
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic liquid cations as methylation agent for extremely weak chalcogenido metalate nucleophiles†
Bertram
Peters
a,
Silke
Santner
a,
Carsten
Donsbach
a,
Pascal
Vöpel
b,
Bernd
Smarsly
 b and
Stefanie
Dehnen
*a
b and
Stefanie
Dehnen
*a
aFachbereich Chemie, Wissenschaftliches Zentrum für Materialwissenschaften (WZMW), Philipps-Universität Marburg, Hans-Meerwein-Straße 4, 35043 Marburg, Germany. E-mail: dehnen@chemie.uni-marburg.de
bPhysikalisch-Chemisches Institut, Justus-Liebig-Universität Gießen, Heinrich-Buff-Ring 17, 35392 Gießen, Germany
First published on 22nd April 2019
Abstract
Selective in situ methylation of terminal chalcogenide ligands of molecular chalcogenido metalate anions in ionothermal reactions with alkylimidazolium-based ionic liquids yields a series of organo-functionalized chalcogenido metalate compounds. We present the syntheses and crystal structures of (C4C1C1Im)4+x[Sn10S16O4(SMe)4][An]x (1a–1f), (dmmpH)6[Mn4Sn4Se13(SeMe)4] (2), and (CnC1Im)6[Hg6Te10(TeMe)2] (3a, 3b). The methylation was confirmed by Raman spectroscopy, and the optical absorption properties of the methylated compounds were determined and compared to purely inorganic analogs.
Introduction
Chalcogenido metalate clusters represent low-dimensional fragments of metal chalcogenide semiconductor materials, which can resemble exact extracts of these or may exhibit very different architectures.1–5 Such units are intensively investigated for a variety of potential uses, such as for opto-electronic applications,6 molecular trapping7 and photocatalytic hydrogen activation,8 or for selective ion capture and exchange.9 Owing to the high anionic charge of discrete clusters, or the polymeric nature of cluster networks (of lower charge per cluster unit), these compounds form solid salts-like structures. The transfer of these highly anionic metal chalcogenide species into lower-charged derivatives is highly desired for eventually enhancing the solubility of such semiconductor-derived molecular units. However, in order to maintain the opto-electronic properties, it is necessary to selectively alkylate selected chalcogenide ligands only. This cannot be realized selectively by conducting the synthesis with chalcogenolate sources, which leads to fully ligands-decorated clusters.10–14 Owing to the extremely low nucleophilicity of the terminal chalcogenide ligands of such clusters, it has also not been possible so far to introduce organic ligands post-synthetically by means of common alkylation reagents.Ionic liquids, although regarded as relatively inert, “green” solvents that usually behave extremely indifferent in many types of reactions, have shown in recent years to be reactive to some extent, particularly towards chalcogenides.4 Besides the well-known incorporation of ionic liquid cations as a whole,15–21 methylation of amines22 was observed upon their addition as auxiliary agents for controlling the basicity of the reaction medium or as structure-directing agents.23 Recently, we have furthermore reported the distinct influence of the ionic liquid anion on the course of the reactions, though the incorporation of the anions in the finally isolated reaction products has remained rare so far.24
These results prompted us to systematically investigate their potential use as reactive solvents for the formation of organic derivatives of chalcogenido metalate anions, also extended to still rarely studied class of corresponding sulfides and tellurides for exploring the effect of different nucleophilicities. These investigations demonstrate that ionic liquids can be purposefully used for the formation of methyl derivatives of sulfido, selenido, and tellurido metalate cluster anions.
Treatment of the chalcogenido metalate salts Li4[SnS4]·13H2O,25 Na4[SnS4]·14H2O,26 K2[Sn2Se5],27 and Na2[HgTe2],28 respectively, in ionic liquids of the type (Cm(Cn)C1Im)[BF4] and (Cm(Cn)C1Im)Cl‡ afforded nine new salts comprising molecular chalcogenido metalate anions: [Sn10S16O4(SMe)4]4− (in 1a–1f), [Mn4Sn4Se13(SeMe)4]6− (in 2), and [Hg6Te10(TeMe)2]6− (in 3a and 3b). Scheme 1 summarizes the syntheses of compounds 1–3.
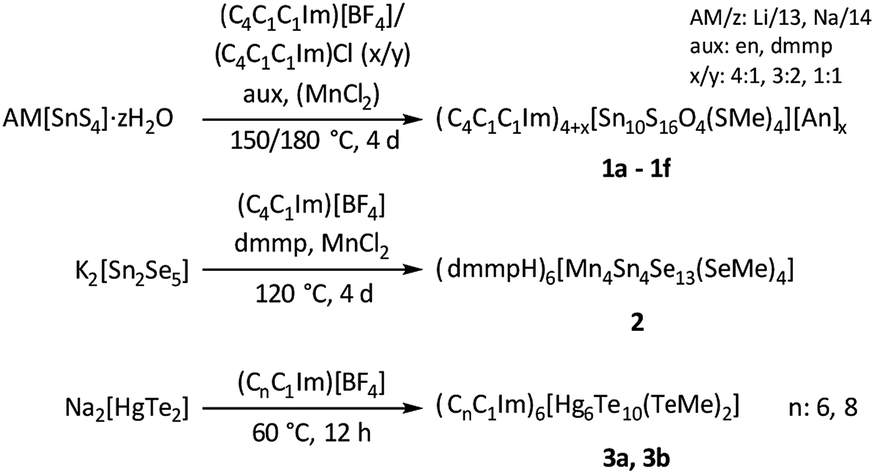 | ||
| Scheme 1 Overview of the ionothermal synthesis of salts comprising different methylated chalcogenido metalate anions [MxTMyEz]q− in 1–3 (M = Sn, E = O, S; M = Sn, TM = Mn, E = Se; M = Hg, E = Te), performed in imidazolium-based ionic liquids with different substitution pattern.‡ The scheme indicates the respective reaction conditions, that is, the presence of auxiliaries (aux) and/or additives (TMCl2), temperatures (T), and reaction durations (t).§ | ||
The title compounds are exceptional in several ways. First, they belong to the rare examples of molecular cluster anions obtained from ionic liquids. Second, the anion in 1a–1f comprises the first known sulfido stannate anions synthesized by ionothermal treatment, and with this contributes to the still small collection of sulfido metalate anions obtained via this route. Third, the anion in 2 represents the first ternary TM/Sn/E anion (TM = transition metal) that was formed in ionic liquids and not in solution or by flux techniques. Finally, the anion in 3a and 3b belongs to the rare family of molecular Hg/Te clusters, and it adopts a so far unknown composition and architecture. The most important aspect of all of the title compounds, however, is the presence of the terminal methyl groups. Based on unsuccessful experiments in solution using common alkylation reagents such as alkyl halides, it was previously assumed that such cluster anions are generally not nucleophilic enough for being alkylated. However, our new results indicate that this seems not to be the case under ionothermal conditions, which allows a methylation of the terminal S, Se, and Te atoms.
The only reasonable source of the Me groups in all of these compounds is the ionic liquid cation, although it has not been possible to identify the unmethylated cations within the large excess of original cations by spectroscopic methods of within the crystal structures. Previously, the transfer of one Me group onto a molecule of 1,2-diaminoethane was reported.22 In another case, the de-methylated fragment of an imidazolium cation was found to coordinate Ga or In atoms in cluster molecules.21 Hence, the fragmentation of the imidazolium cations has been described, yet the methylation of chalcogen atoms has not been observed so far. Below, we describe the structures of the new chalcogenido metalate anions along with some further properties of the title compounds.
The six compounds obtained from the sulfido stannate precursor are summarized as (C4C1C1Im)4+x[Sn10S16O4(SMe)4][An]x (1a–1f). All of them contain the unprecedentedly methylated sulfido–oxido stannate cluster [Sn10S16O4(SMe)4]4−, shown in Fig. 1 for the anion in 1b as an example, yet embedded in different crystal structures.
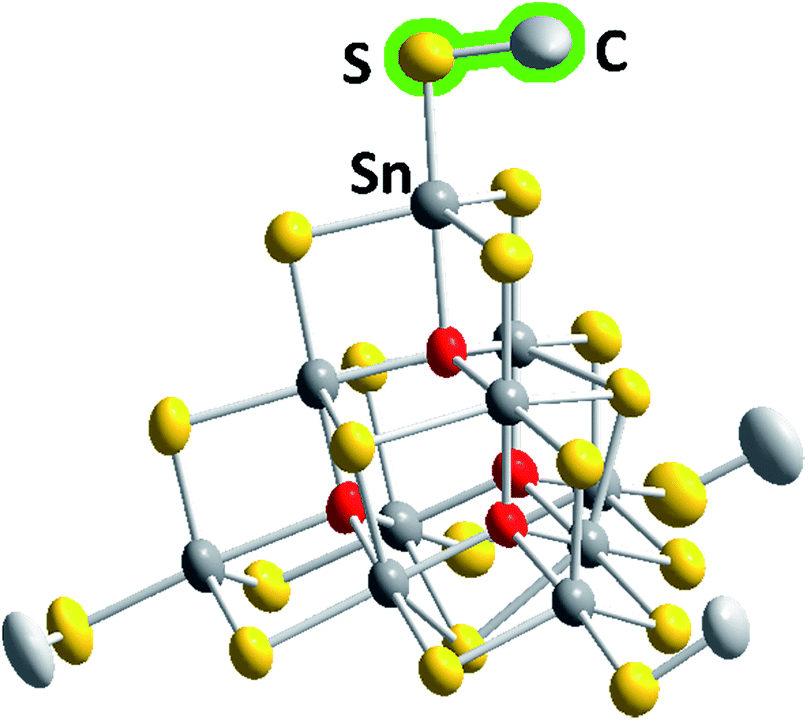 | ||
| Fig. 1 Molecular structure of the cluster anion in 1b. Thermal ellipsoids are drawn at the 50% probability level; hydrogen atoms are omitted for clarity. Color code: grey – Sn, yellow – S, red – O, light grey – C. More structural details are provided in the ESI.† | ||
The inorganic cluster core represents the “oxide-filled” T3-type supertetrahedron, [Sn10S20O4]8−,29,30 the molecular symmetry of which is reduced by the methyl substituents. However, while this does not generally cause lower crystallographic symmetry, the respective hexagonal or cubic structures possess large unit cell volumes of up to approximately 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å3 with up to 136 molecules per unit cell (see Table 1).
000 Å3 with up to 136 molecules per unit cell (see Table 1).
| Compound | 1a | 1b | 1c | 1d | 1e | 1f |
|---|---|---|---|---|---|---|
| a α, β, γ = 79.966(4), 79.823(3), 84.995(3). | ||||||
| Crystal system space group | Orthorhombic Pbcm | Triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
Cubic Fdm |
Tetragonal I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) c2 c2 |
Hexagonal P63/mmc |
| a, b, c/Å | 17.727(4), 35.611(7), 31.733(6) | 13.1046(5), 21.8727(10), 27.9377(12)a | 73.616(2) | 27.266(11) | 29.1874(4), 29.1874(4), 57.5222(14) | 18.9987(7), 18.9987(7), 31.8798(11) |
| V/Å3, Z | 20032(7), 8 |
7748(1), 4 | 398948(28), 136 |
20270(25), 8 |
49003(2), 24 |
9966(1), 4 |
| Sn–μ-S/Å | 2.398(4)–2.479(4) | 2.380(5)–2.482(3) | 2.373(3)–2.461(3) | 2.407(9)–2.466(8) | 2.355(14)–2.513(16) | 2.378(5)–2.464(4) |
| Sn–μ3-S/Å | 2.580(4)–2.635(4) | 2.592(3)–2.646(3) | 2.581(3)–2.618(2) | 2.575(8) | 2.580(14)–2.643(17) | 2.602(3)–2.625(4) |
| Sn–Sterm/Å | 2.438(3)–2.457(5) | 2.427(6)–2.454(3) | 2.428(3)–2.467(5) | 2.480(14) | 2.419(17)–2.468(18) | 2.440(6)–2.511(11) |
| Snapex–O/Å | 2.429(9)–2.532(5) | 2.423(8)–2.508(9) | 2.370(9)–2.479(4) | 2.52(3) | 2.49(3)–2.53(3) | 2.450(9)–2.522(12) |
While it is typically difficult to localize ionic liquid counterions in such complex structures, the co-crystallizing constituents of the ionic liquid could be identified and refined for compounds 1a and 1b (Fig. 2). In the case of 1a, the ionic liquid cations compensate for the charge of the cluster anions and additionally co-crystallizing [BF4]− anions. Co-crystallization of the entire ionic liquid is rare; however, it was recently observed in a similar way in the selenido stannate salt (C4C1C1Im)5[Sn9Se20][BF4].24b The cations of compound 1b are located in close proximity of the anions, with the imidazolium rings oriented parallel to the cluster faces; thereby, they serve to effectively separate the anions from each other.
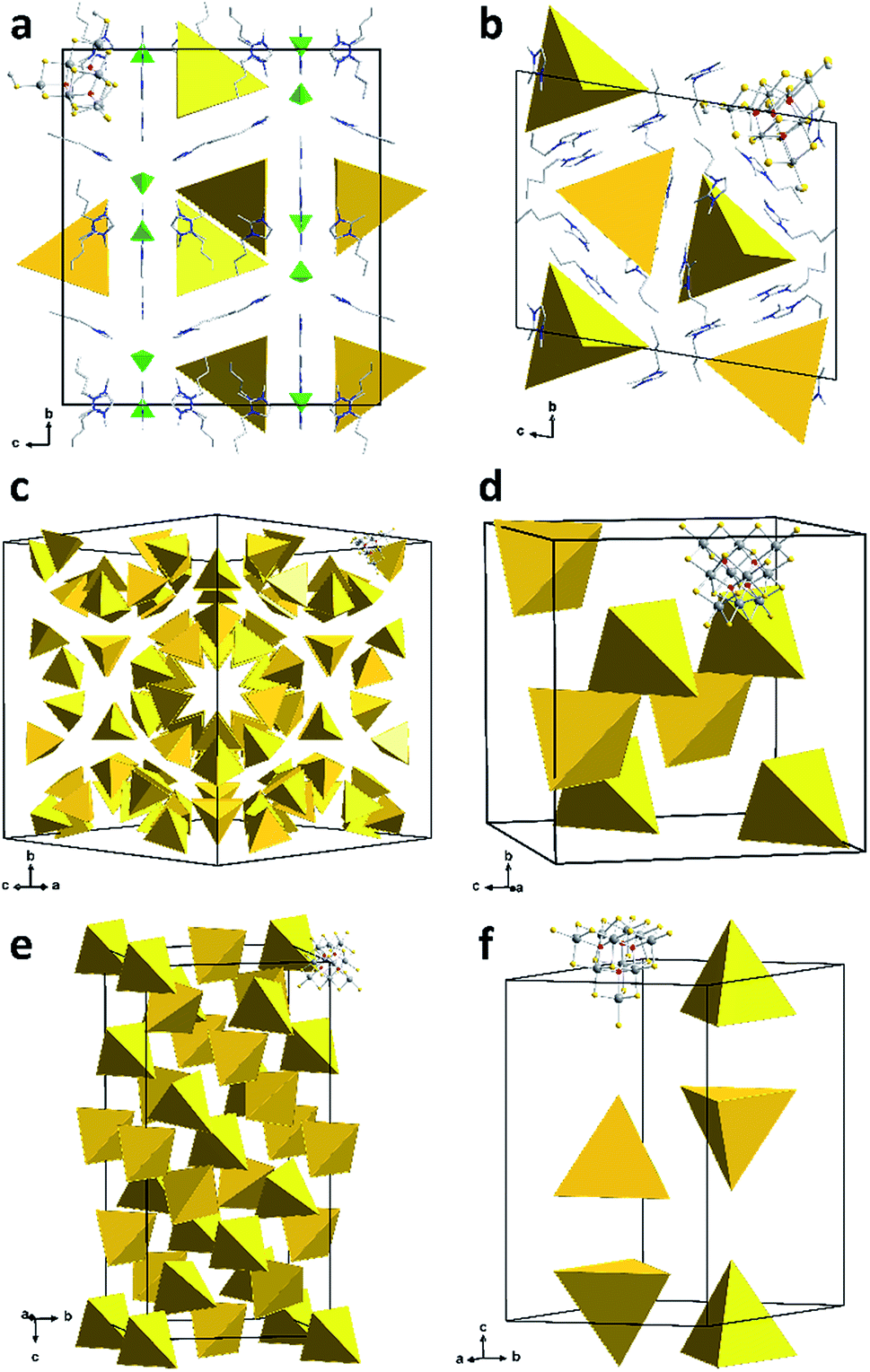 | ||
| Fig. 2 Sections of the crystal structures of 1a (a), 1b (b), 1c (c), 1d (d), 1e (e) and 1f (f). [Sn10S16O4(S–Me)4]4− anions, except one, are shown as yellow tetrahedra; green tetrahedra represent [BF4]− anions. Counterions are shown in wire representation. Color code: grey – Sn, yellow – S, red – O, light grey – C, blue – N. | ||
Due to their heavy positional disorder, not all of the butyl substituents of the cations in 1b could be refined. For the same reason, neither the organic cations nor the methyl groups of the cluster anion were refined in 1c and 1d. In 1e and 1f, some of the methyl groups were localized on the Fourier map, but the position of the organic cations could not be refined. Similar observations have been reported in related previous work, which also indicated that the co-crystallization of alkali metal counterions cannot be strictly excluded.24a,31
However, the unprecedented methylation of the terminal S atoms has a notable impact on the cluster structures of all variants of compound 1, which was used as an indirect – yet unambiguous – proof of their existence: the methylation leads to significant elongation of the terminal Sn–S bonds as compared to the corresponding bond lengths in the purely inorganic analogs (2.355–2.374 Å): in 1a–1f, the Sn–Sterm distances are 2.419(17)–2.511(11) Å (see Table 1); the relatively broad range is attributed to different environments and thus interactions of the S and SMe ligands by their surrounding (counter)ions. In turn, the respective Sn atoms are pulled inside, towards the cluster center, with Sn–O distances that are smaller than those of the purely inorganic structures (2.604–2.713 Å).29,30 Consequently, the (μ-S)–Sn–(μ-S) bond angles are more obtuse (114.86(19)–118.55(16)°), while the (μ-S)–Sn–S(Me) angles are more acute. The Sn–S–C angles possess typical values of 98.5(8)–100.5(7)°.
In the most complex structure of the six related compounds, 1c, 136 cluster anions are located in the unit cell. They are arranged in pseudo-tubes parallel to [101] direction. In contrast, the unit cell of 1d comprises eight anions only.
To confirm the existence of the terminal methyl groups, crystals of 1a–1f were additionally investigated by means of Raman spectroscopy. Fig. 3 shows the spectrum of compound 1c as an example; a full set of spectra of 1a–1f is provided in the ESI.† The characteristic bands of the lattice vibrations of the sulfido–oxido stannate cluster core (most intensive bands at approximately 175, 320–330, and 350 cm−1) show a slight red-shift in comparison to the values reported for the non-methylated cluster (ca. 193, 327, 336, 357 and 385 cm−1),30 which is in agreement with the expectations owing to the slight increase of average Sn–S bond lengths. One additional signal group is observed at 698 and 724 cm−1, which is completely missing in the vibrational spectrum of the purely inorganic cluster anion. The signals can therefore be assigned to the symmetric valence vibration of the S–CMe bonds, being slightly different owing to the slightly different orientation of the Me groups with respect to the cluster core. Furthermore, the signal of the symmetric valence vibration of the CMe–H bond can be found at 1515 cm−1. Another signal group, assigned to the imidazolium counter ion, is located between 2850 and 3000 cm−1. A characteristic signal profile of this signal is observed in all six spectra, which proves the presence of imidazolium groups as cations in these compounds. B–F vibrations (in 1a) were not identified.
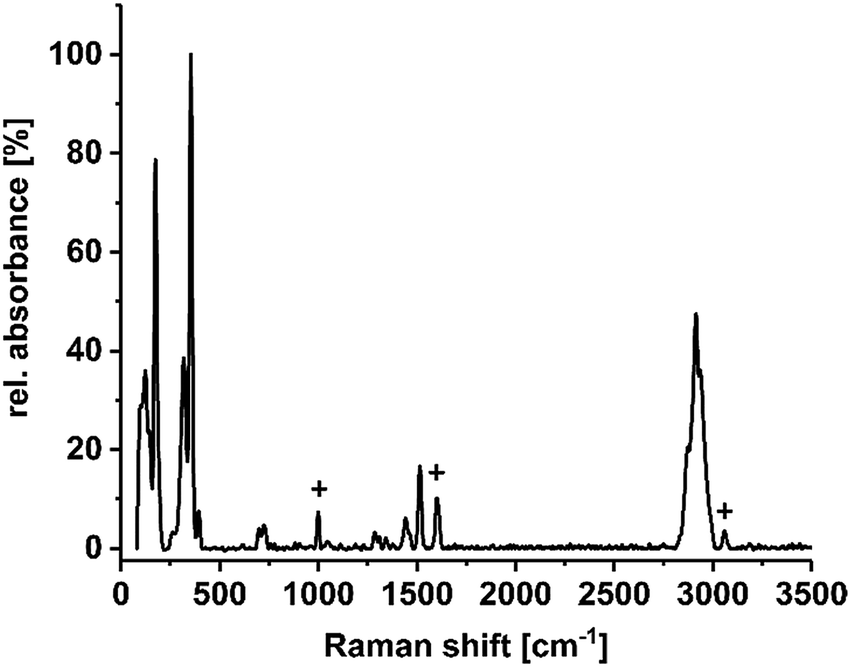 | ||
| Fig. 3 Raman spectrum of compound 1c, measured on suspension of single crystals in NVH® oil (low intensity signals caused by the oil are marked by +). Spectra of all compounds 1a–1f are provided in the ESI.† | ||
Compounds 1a–1f contribute to the rare examples of crystalline sulfides gained from ionic liquids. Further examples are (C2C1Im)3[Re3S4Br9]Br,19 besides various nickel thiophosphate compounds,20 and several compounds containing supertetrahedral clusterions like [Cu5Tr30S52X4−n(C4Im)n](13−n)− (Tr = Ga, In; X = SH/Cl).21 To explore whether this unprecedented methylation occurs only due to the presence of more nucleophilic sulfide ligands, or whether it can also occur to other chalcogenido metalates under the given reaction conditions, we also investigated syntheses of related selenido and tellurido metalates.
Compound 2, which forms from the selenidostannate salt K2[Sn2Se5], crystallizes in the trigonal crystal system (space group R) with six formula units within the unit cell. 2 contains the molecular anion [Mn4Sn4Se13(SeMe)4]6−, which is the first known methyl derivative of the otherwise well-known [Mn4Sn4Se17]10− penta-supertetrahedral P1 cluster anion.22,32,33 This type of anion consists of an [SeMn4]6+ tetrahedral cluster core, where each face of the tetrahedron is capped by an [SnSe4]4− tetrahedron. As for 1a–1f, the methylation takes place selectively at the terminal chalcogen atoms, suggesting that it affects the bond lengths within the cluster anion. The bond between the Sn atom and the formerly terminal Se atom is elongated (by about 0.05 Å), while unlike 1a–1f, all other bonds are unaffected (Fig. 4). For further information on the bond lengths see ESI.†
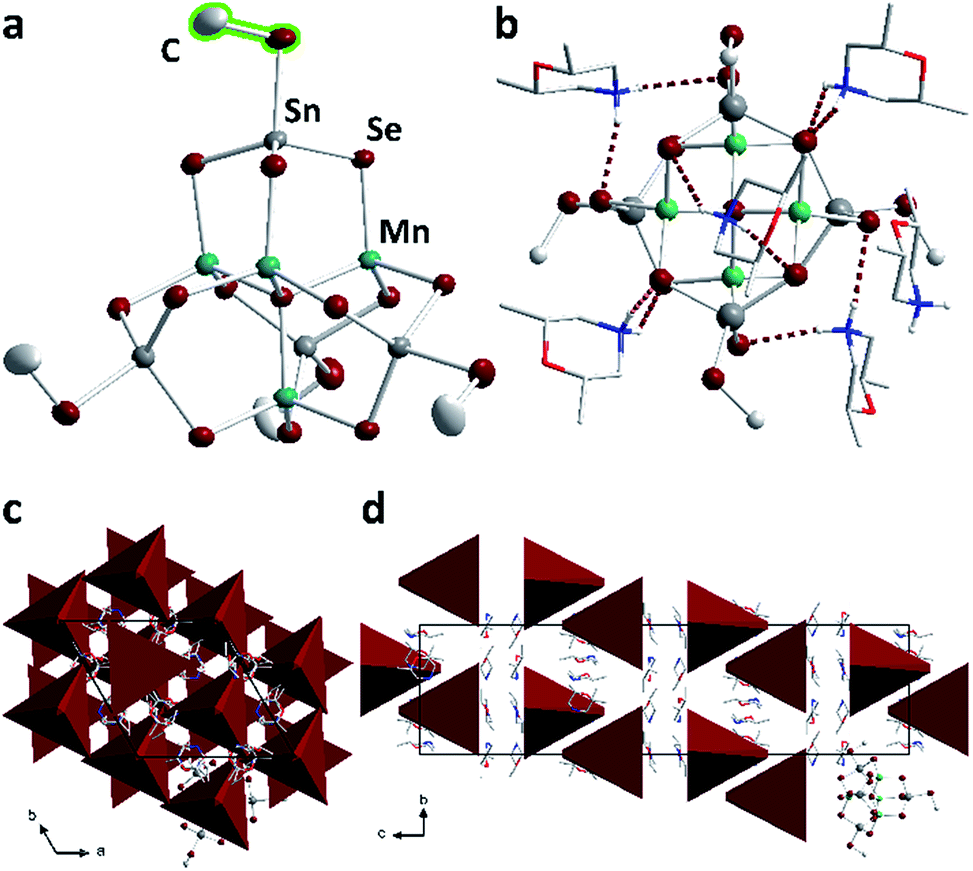 | ||
| Fig. 4 (a) Molecular structure of the anion in 2. (b) N–H⋯Se bridges (dark red) between the protons of the (dmmpH)+ cations and the cluster anion in 2 (methyl H atoms not drawn). Arrangement of the cluster anions (in polyhedral representation) in the unit cell of 2, viewed along the crystallographic a axis (c) and c axes (d). Color code: grey – Sn, dark red – Se, teal – Mn, red – O, blue – N, light grey – C. | ||
Above each edge of the supertetrahedra, one (dmmpH)+ (dmmp = 2,6-dimethylmorpholine) cation is located. The positions of the H atoms were calculated such that opposite μ-Se atoms are connected by N–H⋯Se hydrogen bonds (2.5574(12)–2.8760(8) Å, Fig. 4b). The cluster anions are arranged within {110} planes in pairs, and twisted against each other. Their center of gravity is located at nb/3 (n = 1, 2); two adjacent anion pairs are shifted about c/3. The anions possess the same orientation along a and b and are twisted along c, as illustrated in Fig. 4 with simplified tetrahedral representation. The cations are located along the tetrahedral faces.
Compound 2 (orange crystals), comprising the inorganic–organic hybrid P1 cluster anion, as well as the unmethylated cousin [Cs10(H2O)15.5][Mn4Sn4Se17] (red crystals) comprising the purely inorganic P1 cluster anion, were investigated by UV-Vis spectroscopy (Fig. S6†). The optical excitation energy, Eg, of 2 (2.18 eV) is positioned between the one of the Cs+ salt (1.90 eV) and the respective values reported for the related Na+ salt [Na10(H2O)34][Mn4Sn4Se17] (2.30 eV, yellow-orange crystals)32 as well as the related K+ salt [K10(H2O)16(MeOH)0.5][Mn4Sn4Se17] (2.27 eV, orange-red crystals).33 All of these Eg values correlate with the observed and described crystal colors. Hence, the overall optical absorption properties are not altered by this selective methylation of the terminal chalcogenide ligands, which is in stark contrast to the findings with fully organo-substituted clusters of this size, in which the optical gap is notably larger.34
The observed trend of the Eg values is the result of a variety of factors. One is the density of clusters in the crystal, which is controlled by the number, size and nature of the counter ions and their respective solvate shells. In the current series, the following densities (cluster per Å3) are found: 1/1735.45 (Na+ salt), 1/1530.95 (K+ salt), 1/2127.33 (2), 1/1755.88 (Cs+ salt). However, this does not correlate with the Eg trend. Another factor is the inter-cluster interaction, which is also dependent on nearest cluster distances (e.g., of a cluster pair), irrespective of the overall density. Indeed, while the alkali metal salts form relatively regular packing schemes with a central cluster being surrounded by others in similar distances (given as closest μ4-Se⋯μ4-Se distances; Na+ salt: 14 next neighbors, 12.9–15.3 Å; K+ salt: 13 next neighbors, 10.1–16.9 Å; Cs+ salt: 8 next neighbors, 12.4–17.3 Å), the packing of clusters in 2 is different: there are only 7 adjacent clusters (μ4-Se⋯μ4-Se 12.8–20.8 Å) in a wide range of distances, besides one much closer cluster neighbor (9.4 Å). Hence, we understand the relatively small Eg value based on the formation of cluster pairs. A third factor is the linkage of clusters through the counterions, which seems to be very effective for 2 exhibiting the second smallest Eg value in spite of the lowest cluster density. Obviously, the organic counterions and H-bonds are better linkers in terms of band gap reduction than hydrated alkali metal cations.
The subtle balance of influences by the nature of the ligands versus the charge and thus number of counterions on the optical properties of the crystalline compounds can be gathered from another example in the literature, in which the optical absorption energy increased with decreasing charge owing to the significant steric demand of the charge-reducing ligands at [Cu5Ga30S52X]q− (X/q/Eg = (SH)4/−13/3.04 eV, (SH)1.5Cl(C4Im)1.5/−11.5/3.62 eV, (SH)2(C4Im)2/−11/3.68 eV; C4Im = 1-butyl-2-methylimidazole).21 Finally, also mere differences in the packing pattern of identical compounds may affect the density of the crystal structures and thus can cause different excitation energies.4
As the third system to be studied, the salt of a polymeric tellurido mercurate anion, Na2[HgTe2],35 was treated with ionic liquids comprising hexyl or octyl chains. Recently, we reported about very uncommon tellurido mercurate anions [Hg8Te16]8− to be stabilized by imidazolium salts with decyl or dodecyl chains,36 so we were eager to probe the effect of counterions with medium-size alkyl chain lengths. Indeed, molecular cluster anions were received again, yet with another cluster structure and methylated terminal Te ligands. Both products, (C6C1Im)6[Hg6Te10(TeMe)2] (3a) and (C8C1Im)6[Hg6Te10(TeMe)2] (3b), crystallize in the triclinic crystal system (space group P) with one formula unit per unit cell. While they differ in their imidazolium counterion, both 3a and 3b contain the same anion [Hg6Te10(TeMe)2]6−.
As observed with compounds 1 and 2, methylation of the cluster molecules occurred in situ during their formation under mild ionothermal conditions. This indicates a significant nucleophilicity of the underlying, highly charged tellurido metalate anions: these would possess a −8 charge without methyl groups, hence the same charge as found in [Hg8Te16]8−, yet distributed over only 18 instead of 24 atoms. We ascribe the fact that a smaller anion is observed in 3 to the size of the imidazolium counterions, which perfectly embed and stabilize this cluster, while the longer-chain cations match the larger size of [Hg8Te16]8− units. As in the latter, the anion in 3 comprises ditelluride units upon partial oxidation (by residues of oxygen in the ionic liquid or by the ionic liquid itself). This seems to be an intrinsic property of the imidazolium-based ionic liquids used at the treatment of selenido or tellurido metalate structures.37 The unprecedented hexanuclear tellurido mercurate anion has C2 symmetry and comprises four tetrahedral [HgTe4]6− and two trigonal planar [HgTe3]4− units (Fig. 5). As illustrated in Fig. 5a, each half of the anion is constructed by two edge-sharing [HgTe4]6− tetrahedra. These are further connected by another Hg atom to form a trigonal planar [HgTe3]4− fragment, which is terminated by a methyl ligand. The two cluster halves are fused by two μ-bridging Te atoms and two μ4,η2:η2-bridging ditelluride units. All atoms including the terminal methyl groups are situated approximately in one plane – except for the two ditelluride units, which are located above and below this plane. The central [Hg4Te2(Te2)2] unit is the same as reported for the tellurido mercurate anion, [Hg4Te12]4−,35,38 in which tritelluride ligands are present instead of the [HgTe3Me] units in 3.
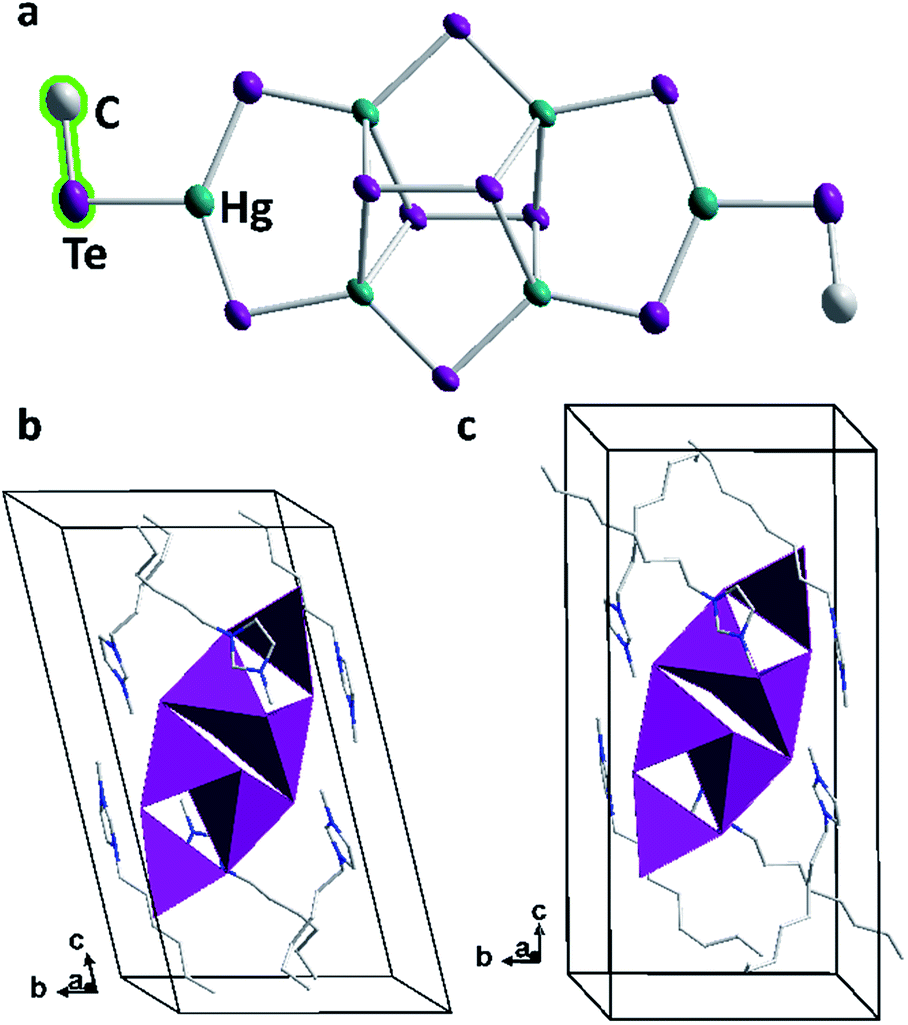 | ||
| Fig. 5 (a) Molecular structure of the [Hg6Te10(TeMe)2]6− anions in 3b. Thermal ellipsoids are drawn at the 50% probability level. H atoms are omitted for clarity. Sections of the crystal structures of 3a ((b) without disorder of two Te atoms and the methyl group) and 3b (c). Anions are given in polyhedral representation, cations are shown in wire representation, and H atoms are omitted for clarity. Color code: teal – Hg, violet – Te, light grey – C, blue – N. | ||
The Hg–μ-Te bonds lengths in 3 are in the range 2.656(6)–2.713(9) Å, those between Hg atoms and μ-Te2 units are slightly longer (2.935(1)–2.962(1) Å). These values, and also those of the Te–Hg–Te bond angles at the tetrahedrally coordinated Hg atoms, accord well with those found in the [Hg4Te12]4− anion. Hence, the replacement of terminal tritelluride units by the [HgTe3Me] fragment does not notably affect the cluster core. The trigonal planar [HgTe3] unit itself is distorted with μ-Te–Hg–Te angles of 136.16(17)–137.08(3)° to match the metrics of the cluster core to which it binds. In turn, the μ-Te–Hg–Te(Me) bond angles are only 103.83(26)–119.67(13)°, with Hg–Te(Me) distances of 2.767(7)–2.812(7) Å. As observed in compounds 1 and 2, the methylation leads to increased metal–chalcogen bond lengths; however, while the increase was about 0.10 Å for the sulfide clusters, and about 0.05 Å for the selenide cluster, it is only marginal here (below 0.04 Å), as compared with terminal Te atoms in purely inorganic trigonal planar [HgTe3] arrangements (cf. K4[HgTe3]·H2O or Na2[HgTe2], 2.745(1)–2.778(1) Å).28,39 The Te–C bond length of 2.174(34)–2.187(46) Å is fairly typical (2.184(5)–2.194(4) Å).40
The position of the anion in the unit cell is body-centered with its mean plane tilted around (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 11) and (211) in the crystal structures of 3a or 3b, respectively. The imidazolium rings of the cations are located in the vicinity of the anions to effectively compensate for their charge. The hexyl or octyl chains, respectively, serve to separate the cations from each other; they are heavily disordered in both 3a and 3b. Still, a preferred arrangement of the alkyl chains can be noticed as they are orientated roughly along the crystallographic c axis and thus close the anion in a capsule-like cavity. This way, ionic layers of (tilted) anions and imidazolium cations parallel to the ab plane alternate with double layers of weakly interacting alkyl chains parallel to the c axis (Fig. 5b and c).
11) and (211) in the crystal structures of 3a or 3b, respectively. The imidazolium rings of the cations are located in the vicinity of the anions to effectively compensate for their charge. The hexyl or octyl chains, respectively, serve to separate the cations from each other; they are heavily disordered in both 3a and 3b. Still, a preferred arrangement of the alkyl chains can be noticed as they are orientated roughly along the crystallographic c axis and thus close the anion in a capsule-like cavity. This way, ionic layers of (tilted) anions and imidazolium cations parallel to the ab plane alternate with double layers of weakly interacting alkyl chains parallel to the c axis (Fig. 5b and c).
In conclusion, we presented the products of an unprecedented reaction occurring in ionothermal syntheses involving methyl-(di-)alkylimidazolium-based ionic liquids, namely the selective in situ methylation of terminal chalcogenide ligands of sulfido, selenido, and tellurido metalate cluster anions, which could so far not be realized in common solvent-based reactions. Moreover, in case of the telluride, the reaction yielded a cluster architecture that has so far been unknown at all. Structural and spectroscopic analyses confirmed the presence of the terminal Me groups, and also served to demonstrate and explain the influence of the methylation on optical absorption properties. We summarize that unlike common methylation reagents, ionic liquids serve to alkylate the very weakly nucleophilic chalcogenido metalate anions under ionothermal conditions. This indicates the role of ionic liquids as non-innocent reaction media, and provides access to new strategies in the functionalization of chalcogenido metalates which will be the scope of future investigations in a more general sense.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of SPP 1708. We thank K. Kirchberg for her help with measuring the vibrational spectra, Dr N. Rinn for help with the manuscript, and M. Hoyland (IUCr) for assistance with the checkCIF run of compound 1c.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Feng, X. Bu and N. Zheng, Acc. Chem. Res., 2005, 38, 293–303 CrossRef CAS PubMed.
- M. J. Manos, K. Chrissafis and M. G. Kanatzidis, J. Am. Chem. Soc., 2006, 128, 8875–8883 CrossRef CAS PubMed.
- T. Wu, X. Bu, P. Liao, L. Wang, S.-T. Zheng, R. Ma and P. Feng, J. Am. Chem. Soc., 2012, 134, 3619–3622 CrossRef CAS PubMed.
- S. Santner, J. Heine and S. Dehnen, Angew. Chem., Int. Ed., 2016, 54, 876–893 CrossRef PubMed.
- H. Yang, J. Zhang, M. Luo, W. Wang, H. Lin, Y. Li, D. Li, P. Feng and T. Wu, J. Am. Chem. Soc., 2018, 140, 11189–11192 CrossRef CAS PubMed.
- (a) N. Zheng, X. Bu, B. Wang and P. Feng, Science, 2002, 298, 2366–2369 CrossRef CAS PubMed; (b) N. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2003, 125, 1138–1139 CrossRef CAS PubMed; (c) X. Xu, W. Wang, D. Liu, D. Hu, T. Wu, X. Bu and P. Feng, J. Am. Chem. Soc., 2018, 140, 888–891 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, J. Am. Chem. Soc., 2012, 134, 4497–4500 CrossRef CAS PubMed.
- (a) N. Zheng, X. Bu, H. Vu and P. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS PubMed; (b) Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- (a) M. J. Manos, K. Chrissafis and M. G. Kanatzidis, J. Am. Chem. Soc., 2006, 128, 8875–8883 CrossRef CAS PubMed; (b) M. J. Manos, C. D. Malliakas and M. G. Kanatzidis, Chem.–Eur. J., 2007, 13, 51–58 CrossRef CAS PubMed; (c) M.-L. Feng, D.-N. Kong, Z.-L. Xie and X.-Y. Huang, Angew. Chem., Int. Ed., 2008, 47, 8623–8626 CrossRef CAS PubMed; (d) N. Ding and M. G. Kanatzidis, Nat. Chem., 2010, 2, 187–191 CrossRef CAS PubMed; (e) M. J. Manos and M. G. Kanatzidis, Chem. Sci., 2016, 7, 4804–4824 RSC.
- J. F. Corrigan, O. Fuhr and D. Fenske, Adv. Mater., 2009, 21, 1867–1871 CrossRef CAS.
- O. Fuhr, S. Dehnen and D. Fenske, Chem. Soc. Rev., 2013, 42, 1871–1906 RSC.
- V. N. Soloviev, A. Eichhöfer, D. Fenske and U. Banin, J. Am. Chem. Soc., 2001, 123, 2354–2364 CrossRef CAS PubMed.
- A. Aharoni, A. Eichhöfer, D. Fenske and U. Banin, Opt. Mater., 2003, 24, 43–49 CrossRef CAS.
- C. B. Khadka, B. K. Najafabadi, M. Hesari, M. S. Workentin and J. F. Corrigan, Inorg. Chem., 2013, 52, 6798–6805 CrossRef CAS PubMed.
- Z. Wang, C.-F. Du, C.-C. Cheng, N.-N. Shen, J.-R. Li and X.-Y. Huang, Inorg. Chem. Commun., 2016, 74, 58–61 CrossRef CAS.
- Y. Lin and S. Dehnen, Inorg. Chem., 2011, 50, 7913–7915 CrossRef CAS PubMed.
- J.-R. Li, Z.-L. Xie, X.-W. He, L.-H. Li and X.-Y. Huang, Angew. Chem., Int. Ed., 2011, 50, 11395–11399 CrossRef CAS PubMed.
- Y. Lin, D. Xie, W. Massa, L. Mayrhofer, S. Lippert, B. Ewers, A. Chernikov, M. Koch and S. Dehnen, Chem.–Eur. J., 2013, 19, 8806–8813 CrossRef CAS PubMed.
- H. Sakamoto, Y. Watanabe and T. Saito, Inorg. Chem., 2006, 45, 4578–4579 CrossRef CAS PubMed.
- J. A. Cody, K. B. Finch, G. J. Reynders III, G. C. B. Alexander, H. G. Lim, C. Näther and W. Bensch, Inorg. Chem., 2012, 51, 13357–13362 CrossRef CAS PubMed.
- W.-W. Xiong, J.-R. Li, B. Hu, B. Tan, R.-F. Li and X.-Y. Huang, Chem. Sci., 2012, 3, 1200–1204 RSC.
- S. Santner and S. Dehnen, Inorg. Chem., 2015, 54, 1188–1190 CrossRef CAS PubMed.
- (a) L. Wang, Y. P. Xu, Y. Wei, J. C. Duan, A. B. Chen, B. C. Wang, H. J. Ma, Z. J. Tian and L. W. Lin, J. Am. Chem. Soc., 2006, 128, 7432–7433 CrossRef CAS PubMed; (b) R. S. Xu, W. P. Zhang, J. Guan, Y. P. Xu, L. Wang, H. J. Ma, Z. J. Tian, X. W. Han, L. W. Lin and X. H. Bao, Chem.–Eur. J., 2009, 15, 5348–5354 CrossRef CAS PubMed; (c) J.-R. Li, W.-W. Xiong, Z.-L. Xie, C.-F. Du, G.-D. Zou and X.-Y. Huang, Chem. Commun., 2013, 49, 181–183 RSC.
- (a) S. Santner, S. Yogendra, J. J. Weigand and S. Dehnen, Chem.–Eur. J., 2017, 23, 1999–2004 CrossRef CAS PubMed; (b) S. Santner, J. A. P. Sprenger, M. Finze and S. Dehnen, Chem.–Eur. J., 2018, 24, 3474–3480 CrossRef CAS PubMed.
- T. Kaib, S. Haddadpour, M. Kapitein, P. Bron, C. Schröder, H. Eckert, B. Roling and S. Dehnen, Chem. Mater., 2012, 24, 2211–2219 CrossRef CAS.
- W. Schiwy, S. Pohl and B. Krebs, Z. Anorg. Allg. Chem., 1973, 402, 77–86 CrossRef CAS.
- K. O. Klepp, Z. Naturforsch., B: J. Chem. Sci., 1992, 47, 197–200 CAS.
- C. Donsbach and S. Dehnen, Z. Anorg. Allg. Chem., 2017, 643, 14–19 CrossRef CAS.
- T. Kaib, M. Kapitein and S. Dehnen, Z. Anorg. Allg. Chem., 2011, 637, 1683–1686 CrossRef CAS.
- W. Schiwy and B. Krebs, Angew. Chem., Int. Ed. Engl., 1975, 14, 436 CrossRef.
- (a) X. M. Zhang, D. Sarma, Y. Q. Wu, L. Wang, Z. X. Ning, F. Q. Zhang and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 5543–5546 CrossRef CAS PubMed; (b) Q. Lin, X. Bu, C. Mao, X. Zhao, K. Sasan and P. Feng, J. Am. Chem. Soc., 2015, 137, 6184–6187 CrossRef CAS PubMed; (c) J. Lin, Q. Zhang, L. Wang, X. Liu, W. Yan, T. Wu, X. Bu and P. Feng, J. Am. Chem. Soc., 2014, 136, 4769–4779 CrossRef CAS PubMed; (d) T. Wu, Q. Zhang, Y. Hou, L. Wang, C. Mao, S.-T. Zheng, X. Bu and P. Feng, J. Am. Chem. Soc., 2013, 135, 10250–10253 CrossRef CAS PubMed; (e) T. Wu, R. Khazhakyan, L. Wang, X. Bu, S.-T. Zheng, V. Chau and P. Feng, Angew. Chem., Int. Ed., 2011, 50, 2536–2539 CrossRef CAS PubMed.
- E. Ruzin, C. Zimmermann, P. Hillebrecht and S. Dehnen, Z. Anorg. Allg. Chem., 2007, 633, 820–829 CrossRef CAS.
- S. Dehnen and M. K. Brandmayer, J. Am. Chem. Soc., 2003, 125, 6618–6619 CrossRef CAS PubMed.
- (a) V. Soloviev, A. Eichhöfer, D. Fenske and U. Banin, J. Am. Chem. Soc., 2000, 122, 2673–2674 CrossRef CAS; (b) M. K. Brandmayer, R. Clérac, F. Weigend and S. Dehnen, Chem.–Eur. J., 2004, 10, 5147–5157 CrossRef CAS PubMed.
- (a) R. C. Haushalter, Angew. Chem., Int. Ed. Engl., 1985, 24, 433–435 CrossRef; (b) J. Li, B. G. Rafferty, S. Mulley and D. M. Proserpio, Inorg. Chem., 1995, 34, 6417–6418 CrossRef; (c) P. Sun, S. Liu, S. Li, L. Zhang, H. Sun and D. Jia, Inorg. Chem., 2017, 56, 6152–6162 CrossRef CAS PubMed.
- C. Donsbach, K. Reiter, D. Sundholm, F. Weigend and S. Dehnen, Angew. Chem., Int. Ed., 2018, 57, 8770–8774 CrossRef CAS PubMed.
- Y. Lin, W. Massa and S. Dehnen, Chem.–Eur. J., 2012, 18, 13427–13434 CrossRef CAS PubMed.
- K.-W. Kim and M. G. Kanatzidis, Inorg. Chim. Acta, 1994, 224, 163–169 CrossRef CAS.
- G. Thiele, C. Donsbach, R. Riedel, M. Marsch, K. Harms and S. Dehnen, Dalton Trans., 2016, 45, 5958–5967 RSC.
- J. Ścianowski, A. J. Pacuła and A. Wojtczak, Tetrahedron: Asymmetry, 2015, 26, 400–403 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses, experimental methods, crystallographic data, Raman spectra of compounds 1a–1f and the UV-Vis spectrum of compound 2. CCDC 1902968–1902973 and 1902904–1902906. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01358j |
| ‡ In the formulae, (Cm(Cn)CoIm)+ denote tri(/di)-alkylated imidazolium cations, with n, m and o specifying the chain lengths of the alkyl substituents in the respective 1, 2 and 3 positions of the imidazole ring. |
| § en = 1,2-diaminoethane, dmmp = 2,6-dimethylmorpholine. |
| This journal is © The Royal Society of Chemistry 2019 |