAS PubMed; (b) L. Cao, M. J. Meziani, S. Sahu and Y.-P. Sun, Acc. Chem. Res., 2013, 46, 171 CrossRef CAS PubMed; (c) S. Zhu, L. Wang, B. Li, Y. Song, X. Zhao, G. Zhang, S. Zhang, S. Lu, J. Zhang, H. Wang, H. Sun and B. Yang, Carbon, 2014, 77, 462 CrossRef CAS; (d) M. Daigle, A. Picard-Lafond, E. Soligo and J.-F. Morin, Angew. Chem., Int. Ed., 2016, 55, 2042 CrossRef CAS PubMed; (e) H. Hayashi, N. Aratani and H. Yamada, Chem.–Eur. J., 2017, 23, 7000 CrossRef CAS PubMed.
- (a) A. M. van de Craats, J. M. Warman, A. Fechtenkötter, J. D. Brand, M. A. Harbison and K. Müllen, Adv. Mater., 1999, 11, 1469 CrossRef CAS; (b) M. G. Debije, J. Piris, M. P. de Haas, J. M. Warman, Ž. Tomović, C. D. Simpson, M. D. Watson and K. Müllen, J. Am. Chem. Soc., 2004, 126, 4641 CrossRef CAS PubMed; (c) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed.
- (a) C. Soldano, A. Mahmood and E. Dujardin, Carbon, 2010, 48, 2127 CrossRef CAS; (b) X. Li, M. Rui, J. Song, Z. Shen and H. Zeng, Adv. Funct. Mater., 2015, 25, 4929 CrossRef CAS.
- V. V. Jarikov, J. Appl. Phys., 2006, 100, 014901 CrossRef.
- (a) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS PubMed; (b) S. J. Kang, S. Ahn, J. B. Kim, C. Schenck, A. M. Hiszpanski, S. Oh, T. Schiros, Y.-L. Loo and C. Nuckolls, J. Am. Chem. Soc., 2013, 135, 2207 CrossRef CAS PubMed; (c) J. Cao, Y.-M. Liu, X. Jing, J. Yin, J. Li, B. Xu, Y.-Z. Tan and N. Zheng, J. Am. Chem. Soc., 2015, 137, 10914 CrossRef CAS PubMed; (d) K. P. Loh, S. W. Tong and J. Wu, J. Am. Chem. Soc., 2016, 138, 1095 CrossRef CAS PubMed.
- (a) W. Pisula, A. Menon, M. Stepputat, I. Lieberwirth, U. Kolb, A. Tracz, H. Sirringhaus, T. Pakula and K. Müllen, Adv. Mater., 2005, 17, 684 CrossRef CAS; (b) I. O. Shklyarevskiy, P. Jonkheijm, N. Stutzmann, D. Wasserberg, H. J. Wondergem, P. C. M. Christianen, A. P. H. J. Schenning, D. M. de Leeuw, Ž. Tomović, J. Wu, K. Müllen and J. C. Maan, J. Am. Chem. Soc., 2005, 127, 16233 CrossRef CAS PubMed; (c) A. N. Abbas, B. Liu, A. Narita, L. F. Dössel, B. Yang, W. Zhang, J. Tang, K. L. Wang, H. J. Räder, X. Feng, K. Müllen and C. Zhou, J. Am. Chem. Soc., 2015, 137, 4453 CrossRef CAS PubMed.
- (a) J. Wu, M. D. Watson and K. Müllen, Angew. Chem., Int. Ed., 2003, 42, 5329 CrossRef CAS PubMed; (b) H. Seyler, B. Purushothaman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Pure Appl. Chem., 2012, 84, 1047 CAS.
- (a) M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed; (b) K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192 CrossRef CAS PubMed; (c) D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC; (d) S. Gambhir, R. Jalili, D. L. Officer and G. G. Wallace, NPG Asia Mater., 2015, 7, e186 CrossRef CAS; (e) S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS; (f) J. Shen, Y. Zhu, X. Yang and C. Li, Chem. Commun., 2012, 48, 3686 RSC.
- (a) W. W. H. Wong, T. Khoury, D. Vak, C. Yan, D. J. Jones, M. J. Crossley and A. B. Holmes, J. Mater. Chem., 2010, 20, 7005 RSC; (b) J. M. Englert, J. Malig, A. V. Zamolo, A. Hirsch and N. Jux, Chem. Commun., 2013, 49, 4827 RSC; (c) D. Lungerich, J. F. Hitzenberger, M. Marcia, F. Hampel, T. Drewello and N. Jux, Angew. Chem., Int. Ed., 2014, 53, 12231 CrossRef CAS PubMed; (d) D. Lungerich, J. F. Hitzenberger, W. Donaubauer, T. Drewello and N. Jux, Chem.–Eur. J., 2016, 22, 16755 CrossRef CAS PubMed; (e) B. Limburg, J. O. Thomas, G. Holloway, H. Sadeghi, S. Sangtarash, I. C.-Y. Hou, J. Cremers, A. Narita, K. Müllen, C. J. Lambert, G. A. D. Briggs, J. A. Mol and H. L. Anderson, Adv. Funct. Mater., 2018, 28, 180326 CrossRef; (f) D. Lungerich, J. F. Hitzenberger, F. Hampel, T. Drewello and N. Jux, Chem.–Eur. J., 2018, 24, 15818 CrossRef CAS PubMed.
- (a) J. Wu, J. Qu, N. Tchebotareva and K. Müllen, Tetrahedron Lett., 2005, 46, 1565 CrossRef CAS; (b) L. F. Dössel, V. Kamm, I. A. Howard, F. Laquai, W. Pisula, X. Feng, C. Li, M. Takase, T. Kudernac, S. D. Feyter and K. Müllen, J. Am. Chem. Soc., 2012, 134, 5876 CrossRef PubMed.
- (a) W. W. H. Wong, D. Vak, T. B. Singh, S. Ren, C. Yan, D. J. Jones, I. I. Liaw, R. N. Lamb and A. B. Holmes, Org. Lett., 2010, 12, 5000 CrossRef CAS PubMed; (b) A. Kratzer, J. M. Englert, D. Lungerich, F. W. Heinemann, N. Jux and A. Hirsch, Faraday Discuss., 2014, 173, 297 CAS.
- A. Keerthi, I. C.-Y. Hou, T. Marszalek, W. Pisula, M. Baumgarten and A. Narita, Chem.–Asian J., 2016, 11, 2710 CrossRef CAS PubMed.
- (a) T. Böhme, C. D. Simpson, K. Müllen and J. P. Rabe, Chem.–Eur. J., 2007, 13, 7349 CrossRef PubMed; (b) X. Yang, X. Dou, A. Rouhanipour, L. Zhi, J. H. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216 CrossRef CAS PubMed.
- (a) C. D. Simpson, J. D. Brand, A. J. Berresheim, L. Przybilla, H. J Räder and K. Müllen, Chem.–Eur. J., 2002, 8, 1424 CrossRef CAS PubMed; (b) K. Müllen, ACS Nano, 2014, 8, 6531 CrossRef PubMed.
- R. Yamaguchi, S. Hiroto and H. Shinokubo, Org. Lett., 2012, 14, 2472 CrossRef CAS PubMed.
- (a) Y. Xu, Z. Liu, X. Zhang, Y. Wang, J. Tian, Y. Huang, Y. Ma, X. Zhang and Y. Chen, Adv. Mater., 2009, 21, 1275 CrossRef CAS; (b) Z.-B. Liu, Y.-F. Xu, X.-Y. Zhang, X.-L. Zhang, Y.-S. Chen and J.-G. Tian, J. Phys. Chem. B, 2009, 113, 9681 CrossRef CAS PubMed.
- (a) P. Guo, P. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 5336 CrossRef CAS PubMed; (b) S. K. Das, C. B. Kc, K. Ohkubo, Y. Yamada, S. Fukuzumi and F. D'Souza, Chem. Commun., 2013, 49, 2013 RSC; (c) S. Su, J. Wang, E. Vargas, J. Wei, R. Martinez-Zaguilan, S. R. Sennoune, M. L. Pantoya, S. Wang, J. Chaudhuri and J. Qiu, ACS Biomater. Sci. Eng., 2016, 2, 1357 CrossRef CAS.
- (a) Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490 CrossRef CAS PubMed; (b) A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697 CrossRef CAS PubMed; (c) M. Jahan, O. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707 CrossRef CAS PubMed.
- (a) Q. Lu, Y. Zhang and S. Liu, J. Mater. Chem. A, 2015, 3, 8552 RSC; (b) Y. Liu, S. Li, K. Li, Y. Zheng, M. Zhang, C. Cai, C. Yu, Y. Zhou and D. Yan, Chem. Commun., 2016, 52, 9394 RSC; (c) Y. Cao, H. Dong, Z. Yang, X. Zhong, Y. Chen, W. Dai and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 159 CrossRef CAS PubMed.
- (a) T. Umeyama and H. Imahori, J. Phys. Chem. C, 2013, 117, 3195 CrossRef CAS; (b) T. Umeyama and H. Imahori, Nanoscale Horiz., 2018, 3, 352 RSC.
- (a) N. Karousis, A. S. D. Sandanayaka, T. Hasobe, S. P. Economopoulos, E. Sarantopouloua and N. Tagmatarchis, J. Mater. Chem., 2011, 21, 109 RSC; (b) H.-X. Wang, K.-G. Zhou, Y.-L. Xie, J. Zeng, N.-N. Chai, J. Li and H.-L. Zhang, Chem. Commun., 2011, 47, 5747 RSC; (c) M.-E. Ragoussi, G. Katsukis, A. Roth, J. Malig, G. de la Torre, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2014, 136, 4593 CrossRef CAS PubMed.
- (a) T. Umeyama, J. Mihara, N. Tezuka, Y. Matano, K. Stranius, V. Chukharev, N. V. Tkachenko, H. Lemmetyinen, K. Noda, K. Matsushige, T. Shishido, Z. Liu, K. Hirose-Takai, K. Suenaga and H. Imahori, Chem.–Eur. J., 2012, 18, 4250 CrossRef CAS; (b) T. Umeyama, T. Hanaoka, J. Baek, T. Higashino, F. Abou-Chahine, N. V. Tkachenko and H. Imahori, J. Phys. Chem. C, 2016, 120, 28337 CrossRef CAS.
- N. V. Tkachenko, L. Rantala, A. Y. Tauber, J. Helaja, P. H. Hynninen and H. Lemmetyinen, J. Am. Chem. Soc., 1999, 121, 9378 CrossRef CAS.
- D. Sirbu, C. Turta, A. C. Benniston, F. Abou-Chahine, H. Lemmetyinen, N. V. Tkachenko, C. Wood and E. Gibson, RSC Adv., 2014, 4, 22733 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- K.-Y. Kim, S. Liu, M. E. Köse and K. S. Schanze, Inorg. Chem., 2006, 45, 2509 CrossRef CAS PubMed.
- J. G. Lanese and G. S. Wilson, J. Electrochem. Soc., 1972, 119, 1039 CrossRef CAS.
- B. E. Hamaoui, F. Laquai, S. Baluschev, J. Wu and K. Müllen, Synth. Met., 2006, 156, 1182 CrossRef.
- S. K. Das, B. Song, A. Mahler, V. N. Nesterov, A. K. Wilson, O. Ito and F. D'Souza, J. Phys. Chem. C, 2014, 118, 3994 CrossRef CAS.
- (a) M. Andersson, J. Davidsson and L. Hammarström, J. Phys. Chem. B, 1999, 103, 3258 CrossRef CAS; (b) N. Mataga, H. Chosrowjan, Y. Shibata, N. Yoshida, A. Osuka, T. Kikuzawa and T. Okada, J. Am. Chem. Soc., 2001, 123, 12422 CrossRef CAS; (c) N. V. Tkachenko, H. Lemmetyinen, J. Sonoda, K. Ohkubo, T. Sato, H. Imahori and S. Fukuzumi, J. Phys. Chem. A, 2003, 107, 8834 CrossRef CAS; (d) R. T. Hayes, C. J. Walsh and M. R. Wasielewski, J. Phys. Chem. A, 2004, 108, 2375 CrossRef CAS; (e) K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, J. Phys. Chem. C, 2009, 113, 11667 CrossRef CAS.
- T. Kesti, N. Tkachenko, H. Yamada, H. Imahori, S. Fukuzumi and H. Lemmetyinen, Photochem. Photobiol. Sci., 2003, 2, 251 RSC.
- (a) R. Croce, M. G. Müller, R. Bassi and A. R. Holzwarth, Biophys. J., 2001, 80, 901 CrossRef CAS PubMed; (b) C. C. Gradinaru, I. H. M. van Stokkum, A. A. Pascal, R. van Grondelle and H. van Amerongen, J. Phys. Chem. B, 2000, 104, 9330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01538h |
| This journal is © The Royal Society of Chemistry 2019 |