
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable cyclic (alkyl)(amino)carbene (cAAC) radicals with main group substituents
Subrata
Kundu
 a,
Soumen
Sinhababu
a,
Vadapalli
Chandrasekhar
*abc and
Herbert W.
Roesky
*a
a,
Soumen
Sinhababu
a,
Vadapalli
Chandrasekhar
*abc and
Herbert W.
Roesky
*a
aUniversität Göttingen, Institut für Anorganische Chemie, Tammannstrasse 4, D-37077, Göttingen, Germany. E-mail: hroesky@gwdg.de
bTata Institute of Fundamental Research Hyderabad, Hyderabad 500107, India
cDepartment of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India. E-mail: vc@iitk.ac.in
First published on 8th April 2019
Abstract
Isolation and characterization of stable radicals has been a long-pursued quest. While there has been some progress in this field particularly with respect to carbon, radicals involving heavier p-block elements are still considerably sparse. In this review we describe our recent successful efforts on the isolation of stable p-block element radicals particularly those involving aluminum, silicon, and phosphorus.
 Subrata Kundu | Subrata Kundu has obtained his M.Sc. in Chemistry from Indian Institute of Technology Delhi in 2010. He received his Ph.D. in 2015 from Indian Institute of Technology Kanpur, under the supervision of Professor V. Chandrasekhar. After that, he moved to University of Göttingen as a postdoctoral fellow in October 2015 and worked with Professor Herbert W. Roesky on stabilization of compounds containing low valent p-block elements till end of 2018. In the beginning of 2019, he joined the group of Professor Ian Manners at University of Victoria as a postdoctoral fellow. |
 Soumen Sinhababu | Soumen Sinhababu completed his M.Sc. in Chemistry from Indian Institute of Technology Delhi in 2010 and continued his Ph.D in the same institute. He received his Ph.D. in 2017 under the supervision of Professor S. Nagendran. Then, he moved to University of Göttingen as a postdoctoral fellow and working with Professor Herbert W. Roesky. His research interest mainly focuses on the stabilization of compounds containing low valent main group elements. |
 Vadapalli Chandrasekhar | Vadapalli Chandrasekhar obtained his Ph.D. in 1982 from the Indian Institute of Science, Bangalore. He did his postdoctoral work at the University of Massachusetts, Amherst, MA (1983–1986). After briefly working at the Indian Petrochemicals Corporation at Vadodara, he joined the Department of Chemistry at the Indian Institute of Technology Kanpur in 1987 where he has been a full professor since 1995. He was the Director of the National Institute of Science Education and Research (NISER), Bhubaneswar, India (2014–17). Currently he is the Centre Director, Tata Institute of Fundamental Research, Hyderabad. He has been a recipient of several national and international awards and fellowships including S. S. Bhatnagar Award in 2003 and he has published more than 360 peer-reviewed papers, articles, and books. |
 Herbert W. Roesky | Herbert W. Roesky obtained his doctorate from the University of Göttingen. After working at DuPont in the United States, he returned to Göttingen and finished his habilitation. He then became a Professor at the Johann-Wolfgang-Goethe-Universität, Frankfurt am Main in 1971. He moved to the University of Göttingen in 1980 and was the director of the Institute for Inorganic Chemistry until 2004. More than 1250 peer-reviewed papers, articles, patents, and books record his research activities in the areas of inorganic chemistry and materials science. He is also the recipient of several prizes, i.e. Gottfried Wilhelm Leibniz Prize, ACS awards in Inorganic and Fluorine Chemistry and Wacker Silicone Award. |
Introduction
Radical chemistry has long been of interest both from academic and practical points of view.1 Among the most widely used radical chemistry in industrial processes is the low-density polyethylene manufacture. In addition, radical chemistry is widely utilized in organic synthesis and has implications in many biological processes including cell damage.2,3 Indeed, scavenging radicals from cells forms an important aspect of the cell machinery in biological systems.4 In view of this ubiquity and importance of radicals there has naturally been an interest in studying radicals both in their transient and isolated forms. While spectroscopic tools such as electron paramagnetic resonance have been of considerable utility in throwing light on the nature of the radicals, isolating the latter and characterizing them, particularly in the solid-state, has been fraught with considerable challenges in view of the reactivity of these systems. While metal-based radical systems involving coordination complexes have been far more amenable for isolation and characterization similar examples involving carbon and other main-group elements, although known, are still rare. Surprisingly, however, nature stabilizes molecular oxygen in its biradical form, quite readily. Open-shell main-group element compounds containing unpaired electrons are unstable for several reasons including the fact that many of these prefer to adopt closed-shell configurations and they readily react with any available chemical entity.5 Considering this propensity for reactivity, typical methods for stabilizing main group element-based radicals mainly involve delocalizing the unpaired electron on the overall molecular scaffold to achieve thermodynamic stability and/or by employing sterically encumbered groups around the main-group element such that the corresponding radicals are stabilized kinetically and are prevented from reacting with other species.5Fortuitously, during this pursuit of main-group element based radicals, progress in another aspect of reactive species viz., singlet carbenes is aiding the former.6 Thus, singlet carbenes are those that have a lone pair and a vacant orbital and have long been elusive. However, N-heterocyclic carbenes (NHCs) which are accessed by the deprotonation of an N,N′-disubstituted imidazolium (or other azolium) salt are known to be stable and have been shown to be strong σ-donors with negligible π-accepting character.7 Recent reports indicate that the substituents on the carbene center play a crucial role on the σ-donor ability as well as π-electron accepting properties.8 Bertrand et al. in 2005 reported that cyclic (alkyl)(amino)carbenes (cAACs) which have a nitrogen and a carbon flanking the carbene center have better σ-donor ability as well as better π-electron accepting properties than conventional NHCs.9 The empty p-orbital of cAACs can indeed engage in π-back bonding interactions and delocalize the electron density and at the same time stabilize an electron deficient center by σ-donation. This dual nature of the cAACs is becoming extremely promising in catalytic applications. Indeed, there have been reports on the activation of small molecules with the free carbenes themselves.10
In recent years, the electrophilicity of cAACs has been utilized for accessing compounds of low valent transition metals – as well as main group elements, several of them displaying paramagnetic behaviour.11 These cAAC-supported compounds containing low valent elements are an emerging class that have seen considerable advances in the last decade.12 In this review, we summarize the stable radicals of p-block elements by utilizing cAACs as ligands. There is already an elegant review from Bertrand's group on the recent developments on cAAC ligands as well as one on the isolation of stable radicals by using carbene ligands.12 However, this field is fast moving with many new discoveries and we feel the need to highlight the important findings in this area involving stable radicals of p-block elements. We will mainly focus results obtained from our laboratory; however, relevant research from other groups will be mentioned wherever appropriate. Few relevant examples of NHC-stabilized stable radicals of p-block elements are also mentioned. Before we begin the discussion we would like to define the notations used in this article, which will follow recent definitions outlined by Schulz (Scheme 1).13a
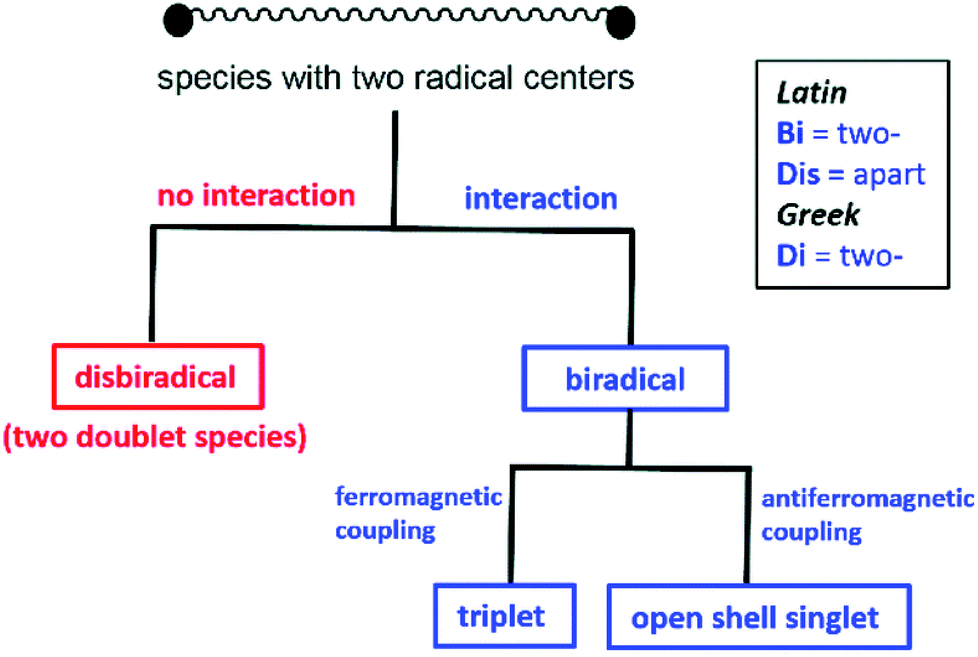 | ||
| Scheme 1 Definition for disbiradicals and biradicals used in this article (reproduced from ref. 13a with permission from the Royal Society of Chemistry). | ||
Monoradical
When an atom or a functional group or a molecule contains an unpaired electron, it is called monoradical. An electron has a spin quantum number S = 1/2 so the spin multiplicity (2S + 1) of a monoradical is two, hence they are also called as doublet species.13bDisbiradical and biradicals (synonym diradicals)
When a molecule contains two unpaired electrons, it is termed as disbiradical or biradical.13 The two electrons of the molecule may interact with each other. For a disbiradical the electron exchange interaction (J) between the two radicals is negligible or nearly negligible (J ≈ 0) due to the significant distance (r) between them or an orthogonal orientation between them. So the disbiradicals are like two doublets within the same molecule.A strong interaction between two radicals leads to two classical situations (a) both spins align in the same direction (↑↑; S = 1, 2S + 1 = 3; triplet) and it is called triplet biradical (b) two spins align in opposite direction to each other (↑↓; S = 0, 2S + 1 = 1; singlet) and it is called as a singlet biradical or open shell singlet (OSS). However, in both the cases the two electrons occupy two separate orbitals. A triplet biradical has a positive exchange interaction (J) while an open shell singlet has a negative exchange interaction. The exchange interaction between two electrons in a closed-shell singlet (CSS) molecule (non radical, both the electron occupies the same orbital) is extremely high and negative in magnitude.
Biradicaloid (synonym diradicaloid)
Often refers to a biradical species in which the two radical centres interact significantly. In general, a relatively small energy gap between their lowest energy singlet and triplet states is observed for singlet biradicals. By increasing the HOMO–LUMO energy gap, the stability of the biradicals increases. When the LUMO occupancy of a molecule reaches zero, they are called as closed-shell molecules. However, the LUMO occupancy is not negligible for biradicaloids due to the small HOMO–LUMO energy gap and biradicaloids are relatively more reactive compared to closed-shell molecules.13bThe singlet biradicals do not have a net magnetic moment, so they are diamagnetic and are EPR silent. Monoradicals show one set of EPR resonance [due to allowed (Δms = ±1) transition] while two sets of EPR signals are observed for a triplet biradical [due to both allowed (Δms = ±1; ms = −1 → 0 and 0 → +1) and forbidden (Δms = ±2; ms = −1 → +1) transitions]. Several theoretical methods are available to study radicals, biradicals and biradicaloids. The energy difference can also be calculated by several theoretical methods. Although, there are some limitations of different levels of theories, they are extremely helpful to study bond and spin density distribution of these species.
Group 13 radicals
To isolate group 13 element-based radicals, the strong σ-donor ability of the N-heterocyclic carbenes (NHCs) has been utilized. A review by Kinjo et al. summarizes all the boron containing radical species.14 In 2007, Gabbaï et al. isolated the first structurally characterized carbene-supported neutral boryl radical (Mes2BMe-acridine)˙ (Mes = 2,4,6-Me3C6H2; acridine = C13H9N) (1) by the reduction of the corresponding borenium cation with magnesium metal (Fig. 1).15a The EPR spectrum of 1 indicates that the unpaired electron is mainly delocalized over the acridinyl moiety with a small contribution on the boron atom (a(11B) = 2.55 G). After 2 years (in 2009), the same group has reported another successful synthesis of NHC-supported neutral boryl radical [(IMe)BMes2]˙ (IMe = 1,3-dimethylimidazol-2-ylidene) (2) (Fig. 1).15b The EPR spectrum of 2 suggested that the delocalization of the spin density over the boron center, mesityl groups, and the entire carbene framework (a(11B) = 7.90 G). Subsequently, a few more NHC-stabilized boryl radicals have been isolated; some examples (3–8) are listed in Fig. 1.16 The EPR spectrum of 6 shows a four-line signal arising from couplings with the two boron isotopes [a(11B) = 3.02 G; a(10B) = 1.02 G], which indicate the delocalization of the unpaired electron over the borolyl ring. The EPR spectra of 7–8 indicate that the spin density is mainly concentrated on the boron–boron bond. It has been observed that the spin density of most of the NHC stabilized boryl radicals is delocalized over the substituents attached to the boron atom.16c The spin density of all the group 13 species discussed here are given in Table 1.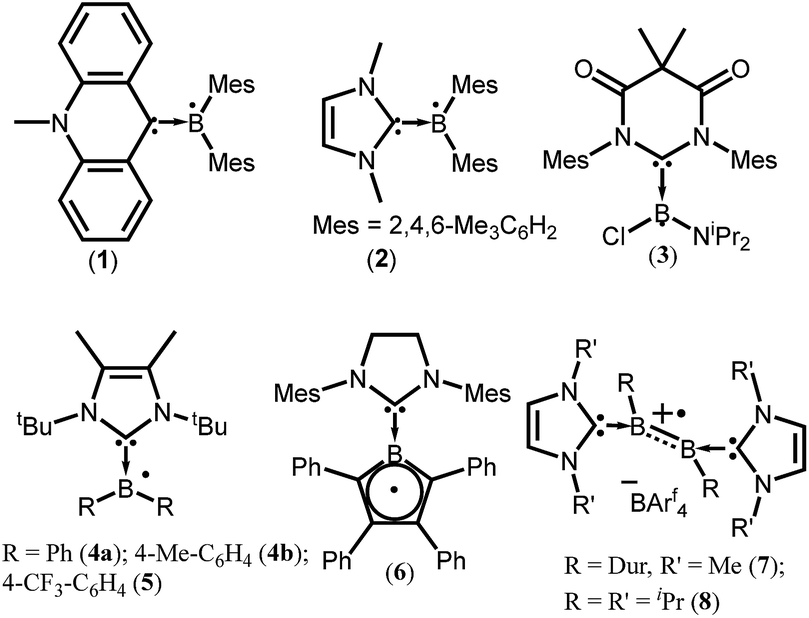 | ||
| Fig. 1 NHC ligated boryl monoradicals. The arrow indicates the donor–acceptor interaction. | ||
| Compounds | Spin density | Type of radical | Ref. |
|---|---|---|---|
| a Unless other wise mentioned all the radicals are neutral. | |||
| 1 | Not given | Monoradical | 15a |
| 2 | Not given | Monoradical | 15b |
| 3 | 90% Ccarbene, 7.8% B | Monoradical | 16a |
| 4a | 87% BR2, 13% NHC, 41% B, 3% Ccarbene | Monoradical | 16b |
| 4b | 86% BR2, 14% NHC, 41% B, 4% Ccarbene | Monoradical | 16b |
| 5 | 90% BR2, 10% NHC, 39% B, 2% Ccarbene | Monoradical | 16b |
| 6 | 47% B, 7% Ccarbene | Monoradical | 16c |
| 7 | Not given | Monoradical cation | 16d |
| 8 | Not given | Monoradical cation | 16e |
| 9 | 49.6% Ccarbene, 27.7% B, 24.1% Ncarbene | Monoradical | 17a |
| 10 | Not given | Monoradical | 17a |
| 11 | Not given | Monoradical | 17b |
| 12 | 57% Ccarbene, 21% Ncarbene | Triplet | 17c |
| 13 | Not given | Monoradical | 17d |
| 14 | Mainly delocalized over the B–CcAAC–NcAAC linkage | Monoradical | 17e |
| 15 | Not given | Triplet | 18a |
| 16 | 27% B, 49% CcAAC, 24% NcAAC | Triplet | 18b |
| 17 | 50% B, 33% NcAAC (total) | Monoradical cation | 18c |
| 18 | 49% B | Monoradical cation | 18d |
| 19 | Not given | Monoradical cation | 17d |
| 20 | 2% Al, 29% CcAAC, 50% CcAAC, 11% NcAAC, 6% NcAAC | Monoradical | 19 |
| 21 | 5.7% Al, 15% CcAAC, 62% CcAAC, 10% NcAAC, 2% NcAAC | Monoradical | 20 |
| 22 | 5.1% Al, 15% CcAAC, 65% CcAAC, 9% NcAAC, 2% NcAAC | Monoradical | 20 |
cAACs could be employed to isolate base-stabilized boryl radical with larger spin density on the boron atom and a large number of stable boryl radicals have been isolated and structurally characterized (9–19; Fig. 2).14,17,18
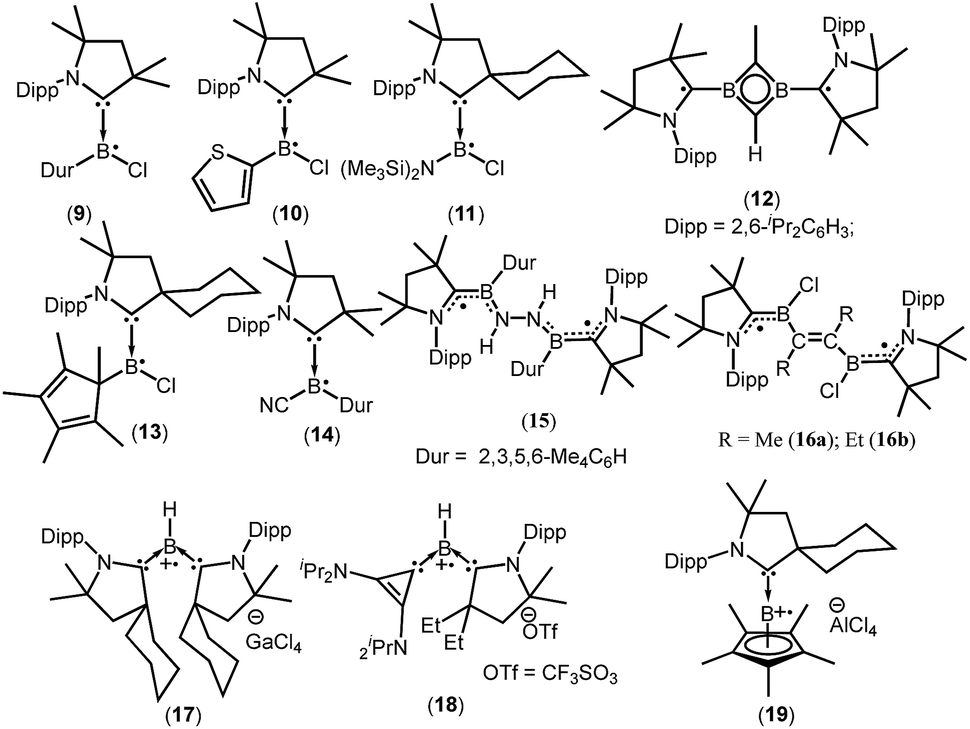 | ||
| Fig. 2 cAAC ligated boryl mono- and biradicals. | ||
The success of cAACs can be attributed to their stronger σ-donating and π-accepting properties, in comparison to NHCs, and also cAACs are sterically more demanding owing to the presence of a quaternary carbon atom adjacent to the carbene center.
In 2014, Braunschweig et al., Bertrand et al. and Stephan et al. independently reported the isolation and structural characterization of cAAC-supported neutral boryl radicals (Me2cAACBClDur)˙ (9),17a (Me2cAACBCl(C4H3S))˙ (10),17a and (cycAACBClN(SiMe3)2)˙ (11),17b respectively (Fig. 2). The EPR signal of 9 was detected as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet due to the hyperfine interaction with the 14N nucleus (a(14N) = 19 MHz) and the weaker hyperfine couplings, including coupling to boron were not resolved due to the spin delocalization onto the cAAC unit. However, attempts to determine the solid state structure of 10 failed. On the other hand, a broad signal in the EPR spectrum of 10 was observed, clearly confirming the presence of the expected radical species.17a Aminochloroboryl radical 11 exhibits similar hyperfine coupling constants in the EPR spectrum [a(11B) = 4.7, a(14N) = 18.4 and a(35Cl) = 2.5 MHz] compared to the arylchloroboryl radical (9). Braunschweig et al. isolated and crystallographically characterized a four membered 2π-aromatic system (12; Fig. 2) from the reaction of bis(Me2cAAC)B2 with propyne. Compound 12 has two unpaired electrons which are localized on the cAACs.17c The EPR spectrum of a frozen-solution sample of 12 in 2-methyltetrahydrofuran shows a weak half-field signal and a four-line spectrum in the g = 2 region which can be attributed to the triplet state of 12. In 2016, Chiu et al. reported the persistent boron-containing radical (cycAACBClCp*)˙ (Cp* = C5Me5) (13) (Fig. 2).17d The EPR signal of 13 at g = 2.0047 can be simulated with the hyperfine couplings to nitrogen (a(14N) = 6.50 G), boron (a(11B) = 1.60 G, a(10B) = 0.54 G), and chlorine (a(35Cl) = 1.50 G, a(37Cl) = 1.25 G) nuclei. The relatively small a(11B) coupling constant suggests that 13 should be better represented as a carbon-centered radical. The same group has reported a cyanide-containing boron radical (Me2cAACBCNDur)˙ (14; Fig. 2) which was characterized by EPR spectroscopy as well as X-ray diffraction.17e EPR spectroscopy of 14 revealed a multiple-line spectrum with giso = 2.0029, indicative of hyperfine interactions with both the nitrogen and boron atoms. The observed hyperfine coupling constants [a(14N) = 18.3 MHz and a(11B) = 10.7 MHz] suggest that the unpaired electron is primarily delocalized over the B–CcAAC–NcAAC linkage. Recently a dipotassium complex {[(Me2cAAC)DurB]2(μ2-N2K2)} was synthesized by Braunschweig et al. by the over-reduction of [(Me2cAAC)BClDur] with an excess of KC8 under nitrogen atmosphere.18a Treatment of the dipotassium complex with distilled water led to formation of the paramagnetic compound {[(Me2cAAC)DurB]2(μ2-N2H2)} (15) (Fig. 2) which was characterized by single crystal X-ray diffraction and EPR spectroscopy.18a The solid state EPR spectrum of 15 at 290 K revealed the typical signature of a triplet state, with a half-field signal at about g = 4 and zero-field splitting parameters of |D/hc| = 0.021 cm−1 and |E/hc| = 0.00083 cm−1. Very recently the neutral two-carbon bridged boron-based biradicals (16a–b) were reported by Braunschweig et al.18b In frozen toluene solution, 16a–b show evidence for a triplet state with a weak half-field signal. Theoretical calculations (using meta-NGA functional MN12Lin conjugation with the 6-311G(d,p) Pople basis set) indicate that singlet–triplet gap for 16a–b is nearly zero and each free electron is effectively delocalised on both sides of the molecule in the NcAAC–CcAAC–B π system; the calculated spin densities are (B 0.27; CcAAC 0.49; NcAAC 0.24).18b
:1:1 triplet due to the hyperfine interaction with the 14N nucleus (a(14N) = 19 MHz) and the weaker hyperfine couplings, including coupling to boron were not resolved due to the spin delocalization onto the cAAC unit. However, attempts to determine the solid state structure of 10 failed. On the other hand, a broad signal in the EPR spectrum of 10 was observed, clearly confirming the presence of the expected radical species.17a Aminochloroboryl radical 11 exhibits similar hyperfine coupling constants in the EPR spectrum [a(11B) = 4.7, a(14N) = 18.4 and a(35Cl) = 2.5 MHz] compared to the arylchloroboryl radical (9). Braunschweig et al. isolated and crystallographically characterized a four membered 2π-aromatic system (12; Fig. 2) from the reaction of bis(Me2cAAC)B2 with propyne. Compound 12 has two unpaired electrons which are localized on the cAACs.17c The EPR spectrum of a frozen-solution sample of 12 in 2-methyltetrahydrofuran shows a weak half-field signal and a four-line spectrum in the g = 2 region which can be attributed to the triplet state of 12. In 2016, Chiu et al. reported the persistent boron-containing radical (cycAACBClCp*)˙ (Cp* = C5Me5) (13) (Fig. 2).17d The EPR signal of 13 at g = 2.0047 can be simulated with the hyperfine couplings to nitrogen (a(14N) = 6.50 G), boron (a(11B) = 1.60 G, a(10B) = 0.54 G), and chlorine (a(35Cl) = 1.50 G, a(37Cl) = 1.25 G) nuclei. The relatively small a(11B) coupling constant suggests that 13 should be better represented as a carbon-centered radical. The same group has reported a cyanide-containing boron radical (Me2cAACBCNDur)˙ (14; Fig. 2) which was characterized by EPR spectroscopy as well as X-ray diffraction.17e EPR spectroscopy of 14 revealed a multiple-line spectrum with giso = 2.0029, indicative of hyperfine interactions with both the nitrogen and boron atoms. The observed hyperfine coupling constants [a(14N) = 18.3 MHz and a(11B) = 10.7 MHz] suggest that the unpaired electron is primarily delocalized over the B–CcAAC–NcAAC linkage. Recently a dipotassium complex {[(Me2cAAC)DurB]2(μ2-N2K2)} was synthesized by Braunschweig et al. by the over-reduction of [(Me2cAAC)BClDur] with an excess of KC8 under nitrogen atmosphere.18a Treatment of the dipotassium complex with distilled water led to formation of the paramagnetic compound {[(Me2cAAC)DurB]2(μ2-N2H2)} (15) (Fig. 2) which was characterized by single crystal X-ray diffraction and EPR spectroscopy.18a The solid state EPR spectrum of 15 at 290 K revealed the typical signature of a triplet state, with a half-field signal at about g = 4 and zero-field splitting parameters of |D/hc| = 0.021 cm−1 and |E/hc| = 0.00083 cm−1. Very recently the neutral two-carbon bridged boron-based biradicals (16a–b) were reported by Braunschweig et al.18b In frozen toluene solution, 16a–b show evidence for a triplet state with a weak half-field signal. Theoretical calculations (using meta-NGA functional MN12Lin conjugation with the 6-311G(d,p) Pople basis set) indicate that singlet–triplet gap for 16a–b is nearly zero and each free electron is effectively delocalised on both sides of the molecule in the NcAAC–CcAAC–B π system; the calculated spin densities are (B 0.27; CcAAC 0.49; NcAAC 0.24).18b
In contrast to anionic and neutral boron radicals, cationic boron radicals are extremely rare due to the intrinsic electron deficient nature of boron. However, the strong electron donating property of cAACs has been exploited to isolate cationic boron radicals. In 2011 Bertrand et al. first reported a bis(cAAC)borylene adduct [(cycAAC)2BH]. The boron atom of [(cycAAC)2BH] is in a formal +1 oxidation state with an active lone pair on boron and it behaves as an electron-donor.18c One-electron oxidation of (cycAAC)2BH by the use of gallium trichloride, quantitatively afforded the first crystallographically characterized radical cation 17 [(cycAAC)2BH]˙+[GaCl4]− (Fig. 2).18c The EPR spectrum (g = 2.0026) of 17 displays couplings with boron (a(11B) = 6.432 G), hydrogen (a(1H) = 11.447 G), and two nitrogen nuclei (a(14N) = 4.470 G). The EPR data and theoretical calculations indicate that the spin density is primarily located on the boron atom (50%) and the nitrogen atoms of the carbenes (33% total). The same research group in 2014 reported a bis-carbene stabilized radical cation 18 [(EtcAAC)(L2)BH] ˙+[CF3SO3]− (L2 = cyclopropenylidene) (Fig. 2) whose radical nature is persistent for several hours at room temperature.18d In contrast to 17, the cationic radical 18 was prepared by a single electron reduction of the bis(carbene) boronium triflate salt. The EPR spectrum of 18 displays couplings with hydrogen (a(1H) = 10.065 G), boron (a(11B) = 4.994 G), and one nitrogen (a(14N) = 6.627 G) which indicates that the unpaired electron is mainly delocalized over the cAAC and BH fragments, with little distribution on the cyclopropenylidene ligand. DFT calculations confirm that the spin density distribution is consistent with the EPR observations. Interestingly, in 2016, the Chiu group reported that treatment of the cAAC-stabilized boron cation [Cp*B(cycAAC)]2+2[AlCl4]− with a half equivalent of tetrakis(dimethylamino)ethylene generated the boron-containing radical cation 19.17d The EPR spectrum (g = 2.0032) can be simulated with hyperfine coupling constants of (a(11B) = 9.00 G), (a(10B) = 3.00 G), and (a(14N) = 6.20 G).17d
In spite of the progress on boron-centered radicals, as discussed above, there has been no report on an aluminum-centered radical till very recently. In 2017, we first reported the neutral radical of aluminum ((Me2cAAC)2AlCl2)˙ (20; Fig. 3), which was synthesized by the reduction of the Me2cAAC → AlCl3 adduct with KC8 in the presence of another equivalent of cAAC (Fig. 3).19 The EPR spectrum is dominated by a sextet that results from coupling of the unpaired electron with one aluminum nucleus (27Al, I = 5/2) (Fig. 4a). The hyperfine splitting [a(27Al) = 12.5 G] indicates a metal-based spin. However, the unusual aluminum radical 20 can be equally well described by the three resonance forms that are shown in Fig. 3 according to the quantum-chemical calculations at the BP86/TZVPP level of theory. Following this result we also reported the analogous cAAC-stabilized mono- and diorgano aluminum radicals [((Me2cAAC)2AlClEt)˙ (21) and ((Me2cAAC)2AlEt2)˙ (22)] (Fig. 3).20 The observed hyperfine splitting for 21 is a(14N) = 4.3 G and a(27Al) = 8.3 G while for 22 it is a(14N) = 4.95 G and a(27Al) = 5.2 G (Fig. 3b and c). Coupling is observed from only one nitrogen atom in both the cases confirming the localization of spin at a single cAAC ligand which is also observed from the asymmetric Ccarbene–Al bond distances [2.1507(13) and 1.9913(13) Å] found in the molecular structure of 21. Thus, with the replacement of chloride substituents by alkyl groups the spin density changes from aluminum to the cAAC carbon. The sequence of the hyperfine coupling constants was reproduced by DFT calculations.
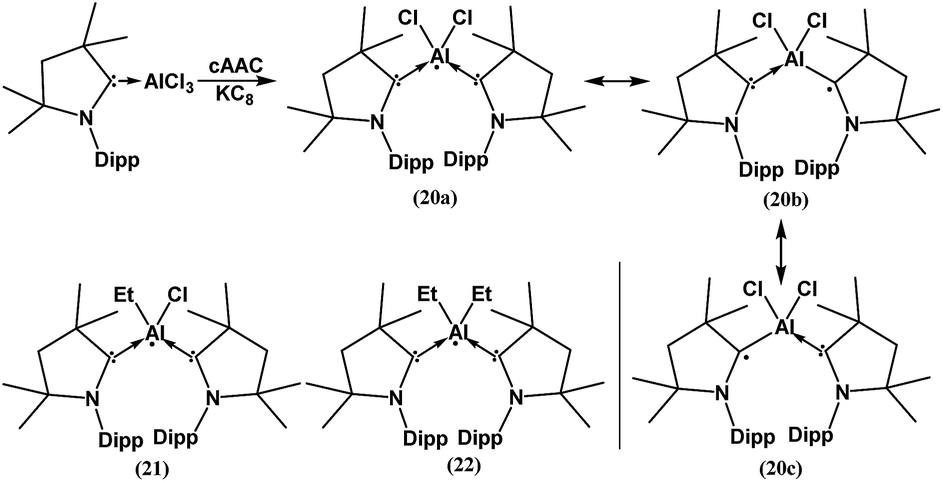 | ||
| Fig. 3 cAAC-supported neutral aluminium monoradicals (20–22). As an example, a general synthesis and possible resonance structures of 20 are given. | ||
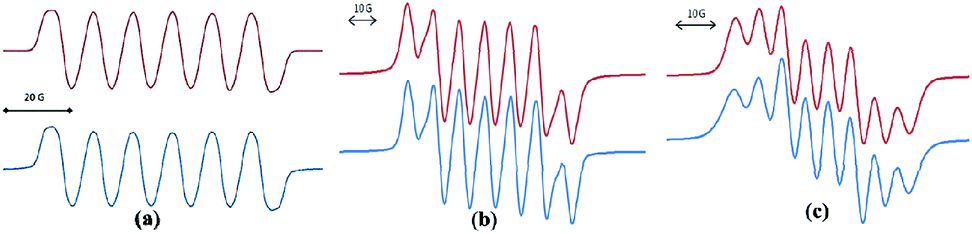 | ||
| Fig. 4 Room-temperature (a) EPR spectrum of 20 simulation with (g = 2.0055), a(27Al) = 12.5 G; a(2 35,37Cl) = 1.9 G; a(2 14N) = 0.25 G; (b) EPR spectrum of compound 21 simulation with a(14N) = 4.3 G, a(27Al) = 8.3 G; (c) EPR spectrum of compound 22 at room temperature simulation with a(14N) = 4.95 G, a(27Al) = 5.2 G (Top: simulated spectrum and Bottom: experimental spectrum). | ||
Group 14 radicals
As mentioned above, cAACs are efficient in the stabilization of paramagnetic species. In this section, persistent and stable group 14 radicals and radical ions associated with cAACs will be discussed. Table 2 summarizes the spin densities of group 14 radical species. Fukuzumi et al. reported the first carbene-stabilized carbon-based radical (Fig. 5) in 1997 which is persistent for several hours at room temperature. As shown in Fig. 5 the carbon-based radicals 23b–c were generated by the electrochemical oxidation of the corresponding thiazolylidene enolate.21 After this, several carbene-stabilized carbon radicals have been reported. Literature data indicate that most of the persistent and stable radicals of carbon contain cAACs because of the delocalization of the spin density over the ligand (24–33) (Fig. 6).12,22 In continuation of the theme on isolation of stable carbon centred radicals our group has contributed by isolating two types of cationic radicals (34–35; Fig. 7 and 8).| Compounds | Spin density | Type of radical | Ref. |
|---|---|---|---|
| 23 | Not given | Monoradical | 21 |
| 24 | 40% CcAAC, 28% O, 24% NcAAC, 6% C | Monoradical | 22a |
| 25 | Not given | Disbiradical | 22a |
| 26a | 42% CcAAC, 28% O | Monoradical | 22b |
| 26b | 39% CcAAC, 29% O | Monoradical | 22b |
| 26c | 37% CcAAC, 31% O | Monoradical | 22b |
| 26d | 31% CcAAC, 33% O | Monoradical | 22b |
| 27 | 19% CcAAC, 36% O | Monoradical | 22b |
| 28 | Not given | Dis-triradical | 22a |
| 29 | Not given | Triplet | 22c |
| 30 | Not given | Triplet | 22c |
| 31 | 41.1% CcAAC, 21.1% NcAAC, 25% (C4F4N) | Monoradical | 22d |
| 32 | 33% CcAAC, 23% NcAAC, 10% CNHC | Monoradical cation | 22e |
| 33 | 25% CcAAC, 20% NcAAC, 9% CNHC | Monoradical cation | 22e |
| 34 | Not given | Monoradical cation | 23 |
| 35a | 60% on C4 unit, 40% NcAAC | Monoradical cation | 24 |
| 35b | 44% NcAAC, 24% CcAAC, 31% CcAAC | Monoradical cation | 25 |
| 36c | 43% CcAAC, 15% NcAAC, 39% CtBu, 2.5% Cment | Monoradical | 26b |
| 36d | 45% CcAAC, 17% NcAAC, 38% CCPh3 | Monoradical | 26b |
| 37a | Not given | Biradicaloid | 26c |
| 37b–c | Not given | Disbiradical | 26c |
| 38a | 58% cAAC, 38% linker | Monoradical cation | 26d |
| 38b | 50% cAAC, 50% linker | Monoradical cation | 26d |
| 38c | 52% cAAC, 48% linker | Monoradical cation | 26d |
| 39 | (37–45%) Ccarbene, (8–11%) Ncarbene, (19–23%) Cortho, (19–29%) Cpara | Monoradical | 26e |
| 40 | Not given | Biradicaloid | 26f and g |
| 41 | Not given | Disbiradical | 27 |
| 42 | Not given | Disbiradical and OSS | 28 |
| 43 | 90% CcAAC, 10% NcAAC | Disbiradical | 29 |
| 44 | 65% CcAAC, 18% NcAAC | OSS | 30 |
| 45a–b | 71% CcAAC, 22% NcAAC, 6% Si | Monoradical | 31 |
| 45c | 66% CcAAC, 25% NcAAC | Monoradical | 30 |
| 46 | 80% CcAAC, 16% NcAAC, 5% Si | Monoradical | 32 |
| 47 | Not given | CSS biradicaloid | 33 |
| 48 | Not given | Monoradical anion | 35 |
| 49 | Not given | OSS | 36 |
| 50 | 95% CcAAC, 5% NcAAC | Disbiradical | 29 |
| 51 | 92% CcAAC, 8% NcAAC | Disbiradical | 29 |
| 52 | 52% Si | Monoradical cation | 37 |
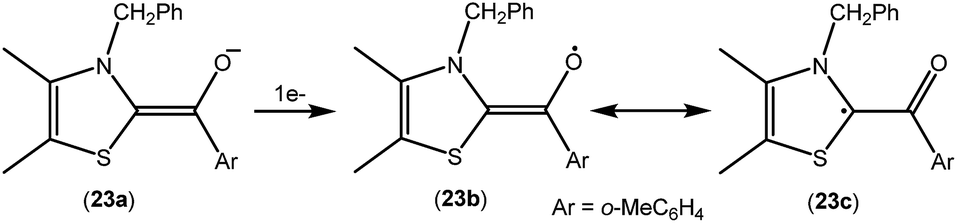 | ||
| Fig. 5 Oxidation of the thiazolylidene based enolate to a carbon-radical. | ||
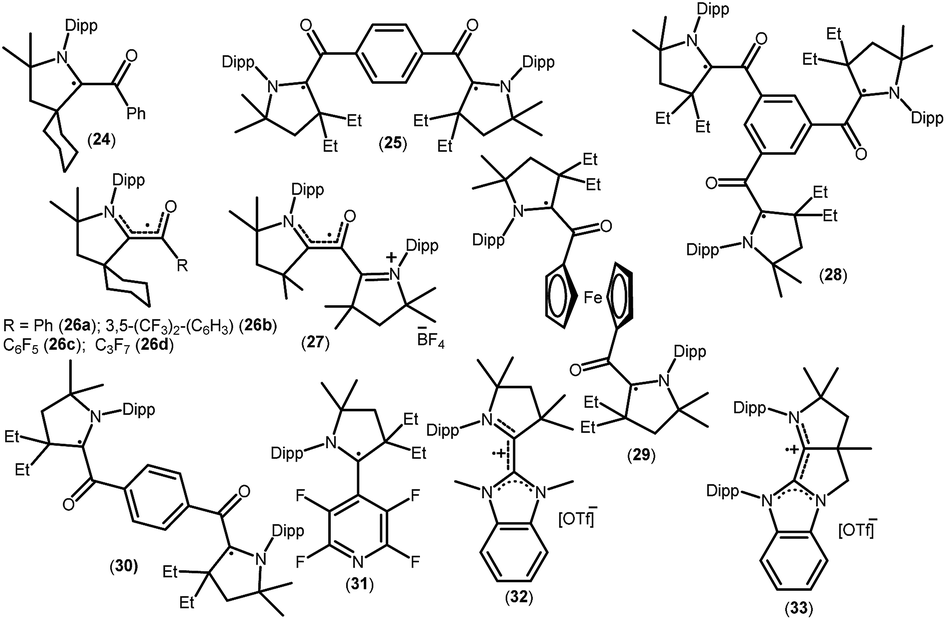 | ||
| Fig. 6 cAAC-derived carbon radicals. | ||
 | ||
| Fig. 7 Monoradical cations derived from cAACs. | ||
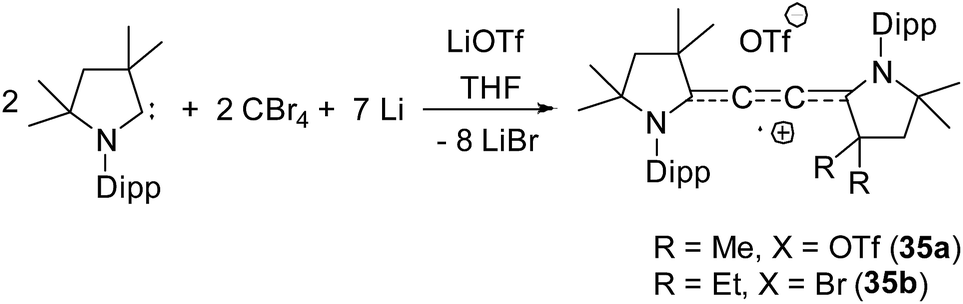 | ||
| Fig. 8 Synthesis of di-cAAC stabilized C2˙+. | ||
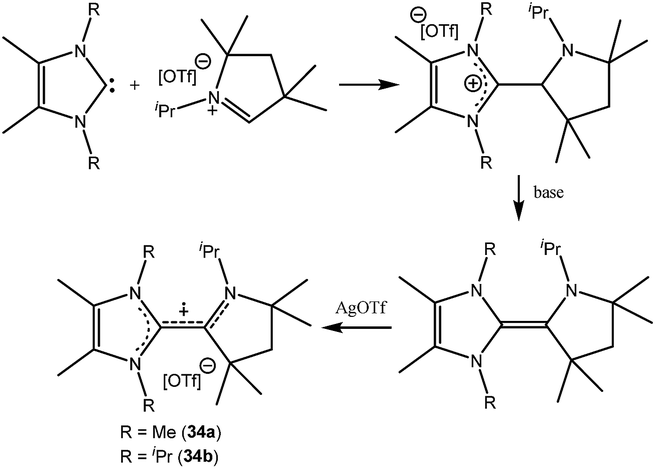
Organic cationic radicals [Me2cAAC-NHC]˙+ OTf− (34, Fig. 7) were obtained from the one-electron oxidation of the NHC-cAAC heterodimers which were synthesized by the reaction of NHC with a cyclic iminium salt, followed by deprotonation.23 X-band EPR measurements of the radical cations (34) in THF at room temperature revealed that the unpaired electron couples with all the three nitrogen atoms as indicated by the presence of hyperfine coupling [34a: 16.8, 13.2, and 12.0 MHz; 34b: 19.8, 12.7, and 10.4 MHz]. We and Bertrand and co-workers independently reported the radical cations [(Me2cAAC)2C2]˙+ OTf− (35a) and [(Et2cAAC)2C2]˙+Br− (35b) respectively, which can be viewed as a C2˙+ fragment, stabilized by two carbenes (Fig. 8).24,25 The synthetic routes used to prepare these di-carbene-stabilized C2˙+ compounds were, however, different. We obtained the dark red coloured cationic radical species [(Me2cAAC)2C2]˙+OTf− (35a) from the reduction of CBr4 with 3.5 equivalents of lithium sand in the presence of one equivalent of Me2cAAC·LiOTf. On the other hand, Bertrand and co-workers isolated [(Et2cAAC)2C2]˙+Br− (35b) from the reaction of [(Et2cAAC)Br]+Br− with LiC2SiMe3. Since the radical cations 35a and 35b are readily prepared and are surprisingly air stable, these results open up the possibility for the preparation of a variety of organic mixed valence systems using cAACs as stabilizing groups along with different radical cation spacers. The relatively small 14N hyperfine coupling [a(14N) = 5.3 G; quintet 1:2:3:2:1 for two equivalent nitrogen atoms] indicates the concentration of spin on the C4 backbone with limited participation of the nitrogen center of the carbenes.
Bertrand and co-workers have reported the synthesis of the allenyl/propargyl radicals {[Et2cAAC(C2Ph)]˙ (36a); [MentcAAC(C2Ph)]˙ (36b); [MentcAAC(C2tBu)]˙ (36c); [Et2cAAC(C2CPh3)]˙ (36d); Fig. 9}, by the one-electron reduction of the corresponding alkynyl-iminium salts [(36+a–d)(SbF6−)] using cobaltocene.26a,b The desired alkynyl-iminium precursors [(36+a–d)(SbF6−)] were prepared in two steps. In the first step, oxidative insertion of the sp-hybridized C–H bonds of the alkyne to the cAAC centre occurred at room temperature. Subsequently, hydride abstraction from the oxidatively inserted product by a stoichiometric amount of DDQ, followed by treatment with NOSbF6, afforded alkynyl-iminium salts in good yield. The stability of these radicals vastly differ, being short-lived, [Et2cAAC(C2Ph)]˙ (36a), or stable, [Et2cAAC(C2CPh3)]˙ (36d), depending on the nature of the cAAC and the alkyne substituents. Single crystal X-ray diffraction studies indicate that 36a–b dimerize in the solid state; however, 36d exists as a monomer in the solid state. The EPR spectrum of 36b shows the hyperfine couplings with N and the ortho, meta and para Hs and with one H atom of the menthyl group [a(14N) = 4.3 G; a(1H) = 2.9 G; a(1H) = 0.5 G; a(1H) = 2.8 G; a(1H) = 2.3 G]. The X-band EPR spectrum of 36d in solution reveals a triplet (a(14N) = 4.45 G).26b
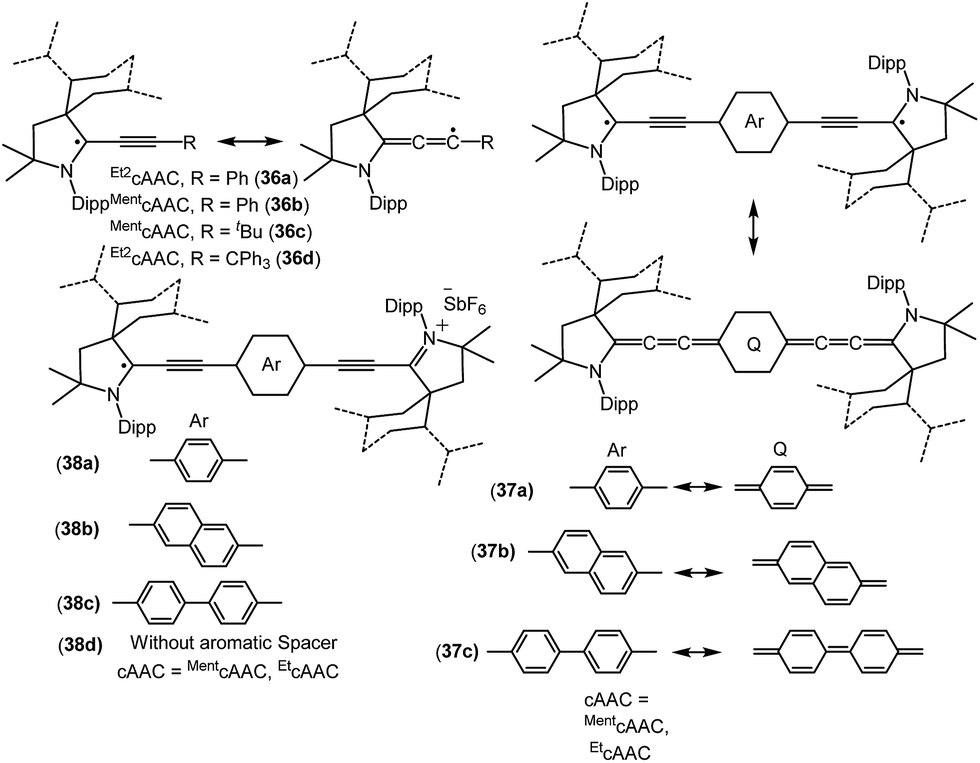 | ||
| Fig. 9 cAAC stabilized mono-, di- and biradicals over extended carbon systems. | ||
Similar to the above, an interesting study on cAAC supported compounds, [1,4-[(cAAC)˙C2]2(Ar) where Ar = C6H4 (37a); C10H6 = (37b); C12H10 (37c)] were recently reported by Bertrand et al. (Fig. 9).26c Analogous to the synthesis of 36, bis(2-acetylenyliminium) salts which were obtained from the reactions of cAACs and bis(acetylenes) in the initial step, undergo reduction with 2 equivalents of cobaltocene to give the respective biradicaloid/disbiradical compounds 37a–c (Fig. 9).26c The (U)CAM-B3LYP/6-31G** and (U)M05-2X/6-31G** calculations indicate that 37a–c possess biradical and quinoid resonance structures as indicated in Fig. 9 and have a OSS ground state with varying degrees of biradical character in combination with small singlet–triplet gaps. It has been observed that upon increasing the length of the spacer, the properties of the compounds approach those of monoradicals. The EPR spectrum of 1,4-[(Et2cAAC)˙C2]2 C6H4 (37a) in pentane at room temperature shows a triplet with isotropic hyperfine coupling constants with nitrogen (a(14N) = 5.6 G). In the solid state, at elevated temperature (325 K), a half-field signal (g = 1682 G) was detected, demonstrating the triplet nature of 37a. Theoretical calculations ((U)CAM-B3LYP/6-31G** and (U)M05-2X/6-31G**) reveal a singlet ground state with considerable triplet character, i.e., population of the antibonding lowest unoccupied molecular orbital (LUMO) by 0.3 electrons (biradical index of 0.3), and a singlet–triplet gap of 15.2 kcal mol−1 were obtained for 37a. With the increased spacer length the radical did not interact, so 37b–c are disbiradicals. Bertrand and co-workers have also reported the related radical-cations 38a–c [(MentcAAC)˙C2(Ar)C2MentcAAC)(SbF6) where Ar = C6H4 (38a); C10H6 (38b); C12H10 (38c) (Fig. 9)] featuring aromatic spacers and acetylenic units, as well as without an aromatic spacer (Et2cAAC)˙C4(Et2cAAC)(SbF6) (38d) (Fig. 9) which have been synthesized from the corresponding bis(2-acetylenyliminium) salts by using 1 equiv. of cobaltocene or zinc as a reducing agent.26d Compounds 38a–d are EPR active and show characteristics of mixed valent compounds. The X-band EPR spectrum of 38d in THF at room temperature can be simulated by involving coupling with two equal nitrogen nuclei, a(14N) = 4.14 G. Interestingly, the EPR spectrum of [(EtcAAC)2C2]+˙(35b) shows similar hyperfine splitting pattern but with larger N-coupling constants (a(14N) = 5.3 G), which indicate a decreased spin density on the nitrogen atom of cAAC in (Et2cAAC)˙C4(Et2cAAC)(SbF6) (38d) and thus a larger electron delocalization. Similarly, the hyperfine coupling constants decrease with the increase of the spacer length [38a (a(14N) = 2.70 G), 38b (a(14N) = 2.30 G), and 38c (a(14N) = 2.22 G] indicating a symmetrical delocalization of the unpaired electron over the central spacer.26d Very recently classical NHCs have been employed by Ghadwal et al. to isolate analogous carbon based radicals (39–40; Fig. 10) where the spin density is delocalized over an extended π system.26e–g
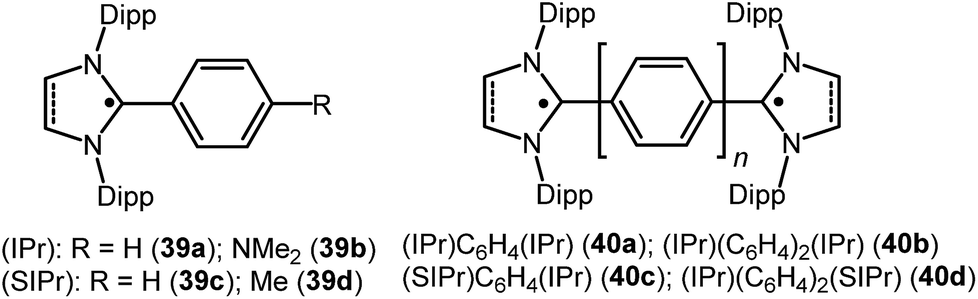 | ||
| Fig. 10 NHC stabilized radicals over extended carbon systems. | ||
As with carbon-centered radicals, the strong σ-donor and π-acceptor properties of cAACs have been exploited to isolate various neutral and ionic radicals of silicon. The most striking examples of the ability of cAACs to stabilize neutral radical species of silicon have been reported by us. Thus, we have reported that the reaction of three equivalents of cAAC with one equivalent of (NHC)SiCl2 adduct gives deep blue coloured biradicals 41a–b [(Me2cAAC˙)2SiCl2 (41a) and (CycAAC˙)2SiCl2 (41b); Fig. 11].27
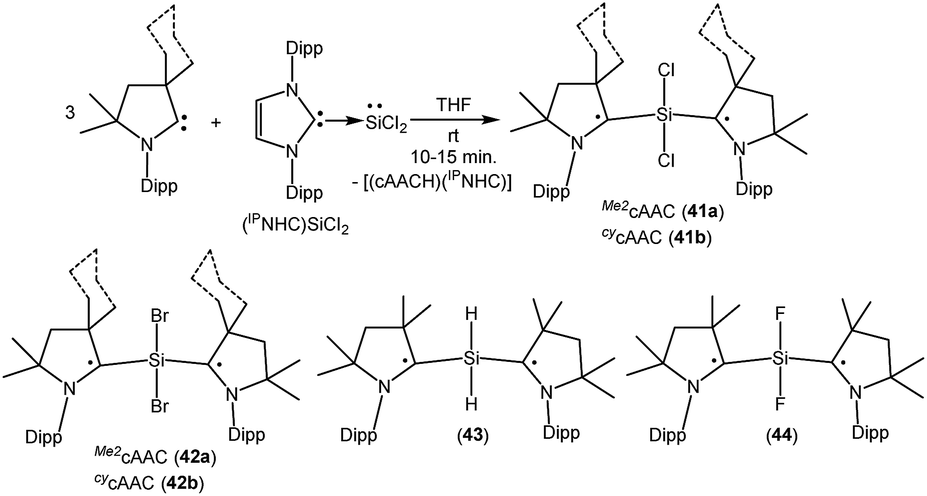 | ||
| Fig. 11 cAAC coordinated SiR2 (R = Cl, Br, H, F) dis- and biradicals. | ||
Unlike (NHC)SiCl2, in compounds 41a–b the two nonbonding electrons are not localized on silicon, rather, an unpaired electron resides on each carbene center. Moreover, we were able to isolate both the disbiradical (polymorph I) and open shell singlet (polymorph II) as two polymorphs via fractional crystallization of 41a, the singlet being air stable for up to a week in the solid state. The EPR spectrum of the diluted C6D6 solution of the polymorph I of 41a shows six hyperfine lines at room temperature (a(14N) = 5.7 G). The EPR spectrum could be simulated as a disbiradical species (two Seff = 1/2), each electron interacting with the closest 14N nucleus. As expected for a disbiradical, the half field (Δms = ±2 forbidden) transition was not observed either in the solid state or in solution of polymorph I of 41a. Theoretical calculations on 41a at the UM05-2X/SVP level using Gaussian 09 revealed that the open shell singlet state is 2.6 kcal mol−1 lower in energy than the triplet (3.2 kcal mol−1 in single-point energy calculations using the TZVPP basis set). By following a similar synthetic strategy used for 41a–b, we also reported the bromide analogues 42a–b [(Me2cAAC˙)2SiBr2 (42a) and (cycAAC˙)2SiBr2 (42b)] (Fig. 11) using (NHC)SiBr2 as a precursor.28 It has been observed that the bromide analogues (42a–b) are comparatively less stable than the chloride analogues (41a–b) and are more prone to decomposition in the solution. No EPR characterization was possible for (42a–b); however, compound (cycAAC˙)2SiBr2 (42b) was characterized by a single crystal X-ray diffraction study.
As there were no suitable precursors available for the synthesis of [(Me2cAAC˙)2SiH2] (43, Fig. 11), we prepared it from a direct reduction of H2SiI2 with two equivalents of KC8, in the presence of two equivalents of cAAC by developing a one step synthetic strategy.29 However unlike 41, polymorph formation was not observed in 43 and the theoretical calculations using density functional theory (DFT) at the M06-2X/def2-TZVPP level using M06-2X/def2-SVP optimized geometries of the two molecules in the electronic singlet and triplet states suggest that the triplet state of 43 is lower in energy than the respective open shell singlet state by 9.3 kcal mol−1. The EPR spectrum of 43 exhibits hyperfine splitting (a(14N) = 6.2 G) and a satellite coupling (a(29Si) = 23 G). The small 14N hyperfine splitting is in agreement with calculations that the bulk of spin density resides on the cAAC carbon. Very recently, we have isolated the elusive SiF2 species as a cAAC coordinated biradical [(Me2cAAC˙)2SiF2 (44); Fig. 11].30 Compound 44 was prepared from the reduction of (Me2cAAC)SiF4 adduct by using two equivalents of KC8 in the presence of one equivalent of cAAC. Theoretical calculations on 44 at M05-2X/def2-TZVPP//M05-2X/def2-SVP level suggest that the open-shell singlet is lower in energy than the triplet and closed-shell singlet by 4.9 kcal mol−1 and 10.2 kcal mol−1 respectively. Previous calculations on (Me2cAAC˙)2SiCl2 (41a) also indicated that the open-shell singlet is lower in energy by 3.2 kcal mol−1 than the triplet. Although both forms were experimentally found as a paramagnetic polymorph I (minor component) and a diamagnetic polymorph II (major component) for 41a, the fluoride homologue 44 appears only as a EPR-silent compound. A triplet at −29.73 ppm and a broad singlet −123.47 ppm were observed in the 29Si{1H} and 19F{1H} NMR spectra respectively at low temperatures.
We have also reported that the (cAAC)SiCl4 adduct can be converted to isolable trichlorosilylcarbene radicals (CycAAC˙)SiCl3 (45a) and (Me2cAAC˙)SiCl3 (45b) (Fig. 12) through a one equivalent KC8 reduction in hexane.31 The use of non-polar solvent (n-hexane) is important for the selective reduction of (cAAC)SiCl4 to (cAAC˙)SiCl3 (45a–b) at room temperature. Theoretical calculations show that the unpaired electron is mainly located on the carbene carbon of cAAC (52%), with a smaller contribution (23%) from the nitrogen atom of cAAC and the remaining 25% electron density is distributed over the Dipp/cAAC units and one of the Cl atoms. The EPR spectra of 45a–b in C6D6 solution show multiple hyperfine lines. The EPR spectrum of 45a reveals hyperfine coupling [a(14N) = 6.4 and 3.4 G (1 Cl) and 2.7 G (2 Cl), with three Cl atoms (I = 3/2, nat. abundance of 35Cl: 75.77%, 37Cl: 24.23%; gyromagnetic ratio = 1.20)] which suggests a partially hindered rotation of the SiCl3 group around the carbon–silicon bond. Very recently we have also reported the synthesis of a silicon trifluoride monoradical (Me2cAAC˙)SiF3 (45c; Fig. 12) from (Me2cAAC)SiF4 adduct by adopting a similar synthetic strategy as used for preparing (cAAC˙)SiCl3 (45a–b).30,31 The EPR spectrum of 45c in hexane at room temperature shows multiple hyperfine coupling constants [a(14N) = 6.9 G, a(29Si) = 10 G, a(19F, 3F) = 16.8 G, a(1H, 3H) = 1.2 G.]. The trichlorosilylcarbene radical (45a) was directly converted to (cycAAC˙)SiPh3 (46; Fig. 12) in 90% yield by substitution of the three chlorine atoms with phenyl groups using PhLi without affecting the radical center adjacent to the silicon atom.32 The X-band EPR spectrum of 46 exhibits three hyperfine lines due to coupling with one nitrogen nucleus (a(14N) = 5.4 G). Additionally, satellites for the silicon and three carbon atoms (a(29Si) = 8.0 G and a(13C) = 25 G), are also identified through simulation of the EPR spectrum of 46.
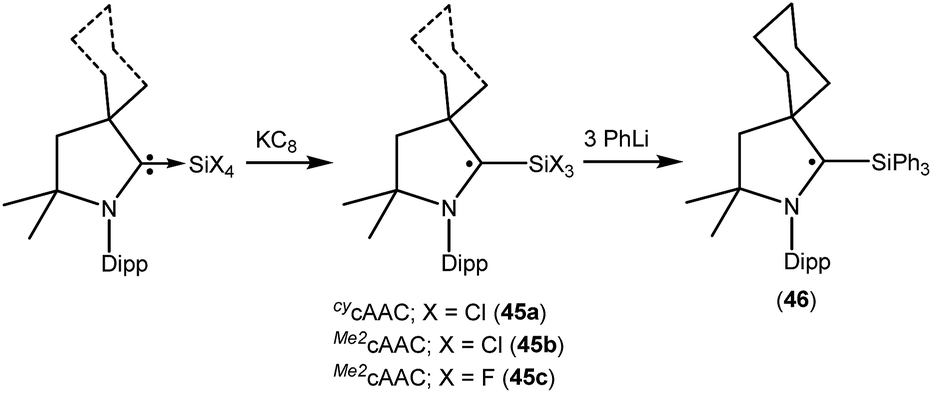 | ||
| Fig. 12 Synthesis of (cAAC˙)SiR3 (R = Cl, F or Ph). | ||
A structurally characterized, stable siladicarbene (Me2cAAC)2Si (47, Fig. 13) was reported by us by the reduction of 41a with two equivalents of KC8.33 Compound 47 has singlet spin ground state which has been confirmed by magnetic susceptibility and EPR measurements. The 29Si NMR spectrum of (Me2cAAC)2Si (47) exhibits a singlet at 66.71 ppm which is downfield shifted when compared with that of the precursor (Me2cAAC˙)2SiCl2 (41a) (4.13 ppm). However, the dark blue color of 47 suggests a small HOMO–LUMO gap. The experimental charge density calculations have confirmed the presence of two pairs of electrons on the silicon atom of 47.34 Calculations at various levels of theory using the M05-2X/SVP optimized geometries predict that the triplet form is between 17.2 kcal mol−1 (M05-2X/TZVPP) and 18.5 kcal mol−1 (B3LYP/TZVPP//M05-2X/SVP) higher in energy than the singlet. CASSCF(2,2)/SVP calculations using the M05-2X/SVP optimized geometry gave coefficients of 0.96 for the closed-shell 2,0 configuration, −0.28 for the 1,1 configuration, and 0.0 for the 0,2 configuration which indicate that 47 has a closed-shell singlet with a non-negligible contribution from the singly excited state.
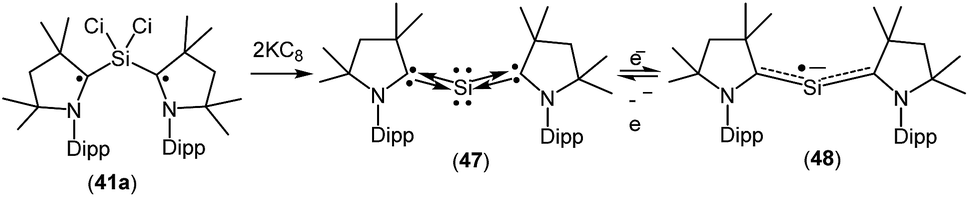 | ||
| Fig. 13 cAAC-stabilized siladicarbene and in situ generation of (Me2cAAC)2Si˙−. | ||
When 47 was stirred in THF at 298 K in the presence of potassium metal for 30 min a radical anion [(Me2cAAC)2Si˙− (48); Fig. 13] was generated.35 The EPR spectrum of (Me2cAAC)2Si˙− (48) reveals five hyperfine lines at g = 2.0058 indicating the coupling of a radical electron with two equivalent nitrogen nuclei (a(14N) = 5.89 G). Two satellites (a(13C) = 40 G; I = 1/2) are observed due to coupling with CcAAC atoms which indicates that the unpaired electron is delocalized in the C–Si–C back bone of 48. The Mulliken spin density plot of the radical anion (48) shows that the unpaired electron is delocalized between the two carbene carbon atoms of cAAC through the vacant d-orbitals of the silicon atom.
Treatment of 45a with one equivalent of KC8 in THF at low temperature generates a green- colored silane-bridged biradical disilicontetrachloride (cycAAC˙)2Si2Cl4 (49, Fig. 14).36 Compound 49 can also be obtained from the direct reduction of (cycAAC)SiCl4 with two equivalents of KC8. Theoretical calculations (M06-2X/TZVP//M06-2X/SVP level) revealed that 49 possesses an open shell singlet ground state with an unpaired electron residing on each CcAAC atom having opposite spins. The open shell singlet state of 49 was found to be lower in energy than the triplet state by 2.8 kcal mol−1. Accordingly, 49 is EPR silent and reveals a chemical shift at 3.3 ppm in its 29Si{1H} NMR spectrum. Very recently the analogous compounds (Me2cAAC˙)SiMe2–SiMe2(Me2cAAC˙) (50; Fig. 14) and (Me2cAAC˙)SiMeCl–SiMeCl(Me2cAAC˙) (51; Fig. 14) were also reported by us.29 Compounds 50 and 51 were synthesized by a direct reduction of the commodity precursors Me2SiCl2 and MeSiCl3, respectively with KC8 in 1:2 molar ratios in the presence of one equivalent of cAAC. It has been observed that the replacement of chlorine atoms of 49 by methyl groups leads to the disbiradical 50. The EPR spectrum of 50 at room temperature in hexane shows a 1:1:1 triplet with weak satellite signals because of hyperfine coupling with one nitrogen atom (a(14N) = 4.8 G) and with a silicon atom (a(29Si) = 25.7 G). No half-field signal was observed for 50. Compound 51, where one methyl and one chlorine atom is attached to each silicon center also shows paramagnetic behavior. EPR spectrum of 51 in hexane at room temperature shows hyperfine splitting with a(14N) = 4.0 G, a(CH3) = 5.3 G, a(35,37Cl) = 4.5 G, and a(29Si) = 16 G.29 Similar to compound 50, EPR data of 51 indicates that there is no significant spin–spin interaction. Theoretical calculation (M06-2X/def2-TZVPP//M06-2X/def2-SVP) of the two molecules in the electronic OSS and triplet states suggest that the triplet states of 50 (−11.3 kcal mol−1), and 51 (−12.1 kcal mol−1) are lower in energy than the respective OSS states. The calculated spin density distributions show that the unpaired electrons are located at the carbene carbon and nitrogen atoms of the cAAC ligands for compounds 50 and 51.
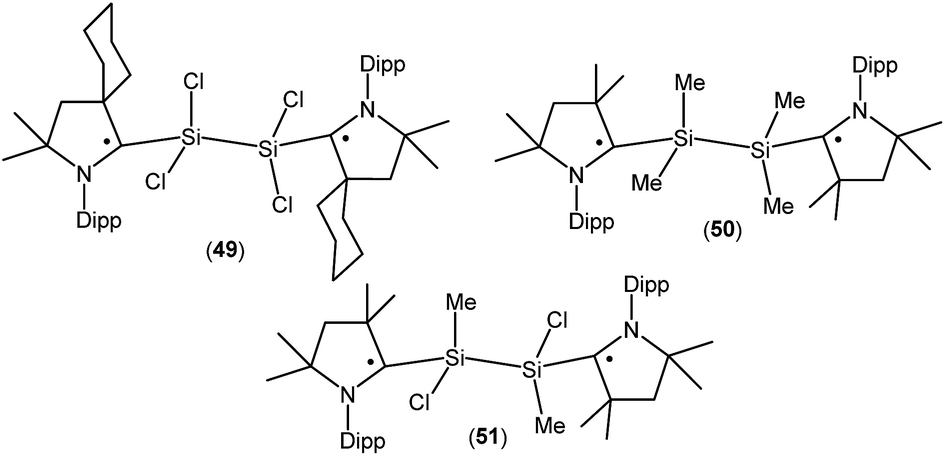 | ||
| Fig. 14 Silane-bridged dis- and biradicals. | ||
Isolation of a purple colored cationic silicon(I) radical [(Me2cAAC)2 Si:˙]+I− (52; Fig. 15) was reported by So et al. in 2017 from the direct reaction of cAAC with H2SiI2 in THF or 1,2-dimethoxyethane solvent.37 Theoretical calculations showed that the unpaired electron is mainly localized on the Si atom (0.52 e) with a slight delocalization on one of the cAAC ligands. Room temperature EPR analysis in 1:1 toluene/THF solvent showed a three line hyperfine multiplet pattern (a(14N) = 18.5 MHz). This characteristic splitting pattern indicates that the radical is delocalized on only one of the cAAC ligands, which is consistent with the calculated spin densities of 52. A pair of weak satellite signals (a(29Si) = 212 MHz) was observed in the EPR spectrum recorded at 115 K.
 | ||
| Fig. 15 cAAC-stabilized monoradical cation of Si(I). | ||
Group 15 radicals
Phosphorus-centered radicals are attracting much attention as ligands for their use in spin-labelling experiments because of their orientation dependence of the large hyperfine coupling with 31P. The large hyperfine coupling constant provides details regarding much faster molecular movements than is possible with the widely used nitroxide probes. A number of cAAC-stabilized group-15 elements-centered radicals have been isolated by various groups which are given in Fig. 16.26a,38Table 3 summarizes the spin densities of group 15 radical species.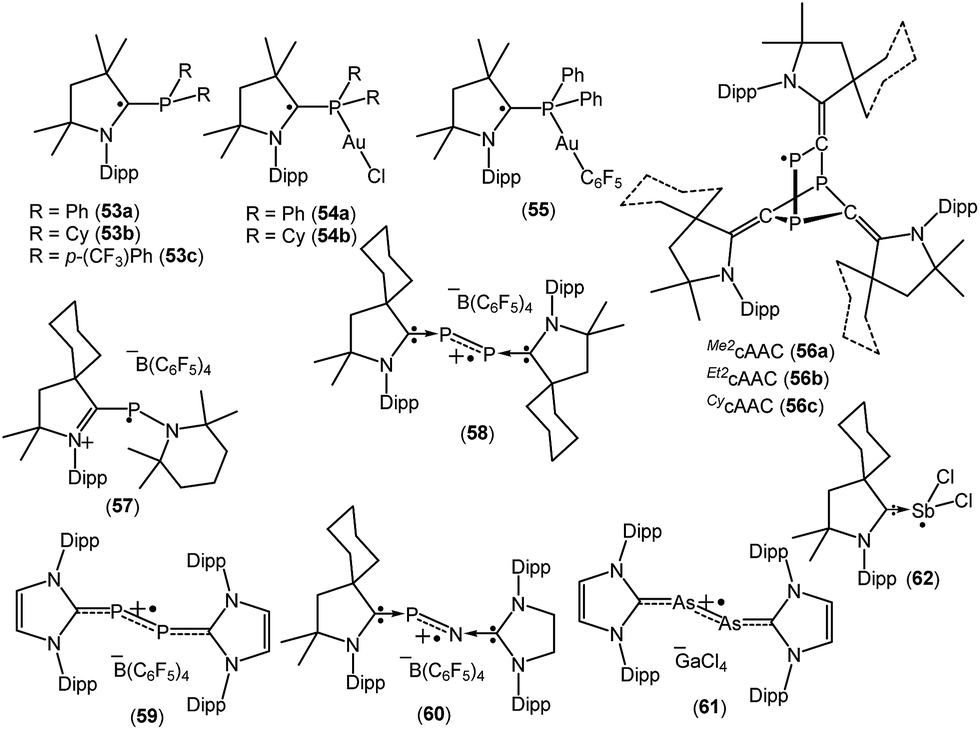 | ||
| Fig. 16 cAAC stabilized group-15 radicals. | ||
| Compounds | Spin density | Type of radical | Ref. |
|---|---|---|---|
| 53a–c | (65–70%) Ccarbene, (20–25%) Ncarbene, 1% P | Monoradical | 38a |
| 54a–b, 55 | (60–67%) CcAAC, (23–28%) NcAAC, (1–2%) P, (0.1–0.5%) Au | Monoradical | 38a |
| 56a–c | 73–76% P | Monoradical | 38b |
| 57 | 67% P, 16% N, 10% Ncarbene | Monoradical cation | 38c |
| 58 | 27% P (each), 14% NcAAC (each) | Monoradical cation | 38d |
| 59 | 33% P, 44% P | Monoradical cation | 38d |
| 60 | 40% P, 18% N, 19% NcAAC | Monoradical cation | 38e |
| 61 | 41% As (each) | Monoradical cation | 38f |
| 62 | 90.7% Sb, 4.6% Cl, 3.9% Cl | Monoradical | 38g |
| 63a–b | 75% CcAAC, 19% NcAAC, 3.6% Si, 5% P | Monoradical | 39 |
| 64 | 81% CcAAC, 9% NcAAC, 6% Si (total), 6% P (total) | Monoradical anion | 40 |
| 65 | 52% CO (40% O and 12% C), 26% Ncarbene (total), 19% Ncarbene (total) | Monoradical cation | 41 |
Recently Alcarazo and co-workers have reported the successful isolation of cAAC-stabilized neutral α-radical phosphines (53a–c; Fig. 16), which were obtained by a one-electron reduction by KC8 from their corresponding cationic precursors.38a These compounds were characterized by X-ray diffraction and EPR spectroscopy. The hyperfine splitting in the EPR spectrum of 53a–c differ and show distinct delocalization of spin density onto the adjacent nitrogen and phosphorus atoms. The extent of delocalization depends on the substituent at the phosphorus centre. Six signals were observed in the EPR spectra of 53a and 53c, which is due to the coupling of the electron spin with the 31P and 14N nuclei [a(31P) = 18.0 G and a(14N) = 6.0 G for 53a, and a(31P) = 14.7 G and a(14N) = 6.1 G for 53c]. In comparison to 53a and 53c, a triplet was observed for 53b which exhibits almost negligible coupling with 31P nuclei (a(31P) = 1.0 G), but similar coupling with 14N (a(14N) = 5.17 G) as in 53a and 53c. Theoretical calculations revealed that the spin density in 53a–c is mainly located at the cAAC ligands (ca. 65–70% on the carbene carbon atoms, ca. 20–25% on the N atoms), but only some residual spin density at the P center (ca. 1%). Interestingly, these α-radical phosphines (53a–b) can form Au(I) complexes [{(Me2cAAC)PPh2}-AuCl]˙ (54a), [{(Me2cAAC)PCy2}-AuCl]˙ (54b) and [{(Me2cAAC)PPh2}AuC6F5]˙ (55) without affecting the radical centre, thus serving as spin-labelled ligands.38a The EPR spectra of 54–55 is entirely dominated by hyperfine coupling with 31P and 14N nuclei and the absence of hyperfine coupling with 197Au (I = 3/2, natural abundance 100%) indicates that the metal and the radical system are electronically isolated from each other. Theoretical calculations confirmed that the unpaired electron in 54a–b and 55 is primarily located on the carbene carbon atom (ca. 60–67%), with significant contributions from the nitrogen atom of cAAC (23–28%), and rather small contributions from phosphorus (ca. 1–2%) and the predicted spin densities at gold centre are only residual (0.1–0.5%).38a
In 2018, Grützmacher et al. reported a series of tricarbontriphosphide tricyclic radicals [(Me2cAAC)3C3P3 (56a); (Et2cAAC)3C3P3 (56b); (CycAAC)3C3P3 (56c)] (Fig. 16). These were synthesized from a one equivalent reduction of the corresponding precursors cAAC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CP–[P(O){(N-Dipp)CH}2] with KC8.38b The reduction of cAACCP–[P(O){(N-Dipp)CH}2] leads to the formation of the carbene-bound CP radical [cAACCP]˙, which further trimerizes to give the tricyclic radical [(cAAC)3C3P3]˙ (56a–c). The EPR spectra of the hexane solutions of 56a–c at room temperature show a doublet because of the large isotropic 31P hyperfine interaction (aiso ≈ 187 MHz), which indicates that a large percentage of the spin density is localized on one of the phosphorus nuclei. Small 14N hyperfine couplings [a(14N) = 11 MHz for 56a; a(14N) = 12 for 56b and a(14N) = 9 MHz for 56c] were observed. These estimations of the spin populations from EPR (0.73–0.74 e on P) agreed well with DFT results (0.76 e on P).38b In 2010, Bertrand reported that CycAAC reacts with R2NPCl2, [(2,2,6,6-tetramethylpiperidino)phosphine dichloride] to generate [(CycAAC)P(Cl)(NR2)]+Cl− which can be reduced to aminophosphinidene [(CycAAC)PNR2] with magnesium metal.38c This aminophosphinidene was converted to its stable radical cation [(CycAAC)(NR2)P]˙+(C6F5)4B− (57) by a one-electron oxidation with Ph3C+B(C6F5)4− in benzene at room temperature (Fig. 16). The radical cation 57+˙ was characterized by single crystal X-ray diffraction and by EPR spectroscopy.38c The EPR spectrum of 57+˙, in fluorobenzene, at room temperature, shows a doublet of multiplets (g = 2.007) due to a large hyperfine coupling with 31P [a(31P) = 99 G] and a small coupling with one or two 14N nitrogen nuclei [a(14N) ≈ 4 G]. Consistent with the EPR data, theoretical calculations confirmed that the spin density in 57+˙ is localized mainly at phosphorus (67%) with small contributions from the nitrogen atoms (16% for piperidino nitrogen and 10% for cAAC nitogen).38c The authors have mentioned that the exceptional stability of 57+˙ is partly due to steric factors but more importantly because of cationic substituent which prevents the dimerization by electrostatic repulsion.
CP–[P(O){(N-Dipp)CH}2] with KC8.38b The reduction of cAACCP–[P(O){(N-Dipp)CH}2] leads to the formation of the carbene-bound CP radical [cAACCP]˙, which further trimerizes to give the tricyclic radical [(cAAC)3C3P3]˙ (56a–c). The EPR spectra of the hexane solutions of 56a–c at room temperature show a doublet because of the large isotropic 31P hyperfine interaction (aiso ≈ 187 MHz), which indicates that a large percentage of the spin density is localized on one of the phosphorus nuclei. Small 14N hyperfine couplings [a(14N) = 11 MHz for 56a; a(14N) = 12 for 56b and a(14N) = 9 MHz for 56c] were observed. These estimations of the spin populations from EPR (0.73–0.74 e on P) agreed well with DFT results (0.76 e on P).38b In 2010, Bertrand reported that CycAAC reacts with R2NPCl2, [(2,2,6,6-tetramethylpiperidino)phosphine dichloride] to generate [(CycAAC)P(Cl)(NR2)]+Cl− which can be reduced to aminophosphinidene [(CycAAC)PNR2] with magnesium metal.38c This aminophosphinidene was converted to its stable radical cation [(CycAAC)(NR2)P]˙+(C6F5)4B− (57) by a one-electron oxidation with Ph3C+B(C6F5)4− in benzene at room temperature (Fig. 16). The radical cation 57+˙ was characterized by single crystal X-ray diffraction and by EPR spectroscopy.38c The EPR spectrum of 57+˙, in fluorobenzene, at room temperature, shows a doublet of multiplets (g = 2.007) due to a large hyperfine coupling with 31P [a(31P) = 99 G] and a small coupling with one or two 14N nitrogen nuclei [a(14N) ≈ 4 G]. Consistent with the EPR data, theoretical calculations confirmed that the spin density in 57+˙ is localized mainly at phosphorus (67%) with small contributions from the nitrogen atoms (16% for piperidino nitrogen and 10% for cAAC nitogen).38c The authors have mentioned that the exceptional stability of 57+˙ is partly due to steric factors but more importantly because of cationic substituent which prevents the dimerization by electrostatic repulsion.
The Bertrand group has shown that white phosphorus (P4) reacts with CycAAC to give 2,3-diphosphabutadiene [(CycAAC)2P2] which can be considered as a diatomic phosphorus molecule stabilized by two cAAC substituents.38d The cyclic voltammogram of [(CycAAC)2P2] in THF solution, containing 0.1 M n-Bu4NPF6 as electrolyte, shows a reversible one-electron oxidation at E1/2 = −0.536 V versus Fc+/Fc, which indicates the formation of the radical cation. The chemical synthesis of the cationic radical [(CycAAC)2P2]˙+(C6F5)4B− (58, Fig. 16) was quantitatively achieved from the reaction of [(CycAAC)2P2] and [Ph3C][B(C6F5)4] in an equimolar ratio in toluene at room temperature.38d The cationic radical 58+˙ was characterized by single crystal X-ray diffraction and EPR measurements. The EPR spectrum of a fluorobenzene solution of 58+˙ at room temperature shows a triplet of quintets due to a large coupling with two equivalent phosphorus nuclei (a(31P) = 42 G) and a small coupling with two nitrogen nuclei (a(14N) = 3 G). Theoretical calculations indicate that the spin density in 58+˙ is distributed between the two phosphorus atoms (0.27 e at each P) and the two nitrogen atoms of the cAAC ligands (0.14 e at each N). It is important to mention that the unpaired electron in a similar radical cation [(NHC)2P2]˙+(C6F5)4B− (59, Fig. 16) is nearly exclusively localized at phosphorus atoms (0.33 e and 0.44 e), with a contribution of less than 0.07 e for any other atoms.38d Phosphorus mononitride (PN) has attracted huge interest because of its existence in the interstellar medium and the atmospheres of Jupiter and Saturn. In 2010, Bertrand and co-workers successfully isolated the phosphorus mononitride (NHC′)NP(CycAAC) in molecular form which was stabilized by two carbene units.38e The precursor, (NHC′)NH [NHC′ = {CH2N(dipp)}2C:] was synthesized by treatment of NHC with bromine, followed by addition of aqueous ammonia (NH4OH). The (NHC′)NH was then deprotonated with n-BuLi and the in situ generated anion (NHC′)N− was treated with PCl3 to obtain (NHC′)NPCl2, which was reduced with magnesium in the presence of one equivalent of cAAC to obtain (NHC′)NP(CycAAC) in good yield.38e The cyclic voltammetry of (NHC′)NP(CycAAC) in a THF solution containing 0.1 M n-BuNPF6 as electrolyte shows a reversible one-electron oxidation at E1/2 = −0.51 V versus Fc+/Fc, indicating the formation of the corresponding radical cation [(NHC′)NP(CycAAC)]+˙ (60+˙, Fig. 16). The chemical synthesis of the 60+˙ was achieved from the 1:1 reaction of (NHC′)NP(CycAAC) with [Ph3C][B(C6F5)4] in toluene at room temperature. The compound [(NHC′)NP(CycAAC)]+˙(C6F5)4B− (60) was characterized by a single crystal X-ray diffraction and EPR spectroscopy.38e The EPR spectrum of a fluorobenzene solution of 60+˙ at room temperature displays a doublet due to a large coupling with phosphorus [g = 2.0048; a(31P) = 44 G] while no coupling was observed with the nitrogen atom. This observation indicates that the major spin density is localized on the phosphorus atom. Theoretical calculations indicated that the spin density in 60+˙ is mainly distributed on the phosphorus atom (0.40 e) along with the central nitrogen atom (0.18 e) and the nitrogen atom of the cAAC ligand (0.19 e).38e Robinson et al. in 2013 reported that the oxidation of a carbene-stabilized diarsenic compound, L:As–As:L [L: = :C{N(2,6-iPr2C6H3)CH}2], with gallium chloride in a 1:2 ratio in Et2O afforded the first arsenic radical [L:AsAs:L]˙+[GaCl4]− (61, Fig. 16) to be structurally characterized in the solid state.38f The room-temperature EPR spectrum of 61˙+ in fluorobenzene displayed a broadened septet (g ≈ 2.05) resulting from a large hyperfine coupling with two equivalent 75As (I = 3/2) nuclei.38f Bertrand et al. in 2014 reported the first molecular example of a neutral antimony centered radical [(CycAAC)SbCl2]˙ (62; Fig. 16) from the one equivalent reduction of (cAAC)SbCl3 adduct by KC8.38g The radical 62 has been characterized by EPR spectroscopy as well as by single-crystal X-ray diffraction. X-band EPR spectrum of 62 in benzene solution at room temperature shows a septet with significant isotropic hyperfine constants due to the coupling with two nearly equivalent chlorine nuclei (S = 3/2). The simulation of the EPR spectrum of 62 shows various hyperfine couplings [a(121Sb) = 0.003 (natural abundance 57%), a(123Sb) = 0.003 (natural abundance 43%), and a(35Cl) = 4.472, a(35Cl) = 4.472 G and calculated values: a(121Sb) = 0.004, a(123Sb) = 0.004, a(35Cl) = 3.217, a(35Cl) = 3.533 G]. The experimental EPR investigations support the theoretical findings, which indicate that the spin density is mostly located on antimony (90.7%) with minor contributions from the two chlorine atoms (4.6% and 3.9%).38g
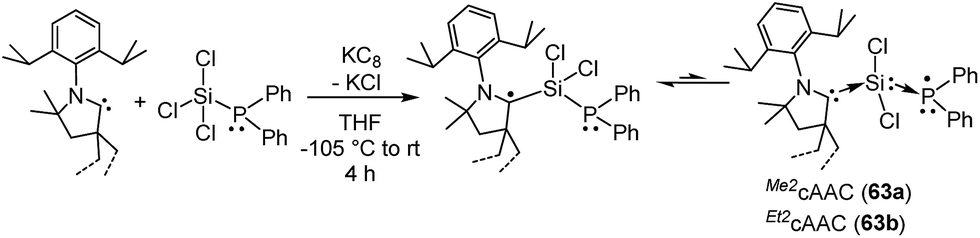
In this regard, we were interested to isolate radicals of mixed group-14 and group-15 elements. Accordingly, we reported two stable radicals [(Me2cAAC˙)Si(Cl2)(PPh2) (63a) and (Et2cAAC˙)Si(Cl2)(PPh2) (63b); Fig. 17] from the commonly used precursors trichlorosilane and diphenylchlorophosphine.39 Compounds 63a and 63b were isolated from the direct reduction of Ph2PSiCl3 by one equivalent of KC8 in the presence of one equivalent of cAAC; all the reactions were initiated at low temperature (∼−105 °C) in THF. Both the compounds were structurally characterized. The calculated Mulliken spin density plots of 63a and 63b suggest that the unpaired electron is mostly located on the carbene carbon (75–76%), with a comparatively lower contribution from the nitrogen atom (18–20%) of the cAAC. Moreover, some finite occupancy over the phosphorus (∼3%) and one of the chlorine atoms (1%) was also observed. The EPR spectrum of 63a in toluene could be simulated [a(31P) = 15.6 G (calc. 20.5 G), a(14N) = 6.5 G (calc. 4.2 G), a(35Cl) = 4.1 G (calc. 3.1 G)]. A 29Si satellite coupling (4.7% nat. abundance, I = 1/2) could be observed for 63b [a(29Si) = 10 G (calcd 13.4 G); 3.6% spin density]. It is worth to note that 63b contains an apparently mobile ethyl substituent of low symmetry which leads to a strong temperature-dependent EPR spectrum between 183 and 340 K. Thus, simulation could not be achieved within the accessible temperature range for 63b. The redox property of 63a was also investigated by cyclic voltammetry measurements in THF solution which shows a one electron quasi-reversible process at E1/2 = −0.86 V against Cp*2Fe/Cp*2Fe+, suggesting the formation of the anion [(Me2cAAC·)Si(Cl2)(PPh2)]−.
 | ||
| Fig. 17 Radicals of silicon and phosphorus stabilized by cAAC. | ||
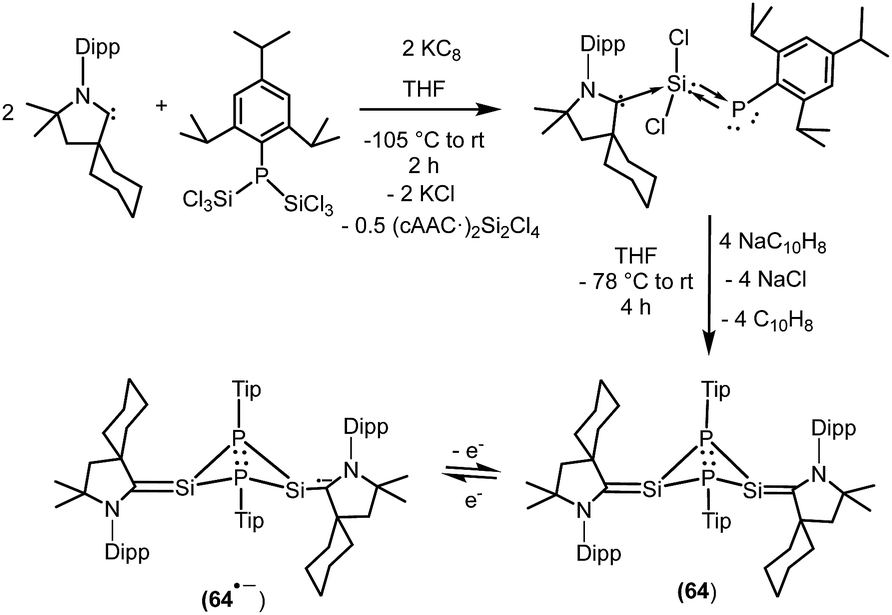
We have also prepared, in a step-wise synthesis, a dimeric heavier analogue of ketenimine with phosphorus and silicon atoms [(CycAAC)Si(P-Tip)]2 (64) (Fig. 18).40 The cyclic voltamogram of 64 revealed a one-electron quasi-reversible process (E1/2 = −0.87 V against Cp*2Fe/Cp*2Fe+) suggesting the formation of the radical anion [(CycAAC)Si(P-Tip)]2˙− (64˙−) which could not be isolated but could be detected by EPR spectroscopy. Thus, the X-band EPR spectrum of the in situ generated radical anion (64˙−) at 285 K in toluene solution shows twelve well-resolved lines of equal intensity (Fig. 19). The splitting pattern shows a doublet of doublets, where each component splits further into three equidistant lines. The latter splitting is assigned to the coupling with one 14N nucleus (a(14N) = 5.9; I = 1) which is in the typical range for cAAC centered radicals. The two larger doublet hyperfine splittings [a(31P) = 44.1 G and a(31P) = 20.6 G] are due to coupling with two inequivalent 31P nuclei (I = 1/2). A satellite for the 29Si nuclei (a(29Si) = 11 G) coupling was also observed in the EPR spectrum of 64˙−. Theoretical calculations and simulation of EPR spectrum of 64˙− reveal the non-equivalence of the two phosphorus nuclei and also the presence of the unpaired electron on one of the two carbene carbon atoms.40
 | ||
| Fig. 18 Radical compounds of silicon and phosphorus stabilized by cAAC. | ||
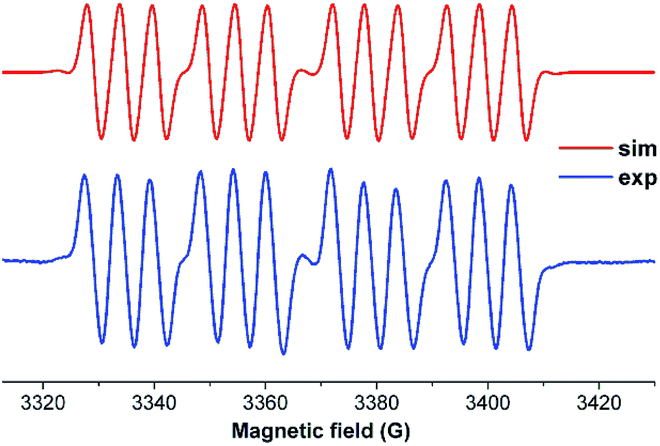 | ||
| Fig. 19 EPR spectrum of radical anion [(CycAAC)Si(P-Tip)]2˙− (64˙−) in toluene at 285 K. | ||
Group 16 radicals
To the best of our knowledge there is no group 16 radical reported in the literature which is stabilized by cAAC. This could be because chalcogens (E = O, S, Se, Te) form strong adducts with carbene and the interaction can be better represented as chalcogen–carbene double bond (carbene = E). As carbene = E is electronically satisfied and has no unpaired electron, chalcogens do not form radical species in presence of carbenes. Chemistry of carbenes with substituted chalcogens (R-E) is not well established and no radical species have been isolated so far. However there is only one report where an air stable oxyallyl radical cation 65˙+ has been isolated by Bertrand and co-workers using a NHC based ligand system (Fig. 20).41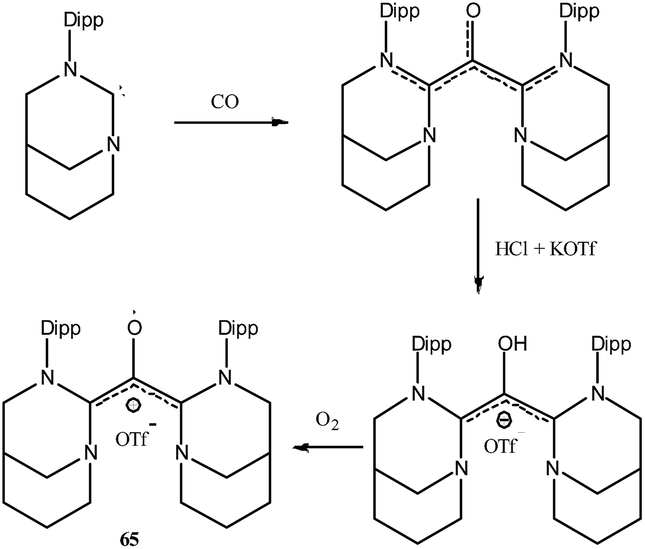 | ||
| Fig. 20 Synthesis of oxyallyl radical cation 65˙+. | ||
Conclusions and perspectives
The area of main-group element radicals is expanding very rapidly with isolation of many exotic and hitherto inaccessible species. It has been observed that all these systems, radicals, radical ions, biradicals, biradicaloids and disbiradicals are stabilized primarily by the steric bulk of the ligands to prevent their dimerization or polymerization. However, recent examples indicate that the radical intermediates can also be stabilized by carbene ligands. NHCs stabilize the radical intermediates mainly by σ-donation whereas cAACs switch their bonding nature depending upon the accumulation of electron density around the radical center because of their additional π-accepting property. The donor and acceptor behavior of the cAACs make it possible to enable the isolation of new types of radicals which were not achievable by conventional methods or by the use of NHC. Thus, the silylene radical anion could be characterized only at low temperatures while the cAAC coordinated silylene radical anion is stable at room temperature. Similarly, dichlorosilylene stabilized by NHC [(NHC)SiCl2] exists in its non-radical monomeric form while SiCl2 stabilized by two cAACs [(cAAC˙)2SiCl2] behaves as biradical. We have reported in this review the successful efforts in the isolation of main-group radical compounds containing carbon, aluminum, silicon and phosphorus stabilized by cAACs. These efforts are the result of development of new synthetic methods involving, first the formation of cAAC main-group adducts, followed by reduction of the adducts by a variety of reducing agents. The role of the cAAC ligand in stabilizing the radical species has been delineated by theoretical methods which throws light on the unique electronic features of this ligand. These results on main-group radical compounds will, we are sure, trigger enormous interest in the coming years not only in the isolation of newer systems but also in finding applications for these new families of stable main-group radicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dedicated to Professor Tobin Marks. V. C. thanks the Alexander von Humboldt Foundation, Bonn, Germany and the Department of Science and Technology, New Delhi, India for financial support. H. W. R. thanks the DFG for financial support (RO224/71-1).Notes and references
- Stable Radicals: Fundamental and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, Wiley, Chichester, 2010 Search PubMed.
- J. Fossey, D. Lefort and J. Serba, Free Radicals in Organic Chemistry, John Wiley and Sons, 1995 Search PubMed.
- P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH, 2008 Search PubMed.
- C. Chatgilialoglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley-VCH, 2012 Search PubMed.
- P. P. Power, Chem. Rev., 2003, 103, 789–810 CrossRef CAS PubMed.
- C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef PubMed.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (b) S. Kundu, P. P. Samuel, A. Luebben, D. M. Andrada, G. Frenking, B. Dittrich and H. W. Roesky, Dalton Trans., 2017, 46, 7947–7952 RSC; (c) S. Kundu, S. Sinhababu, M. M. Siddiqui, A. V. Luebben, B. Dittrich, T. Yang, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2018, 140, 9409–9412 CrossRef CAS PubMed.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- (a) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC; (b) S. Sinhababu, S. Kundu, M. M. Siddiqui, A. N. Paesch, R. Herbst-Irmer, B. Schwederski, P. Saha, L. Zhao, G. Frenking, W. Kaim, D. Stalke and H. W. Roesky, Chem. Commun., 2019, 55, 4534–4537 RSC; (c) W. Li, S. Kundu, C. Köhler, J. Li, S. Dutta, Z. Yang, D. Stalke, R. Herbst-Irmer, A. C. Stückl, B. Schwederski, D. Koley, W. Kaim and H. W. Roesky, Organometallics DOI:10.1021/acs.organomet.9b00041.
- (a) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- (a) A. Schulz, Dalton Trans., 2018, 47, 12827–12837 RSC; (b) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (c) IUPAC Compendium of Chemical Terminology, release 2.3.2, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2012, p. 168 Search PubMed; (d) IUPAC Compendium of Chemical Terminology, release 2.3.2, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2012, p. 427 Search PubMed.
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS.
- (a) C.-W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 1723–1725 CrossRef CAS PubMed; (b) T. Matsumoto and F. P. Gabbaï, Organometallics, 2009, 28, 4252–4253 CrossRef CAS.
- (a) A. D. Ledet and T. W. Hudnall, Dalton Trans., 2016, 45, 9820–9826 RSC; (b) M. F. Silva Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135–1140 CrossRef CAS PubMed; (c) R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed; (d) P. Bissinger, H. Braunschweig, A. Damme, T. Kupfer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 5689–5693 CrossRef CAS PubMed; (e) P. Bissinger, H. Braunschweig, A. Damme, C. Hörl, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2015, 54, 359–362 CrossRef CAS PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed; (b) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed; (c) M. Arrowsmith, J. Bçhnke, H. Braunschweig, M. A. Celik, C. Claes, W. C. Ewing, I. Krummenacher, K. Lubitz and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 11271–11275 CrossRef CAS PubMed; (d) J.-S. Huang, W.-H. Lee, C.-T. Shen, Y.-F. Lin, Y.-H. Liu, S.-M. Peng and C.-W. Chiu, Inorg. Chem., 2016, 55, 12427–12434 CrossRef CAS PubMed; (e) H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS PubMed.
- (a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed; (b) A. Deissenberger, E. Welz, R. Drescher, I. Krummenacher, R. Dewhurst, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 1842–1846 CrossRef CAS PubMed; (c) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed; (d) D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 RSC.
- B. Li, S. Kundu, A. C. Stückl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed.
- S. Kundu, S. Sinhababu, S. Dutta, T. Mondal, D. Koley, B. Dittrich, B. Schwederski, W. Kaim, C. Stückl and H. W. Roesky, Chem. Commun., 2017, 53, 10516–10519 RSC.
- I. Nakanishi, S. Itoh, T. Suenobu and S. Fukuzumi, Chem. Commun., 1997, 1927–1928 RSC.
- (a) J. K. Mahoney, D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef CAS PubMed; (b) J. K. Mahoney, D. Martin, F. Thomas, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (c) J. K. Mahoney, V. Regnier, E. A. Romero, F. Molton, G. Royal, R. Jazzar, D. Martin and G. Bertrand, Org. Chem. Front., 2018, 5, 2073–2078 RSC; (d) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem.–Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (e) D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed.
- D. Mandal, R. Dolai, N. Chrysochos, P. Kalita, R. Kumar, D. Dhara, A. Maiti, R. S. Narayanan, G. Rajaraman, C. Schulzke, V. Chandrasekhar and A. Jana, Org. Lett., 2017, 19, 5605–5608 CrossRef CAS PubMed.
- Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 4168–4172 CrossRef CAS PubMed.
- L. Jin, M. Melaimi, L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351–354 RSC.
- (a) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (b) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (c) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS PubMed; (d) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed; (e) D. Rottschäfer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef PubMed; (f) D. Rottschäfer, N. K. T. Ho, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef PubMed; (g) D. Rottschäfer, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem. Sci., 2018, 9, 4970–4976 RSC.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS PubMed.
- K. C. Mondal, P. P. Samuel, M. Tretiakov, A. P. Singh, H. W. Roesky, A. C. Stückl, B. Niepötter, E. Carl, H. Wolf, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2013, 52, 4736–4743 CrossRef CAS PubMed.
- S. Kundu, P. P. Samuel, S. Sinhababu, A. V. Luebben, B. Dittrich, D. M. Andrada, G. Frenking, A. C. Stückl, B. Schwederski, A. Paretzki, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2017, 139, 11028–11031 CrossRef CAS PubMed.
- S. Sinhababu, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, I. Fernández, G. Frenking, A. C. Steckl, B. Schwederski, W. Kaim and H. W. Roesky, Chem.–Eur. J., 2018, 24, 1264–1268 CrossRef CAS PubMed.
- K. C. Mondal, H. W. Roesky, A. C. Stückl, F. Ihret, W. Kaim, B. Dittrich, B. Maity and D. Koley, Angew. Chem., Int. Ed., 2013, 52, 11804–11807 CrossRef CAS PubMed.
- K. C. Mondal, P. P. Samuel, H. W. Roesky, B. Niepötter, R. Herbst-Irmer, D. Stalke, F. Ehret, W. Kaim, B. Maity and D. Koley, Chem.–Eur. J., 2014, 20, 9240–9245 CrossRef CAS PubMed.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepötter, H. Wolf, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed.
- B. Niepötter, R. Herbst-Irmer, D. Kratzert, P. P. Samuel, K. C. Mondal, H. W. Roesky, P. Jerabek, G. Frenking and D. Stalke, Angew. Chem., Int. Ed., 2014, 53, 2766–2770 CrossRef PubMed.
- S. Roy, K. C. Mondal, L. Krause, P. Stollberg, R. Herbst-Irmer, D. Stalke, J. Meyer, A. C. Stückl, B. Maity, D. Koley, S. K. Vasa, S. Q. Xiang, R. Linser and H. W. Roesky, J. Am. Chem. Soc., 2014, 136, 16776–16779 CrossRef CAS PubMed.
- K. C. Mondal, B. Dittrich, B. Maity, D. Koley and H. W. Roesky, J. Am. Chem. Soc., 2014, 136, 9568–9571 CrossRef CAS PubMed.
- Y. Li, Y.-C. Chan, B.-X. Leong, Y. Li, E. Richards, I. Purushothaman, S. De, P. Parameswaran and C.-W. So, Angew. Chem., Int. Ed., 2017, 56, 7573–7578 CrossRef CAS PubMed.
- (a) L. Gu, Y. Zheng, E. Haldón, R. Goddard, E. Bill, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2017, 56, 8790–8794 CrossRef CAS PubMed; (b) Z. Li, Y. Hou, Y. Li, A. Hinz, J. R. Harmer, C.-Y. Su, G. Bertrand and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 198–202 CrossRef CAS PubMed; (c) O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed; (d) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed; (e) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed; (f) M. Y. Abraham, Y. Wang, Y. Xie, R. J. Gilliard Jr, P. Wei, B. J. Vaccaro, M. K. Johnson, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2013, 135, 2486–2488 CrossRef CAS PubMed; (g) R. Kretschmer, D. A. Ruiz, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 8176–8179 CrossRef CAS PubMed.
- S. Roy, A. C. Stückl, S. Demeshko, B. Dittrich, J. Meyer, B. Maity, D. Koley, B. Schwederski, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 4670–4673 CrossRef CAS PubMed.
- S. Roy, B. Dittrich, T. Mondal, D. Koley, A. Claudia Stückl, B. Schwederski, W. Kaim, M. John, S. K. Vasa, R. Linser and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 6180–6183 CrossRef CAS PubMed.
- D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014–7017 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |