
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-based building blocks from 5-hydroxymethylfurfural via 1-hydroxyhexane-2,5-dione as intermediate
Bartosz
Wozniak
 ,
Sergey
Tin
and
Johannes G.
de Vries
*
,
Sergey
Tin
and
Johannes G.
de Vries
*
Leibniz-Institut für Katalyse e.V., Universität Rostock, Albert-Einstein-Str.29a, 18059 Rostock, Germany. E-mail: Johannes.deVries@catalysis.de
First published on 6th June 2019
Abstract
The limits to the supply of fossil resources and their ever increasing use forces us to think about future scenarios for fuels and chemicals. The platform chemical 5-hydroxymethyl-furfural (HMF) can be obtained from biomass in good yield and has the potential to be converted in just a few steps into a multitude of interesting products. Over the last 20 years, the conversion of HMF to 1-hydroxyhexane-2,5-dione (HHD) has been studied by several groups. It is possible to convert HMF into HHD by hydrogenation/hydrolytic ring opening reaction in aqueous phase using various heterogeneous and homogeneous catalysts. This review addresses both the state of the art of HHD synthesis, including mechanistic aspects of its formation, as well as the recent progress in the application of HHD as a building block for many useful chemicals including pyrroles, cyclopentanone derivatives and triols.
 Bartosz Wozniak | Bartosz Wozniak received his B.S (2013) and M.S (2015) degrees in biological chemistry from the Adam Mickiewicz University, Poznan, Poland, under the supervision of Prof. Bogdan Marciniec. After a four months research project with Prof. Vidar Jensen in Bergen, Norway, he joined the department of Prof. Johannes de Vries at the LIKAT, Rostock, Germany, where he recently obtained his Ph.D degree (2019) in the field of organic chemistry and catalysis. Currently, he is working on the preparation of chemical building blocks from renewables. |
 Sergey Tin | Sergey Tin was born in 1989 in Kazakhstan. He completed his masters in chemistry with an external placement in 2012 at the University of St Andrews. In 2016 he obtained his PhD degree with a thesis entitled “Rh catalysed hydrogenation of enamines: factors affecting the rate and enantioselectivity” in the same place. In 2017 he moved to Germany and started a post-doc in LIKAT. Currently he works as a group leader in the department of Prof. Johannes G. de Vries. |
 Johannes G. de Vries | Johannes G. de Vries received his PhD from the University of Groningen in 1979. After a postdoc at Brandeis University, Waltham, USA, his first job was as a medicinal chemist with Sandoz in Vienna and in London. From 1988–2013 he worked for DSM in Geleen, The Netherlands, lastly as a Principal Scientist in the area of Homogeneous Catalysis. From 1999–2018 he was part-time professor at the University of Groningen. In 2014 he became Department Head Catalysis with Renewables at the Leibniz Institute for Catalysis in Rostock, Germany. In 2013 he received the Paul N. Rylander Award for outstanding contributions in the field of catalysis as it applies to organic synthesis. |
1 HMF as a platform chemical
Extensive utilisation of fossil fuel reserves and as a consequence of this the global warming issues are currently the major challenges our modern society needs to face.1–3 Considering the fact that more than 90% of chemicals our lives depend upon, such as polymers, pharmaceuticals, detergents or food additives are currently derived from crude oil,4 development of new sustainable technologies with the aim to produce chemicals from renewable resources is crucial. Biomass is the most attractive, low-cost feedstock produced by nature and therefore readily available around the world.5–9 Unlike other renewable sources like solar, wind, hydroelectric and geothermal energies, biomass contains organic carbon and has a great potential to substitute oil and coal in the production of fuels and chemicals. Lignocellulosic biomass is one of the most abundant feedstock widely available in the form of nonedible forestry or agro waste.10 Depending on the class and origin of the plant, polymeric lignocellulose is comprised of three basic materials in various ratios (i) 30–50 wt% cellulose, (ii) 20–40 wt% hemicellulose and (iii) 10–20% lignin.11Special attention has been given to the conversion of lignocellulose or its constituents to widely applicable so-called Platform Chemicals of high industrial potential (Fig. 1).8 A platform chemical is an intermediate molecule, which is produced from biomass at a competitive cost and can be transformed to a sufficient number of valuable intermediates.
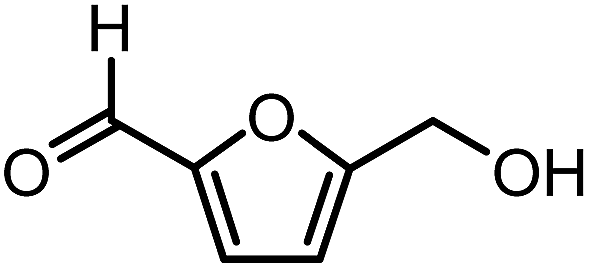 | ||
| Fig. 1 Chemical structure of 5-hydroxymethylfurfural (HMF). | ||
5-Hydroxymethylfurfural (HMF) is one of the most valuable platform chemicals. Although it was initially not included in the “Top 10” list of chemicals obtained from biomass in the report made for the U.S. Department of Energy,5 Bozell did include it in his report on the top ten chemical from biomass ten years later.12 HMF is a furan-type compound which has retained all six carbons from the hexose and contains two functional groups: an aldehyde and a hydroxymethyl group (Fig. 1). It can be obtained in relatively high yields from fructose by dehydration or in lower yields from glucose or even cellulose.13–16
The first reports on HMF synthesis by Düll17 and Kiermayer18 can be dated back to 1895. Since then, a significant growth in interest on HMF formation and applicability resulted in an enormous number of publications with an evident peak in the last years. As a consequence of this, recent advances in the synthesis and utilisation of HMF were reviewed by several groups.13–16,19–21 HMF is a very valuable building block with a wide range of applications. Scheme 1 provides an overview of the conversion of HMF into the most useful follow-up products, such as caprolactam22 2,5-furandicarboxylic acid23,24 or adipic acid25,26 Two main challenges remain for the large-scale production of HMF. One is the use of readily available cheap feedstocks such as cellulose, starch or preferably lignocellulose to reduce the cost price to an acceptable level. Currently, HMF is only produced on a scale of a few hundred tons per year by Ava-Biochem as a side product of sugar carbonisation and hence is relatively expensive.27 The other issue is the instability of HMF, which makes its isolation in high yields problematic.28 To deal with this issue Avantium developed a process for the production of the much more stable methyl ether of HMF29 and Mascal developed a process for the production of stable 5-chloromethyl-furfural in good yield, even from cellulose.30
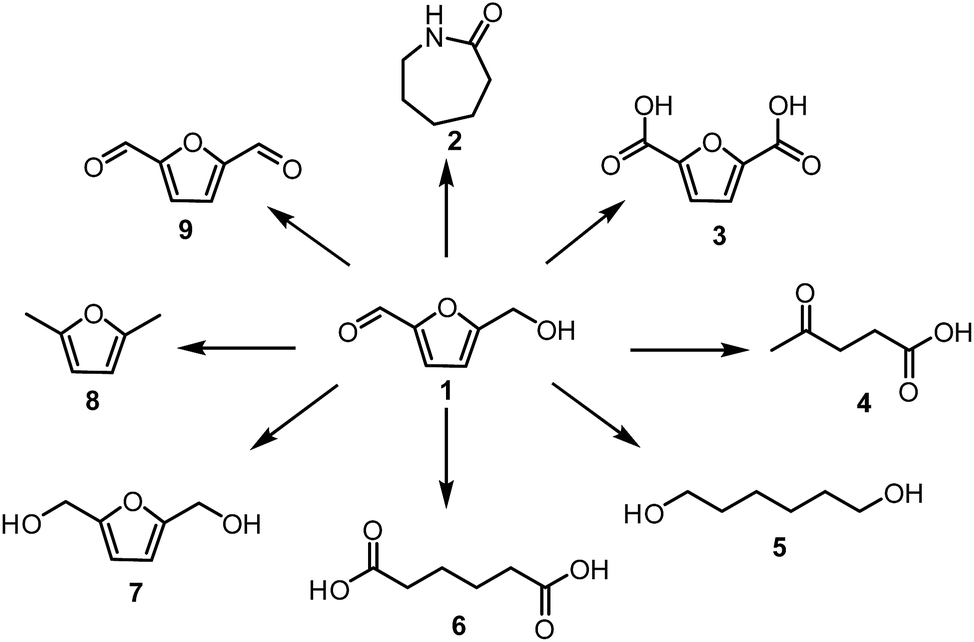 | ||
| Scheme 1 Conversion of HMF to valuable chemicals. | ||
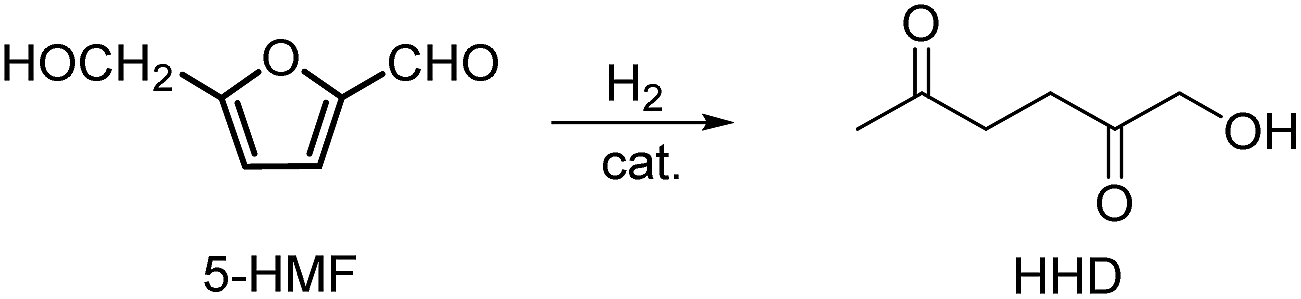
2 Synthesis of 1-hydroxyhexane-2,5-dione
Recently, the synthesis of 1-hydroxyhexane-2,5-dione (HHD) from HMF has attracted the interest of several groups. HHD is a ring-opened product, which has retained all six carbons that were present in HMF (Fig. 2). Moreover, the presence of two carbonyl groups and one hydroxyl group in the alkyl chain makes it an appropriate precursor for the synthesis of valuable follow-up products.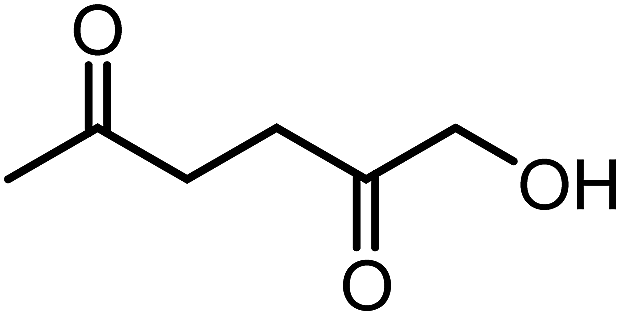 | ||
| Fig. 2 Chemical structure of 1-hydroxyhexane-2,5-dione (HHD). | ||
It is possible to convert HMF into HHD by hydrogenation/hydrolytic ring opening reaction in aqueous phase using various heterogeneous and homogeneous catalysts.22,31–45 In only four of these the isolation and purification of HHD was reported.37,42,45,47 In this chapter, different catalytic strategies for the synthesis of HHD from HMF as well as mechanistic aspects of this transformation will be discussed.
2.1 Hydrogenation/hydrolytic ring opening of HMF catalysed by heterogeneous catalysts
The first publication regarding HHD synthesis dates back to 1991.31 Descotes and co-workers reported the Pt/C catalysed formation of HHD in water containing 15 wt% of oxalic acid. The reaction was carried out at 140 °C under 30 bar of H2 for 3 hours resulting in 60% conversion to HHD. When 70 bar of H2 were applied, a decrease in HHD yield was observed (to 51%). A separate experiment under the same conditions, but in the presence of 75 wt% oxalic acid in water afforded only 31% of HHD. In addition to these experiments a possible mechanism for the conversion of HMF to HHD was proposed (Scheme 2). Under acidic conditions HMF undergoes rehydration followed by ring-opening to form the stable intermediate 10, which was confirmed by 13C NMR. In a subsequent step, the double bond and aldehyde functionalities are hydrogenated selectively in the presence of the ketone groups leading to the formation of HHD. The group of van Bekkum investigated hydrogenolysis of HMF in water using 10% Pd/C under mild reaction conditions (1 bar of H2, 60 °C).32 The experiments showed that adding small amount of concentrated HCl was necessary to obtain HHD. The reported HHD yield after 410 min was 28%, at 98% conversion of HMF. The main product obtained under these conditions was 2,5-bishydroxymethylfuran (BHMF) (47%). Interestingly, when the reaction was performed in a biphasic solvent system, consisting of water and toluene in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, the yield of HHD improved considerably to 68%.
:1 ratio, the yield of HHD improved considerably to 68%.
 | ||
| Scheme 2 Proposed pathway for HHD synthesis by Descotes. | ||
Heeres, de Vries and co-workers reported the use of a bimetallic Rh–Re/SiO2 catalyst for the hydrogenation of HMF to HHD.22 Initially, the reaction was carried out at 120 °C using 10 mol% of the catalyst in water at 10 bar of H2 for 1 hour. After this time, the pressure was raised to 80 bar of H2 and the reaction was stirred for 17 hours resulting in 81% selectivity towards HHD at full conversion of the starting material.
Ohyama et al. reported the hydrogenation of HMF catalysed by gold nanoparticles in aqueous solutions.33 Depending on the metal oxide that was used as a support, BHMF and/or HHD were obtained by applying 38 bar of H2 at 140 °C. The highest HHD yield of 57% was achieved using 1 wt% Au/TiO2 combined with 4 wt% of SO3, which acted as a Brønsted acid. A later publication by the same group reported a 60% yield of HHD at 81% HMF conversion in the reaction catalysed by gold nanoparticles supported on Nb2O5.34 For this reaction, higher pressures (80 bar of H2) and addition of catalytic amounts of H3PO4 were required. In addition, the authors suggested an alternative route for HHD formation (Scheme 3). In the first step, HMF is hydrogenated to BHMF followed by the hydrolytic ring-opening to 1-hydroxyhex-3-en-2,5-dione (11) in the second step and the reduction of 11 to HHD in the last step. In 2014, the group of Jérôme came to the same conclusions concerning the mechanism.35 They were able to convert BHMF to HHD in 60% yield in a reaction catalysed by 2 wt% Pd/C in water at 120 °C under 10 bar of H2 and 30 bar of CO2. The addition of CO2 appears to be necessary to obtain HHD through the formation of carbonic acid, which provides acidic conditions. Further investigations revealed an increase in HHD yield to 77% when the experiment was performed with 7.5 wt% Pd/C and HMF was used as a starting material. It should be noted that different H2/CO2 ratios and higher catalyst loadings showed no significant improvement. The authors also examined direct conversion of fructose and inulin to HHD for the first time. In the first step, carbohydrates are converted to HMF by heating in water at 150 °C under 40 bar of CO2 followed by hydrogenation/hydrolytic ring opening of HMF after addition of Pd/C (7.5 wt%) and applying 10 bar of H2. The overall HHD yields were 36% and 15% from fructose and inulin respectively. In an extended work by the same group, the combination of both solid catalysts Amberlyst-15 and Pd/C was evaluated for the synthesis of HHD.36 Compared to the method described earlier, the reactions were carried out in wet THF at 80 °C. After optimisation, a 77% HHD yield was achieved after 15 hours by the use of 5.5 wt% Pd/C and 20 wt% Amberlyst-15 at 50 bar of H2. Again, this catalytic system was demonstrated to be efficient for the conversion of fructose and inulin towards HHD in 55% and 27% yields respectively.
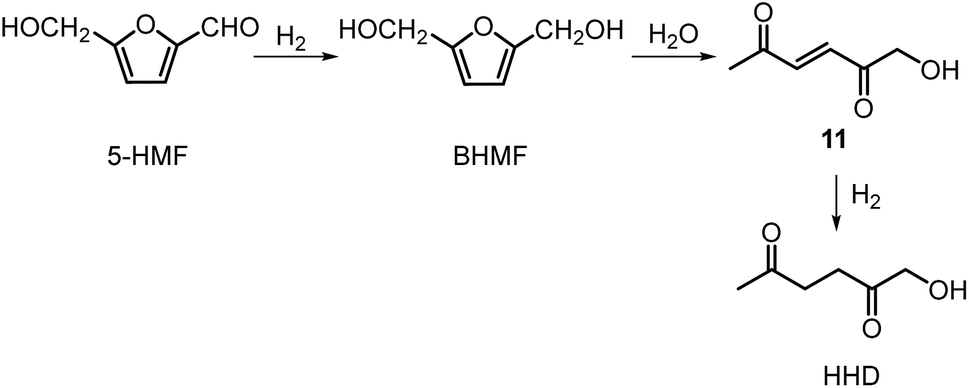 | ||
| Scheme 3 Conversion of HMF to HHD, as proposed by Satsuma. | ||
Xu and co-workers established the hydrogenation of HMF in acidic water (H3PO4, pH 2–3) under 1 bar of H2 at reflux.37 They isolated the product by column chromatography in 37% yield in the form of a yellow solid. In 2017, Yang and co-workers used palladium supported on acidic Nb2O5 for the hydrogenation of HMF in water.38 The highest selectivity towards HHD was 90% under 10 bar of H2 after 2 h, however only 21% conversion of HMF was obtained. After optimising the conditions, 73% selectivity towards HHD was achieved at 97% conversion after 6 hours at 140 °C and 40 bar of H2. In addition, a Pd/Ni2O5-400 catalyst could be recycled up to 4 times without apparent loss in activity.
2.2 Hydrogenation/hydrolytic ring opening of HMF catalysed by homogeneous catalysts
Only a limited number of studies is available on the conversion of HMF to HHD using homogeneous catalysts. Homogeneous catalysts seem to perform better than the heterogeneous catalysts with respect to HHD yields, shorter reaction times and milder conditions (temperature, pressure). Especially, iridium and ruthenium-based metal complexes were found to be efficient in HMF hydrogenation/transfer hydrogenation (Fig. 3). Concerning the use of homogeneous catalyst for the synthesis of HHD, limitations appear to lie mainly in the requirements of water solubility and stability under acidic conditions. Zhang and co-workers were the first to apply a homogeneous catalyst in HHD synthesis.39 Hydrogenation/hydrolytic ring opening of HMF was performed in aqueous solution using bipyridine coordinated Cp*–Ir complexes (Cp* = pentamethylcyclopentadiene) at different temperatures (110–130 °C) and hydrogen pressures (5–20 bar of H2). The highest HHD yield of 86% at full conversion of the starting material was obtained with 0.26 mol% [Ir]-1 at 120 °C after 2 hours at 7 bar H2. | ||
| Fig. 3 Iridium- and ruthenium-based complexes for the synthesis of HHD. | ||
They also performed detailed mechanistic studies using deuterium labelling. Two possible mechanistic pathways were proposed through key intermediate 12 (Scheme 4), which is consistent with the mechanism proposed by Jérôme.35 Pathway A assumes a two-steps reaction to generate 12 from BHMF with an initial water addition followed by water-elimination, whereas pathway B proceeds through a one-step hydrolysis process. In the next step, intermediate 12 is hydrolysed to α,β-unsaturated di-carbonyl compound 11, which then undergoes hydrogenation to form HHD. Hydrogenation of BHMF in D2O catalysed by [Ir]-1 leads to the conclusion that pathway B is more plausible based on the composition of the deuterated HHD ion fragment (GC-MS analysis) and statistical calculations.
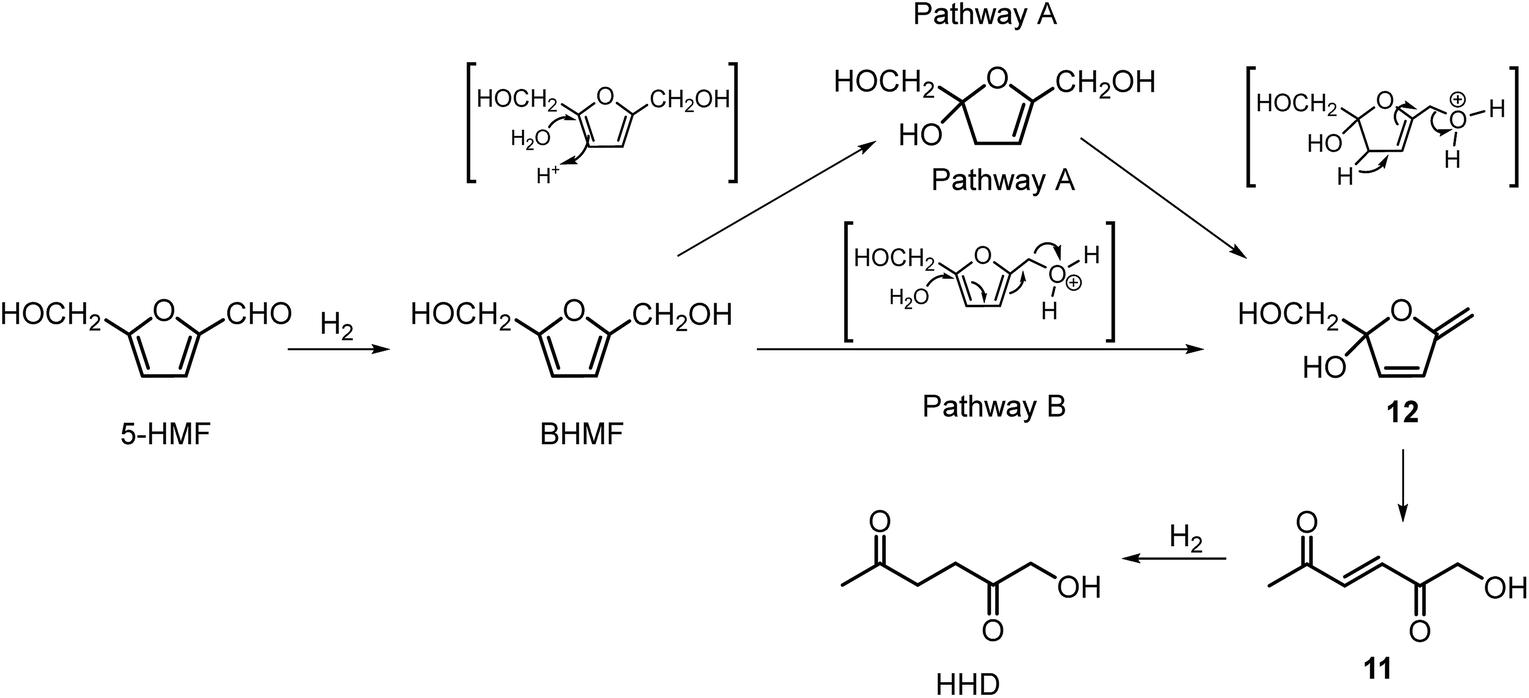 | ||
| Scheme 4 Proposed pathways for HHD synthesis by Zhang. | ||
Singh and co-workers reported water soluble 8-aminoquinoline coordinated arene-ruthenium(II) complexes for the conversion of biomass-derived furans.40 Moreover, formic acid was successfully applied as a hydrogen source. By the use of 12 eq. of formic acid relative to the substrate in the presence of 1 mol% of [Ru]-1, a moderate selectivity to HHD of 52% was obtained from HMF at 80 °C after 48 hours. Apart from HHD, other products were also formed, such as levulinic acid (21%) and 3-hydroxy-2,5-hexanedione (27%). A later publication by the same group reported the use of Ru(II)-arene complexes containing ethylenediamine-based ligands for the synthesis of diketones.41 However, a lower HHD selectivity (44%) was obtained after 8 hours in the reaction catalysed by 5 mol% of [Ru]-2 at 100 °C in the presence of formic acid. In addition, the direct conversion of fructose to HHD was investigated. The optimised conditions afforded 27% selectivity to HHD and 51% to LA. However, longer reaction times (16 h) led to a significantly higher HHD selectivity of 87%.
In 2016, Fu and co-workers made an important step forward in the synthesis of HHD from HMF.42 They evaluated a series of Cp*Ir(III) complexes bearing bipyridine ligands with either electron-donating or electron-withdrawing groups at different positions. They found that the combined influence of temperature and pH is the most relevant factor for the formation of HHD in high yields. Catalyst screening showed relatively high HHD selectivities of around 70% with 0.01 mol% of [Ir]-2 or [Ir]-3 complexes in aqueous formate buffer solution (FBS, pH = 2.5) at 120 °C in 2 hours. Interestingly, an increase in temperature to 130 °C led to remarkably higher HHD selectivity of up to 95%. A large-scale experiment was performed with the aim to isolate HHD and to test the recyclability of the catalyst. The authors reported an isolated HHD yield of 85% (92% GC yield) after the first cycle under the optimised conditions. In a recycling experiment a decrease in activity of the catalyst was observed and HHD was produced in 70% yield. Additionally, another insight to the mechanism of HHD formation was proposed as depicted in Scheme 5. In the first step, HMF is hydrogenated to BHMF as was postulated previously. In the next step, two alternative routes were assessed for the conversion of BHMF to HHD. The formation of 5-methylfurfuryl alcohol as an intermediate is an important distinction between the two catalytic routes. However, when 5-methylfurfuryl alcohol was used as a starting material under the same experimental conditions, the only product obtained was 2,5-hexanedione. On the basis of this result, pathway A was indicated to be the more likely one.
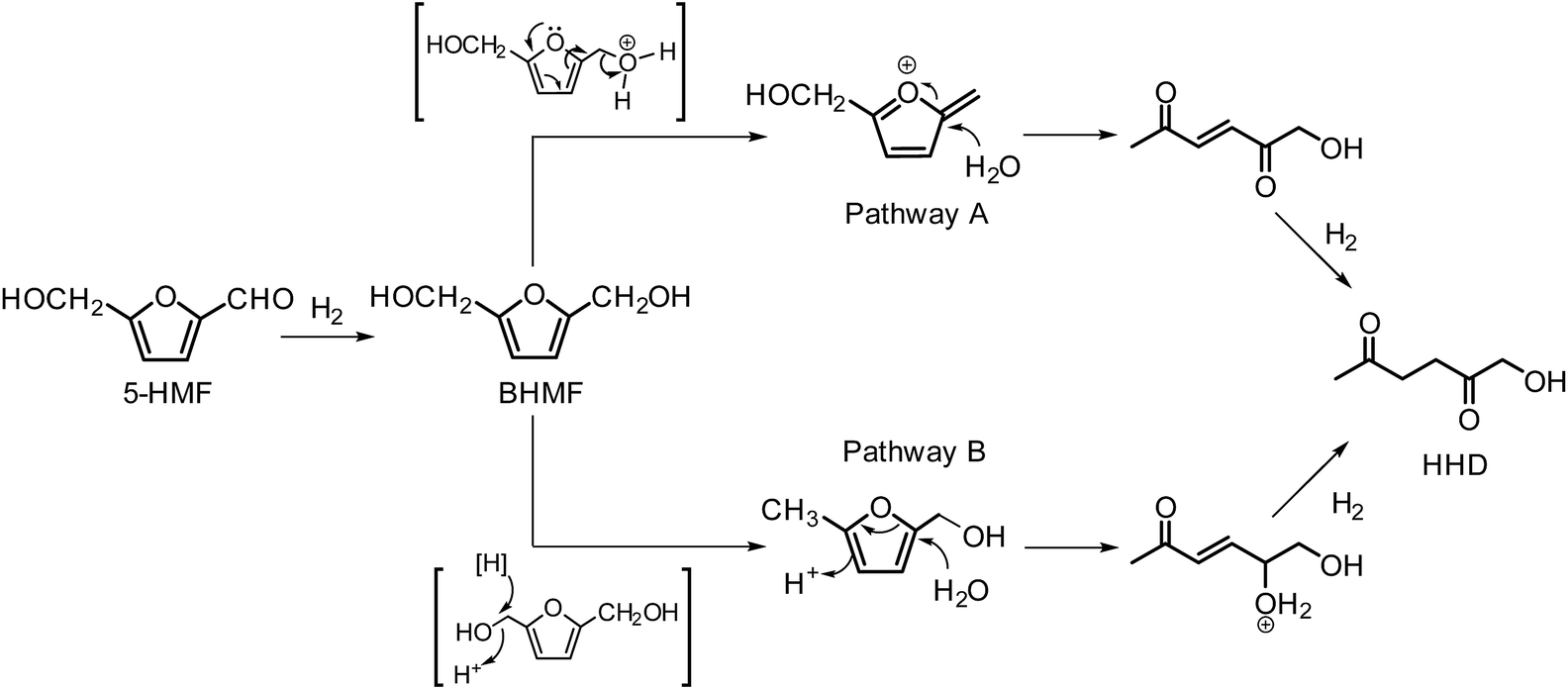 | ||
| Scheme 5 Proposed pathways for HHD synthesis by Fu. | ||
With the same catalyst ([Ir]-2, 0.1 mol%), Xu et al. demonstrated the hydrogenation of HMF in the presence of 5 mol% Al2(SO4)3, which provides acidic conditions in water by generating sulfuric acid.43 A HHD selectivity of 77% was reported in aqueous solution at 30 bar of H2 and 130 °C after 4 hours.
In another publication from Zhang and co-workers, a new series of half-sandwich Cp*–Ir(III) complexes were tested for the synthesis of HHD from HMF.44 Promising results were achieved using complex [Ir]-4 bearing a bipyridine ligand containing both o-hydroxyl and p-N-dimethylamino groups. The effect of the catalyst loading, pH and pressure was investigated. At a catalyst loading of 0.0008 mol% hydrogenation of HMF resulted in 67% selectivity towards HHD at 88% conversion after 6 hours in acidic water (pH = 3.4) at 120 °C under 35 bar of H2. Based on this experiment, the TOF (31560 h−1) and TON (70800) were calculated. The same catalyst was tested for the transfer hydrogenation of HMF using 2 eq. of formic acid at 120 °C. This catalytic system provided 60% HHD yield in the presence of 0.005 mol% of [Ir]-4 after 2 hours.
De Vries and co-workers evaluated a series of air- and moisture-stable iridium complexes for the hydrogenation/hydrolytic ring opening of HMF.45 Depending on the reaction conditions, the highest HHD yields were obtained in the reactions catalysed by half-sandwich iridium complexes [Ir]-5 and [Ir]-6 (Scheme 6). Up to 76% selectivity towards HHD was achieved when HMF was reduced in water in the presence of 0.5 mol% [Ir]-5 catalyst at 120 °C under 10 bar of H2 for 2 h (Scheme 6, conditions A). The major by-product formed under these conditions was an insoluble brown polymeric material, presumably humins, formation of which was also reported with other catalysts.35,42 In addition, the isolation of HHD in 69% overall yield was reported. Moreover, the full characterisation of HHD was reported including its crystal structure, obtained by X-ray diffraction analysis (Fig. 4).
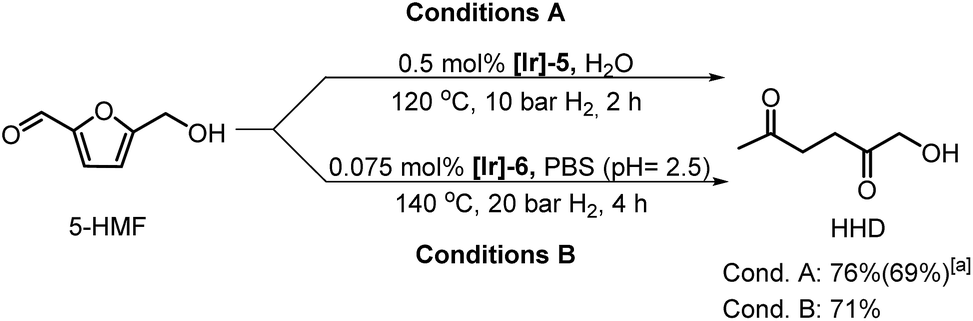 | ||
| Scheme 6 Conversion of HMF to HHD under various reaction conditions; aisolated yield. | ||
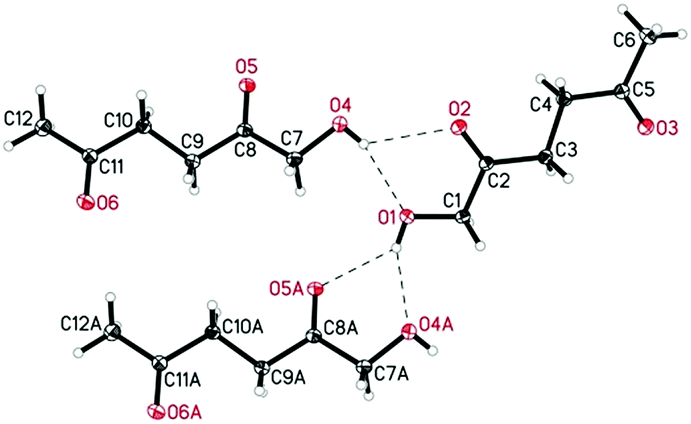 | ||
| Fig. 4 ORTEP representation of HHD showing intermolecular O–H⋯O hydrogen bonds. | ||
Interestingly, the utilisation of phosphate buffer solution (PBS, pH = 2.5) as the reaction medium allowed processing at lower catalyst loading (0.075 mol%) without significant losses in selectivity towards HHD; 71% HHD was obtained in the presence of [Ir]-6 catalyst (Scheme 6, conditions B). In another publication by the same group, the authors aimed to perform a large-scale synthesis of HHD under the optimised conditions in PBS.46 The reaction was conducted under 60 bar of H2 resulting in the full conversion of HMF after 1 hour. Starting from 25 g of HMF, HHD was isolated in 71% yield.
Recently, Dwivedi et al. examined cationic cyclopentadienyl-ruthenium(II)-pyridylamine complexes for the conversion of HMF to HHD.47 The reactions were carried out in water in the presence of formic acid (12 eq. with respect to substrate) at 120 °C. The η-5-Cp–Ru(II) complex bearing N-(pyridin-2-ylmethyl)propan-1-amine as ligand ([Ru]-3) was found to be the most efficient; 69% selectivity towards HHD was achieved after 12 hours in the presence of 5 mol% of the catalyst. In addition, the authors obtained purified product in 42% yield after column chromatography. Direct conversion of fructose under the same conditions resulted in 65% selectivity towards HHD after 6 hours.
2.3 Conclusions on conversion of HMF to HHD
Over the last 20 years, the conversion of HMF to HHD has been studied by several groups. It has been demonstrated that employing acidic conditions was the key parameter affecting selectivity towards HHD for this process. An overview of catalytic systems applied for the production of HHD is summarised in Table 1. In general, the utilisation of half-sandwich Cp*Ir complexes provides the highest selectivities to HHD at the lowest catalyst loadings. However, the requirements of high dilution (to avoid the formation of humines) as well as the poor recyclability of the catalysts remain barriers on the way to large scale application.|
|
||||
|---|---|---|---|---|
| Catalyst (cat. loading) | Conditions | Conv. (%) | Yield (%) | Ref. |
| a Isolated yield; na- not available. | ||||
| Pt/C (0.5 wt%) | 15 mol% C2H2O4, 140 °C, H2O, 30 bar H2, 3 h | na | 60 | 27 |
| Pd/C (50 wt%) | 12.5 mol% HCl, 60 °C, H2O:toluene (2:1), 1 bar H2, 4 h |
97 | 68 | 28 |
| Rh–Re/SiO2 (10 mol%) | 120 °C, H2O, 10 bar H2 for 1 h, then 80 bar H2 for 17 h | 100 | 81 | 22 |
| Au/TiO2 (1 wt%) | 4 wt% SO3, 140 °C, H2O, 38 bar H2, 4 h | 84 | 57 | 29 |
| Au/Nb2O5 (1 wt%) | 8.5 mM H3PO4, 140 °C, H2O, 80 bar H2, 12 h | 81 | 60 | 30 |
| Pd/C (7.5 wt%) | 120 °C, H2O, 10 bar H2, 30 bar CO2, 15 h | 100 | 77 | 31 |
| Pd/C (5.5 wt%) | 20 wt% Amberlyst-15, 80 °C, THF (containing 3.8 wt% H2O), 50 bar H2, 15 h | 100 | 77 | 32 |
| Pd/AC (5 wt%) | Reflux, H2O, 1 bar H2, 72 h | na | 37a | 33 |
| Pd/Nb2O5 (2 wt%) | 140 °C, H2O, 40 bar H2, 6 h | 93 | 73 | 34 |
| [Ir]-1 (0.26 mol%) | 120 °C, H2O, 7 bar H2, 2 h | 100 | 86 | 35 |
| [Ru]-1 (1 mol%) | 80 °C, H2O, 12 eq. HCOOH, 48 h | 100 | 52 | 36 |
| [Ru]-2 (5 mol%) | 100 °C, H2O, 12 eq. HCOOH, 8 h | 100 | 44 | 37 |
| [Ir]-2 (0.01 mol%) | 130 °C, FBS (pH = 2.5), 2 h | 100 | 92 (85)a | 38 |
| [Ir]-2 (0.1 mol%) | 5 mol% Al2(SO4)3, 130 °C, H2O, 30 bar H2, 4 h | 100 | 77 | 39 |
| [Ir]-4 (0.0008 mol%) | 120 °C, H2O (pH = 3.4), 35 bar H2, 6 h | 88 | 67 | 40 |
| [Ir]-5 (0.5 mol%) | 120 °C, H2O, 10 bar H2, 2 h | 100 | 76 (69)a | 41 |
| [Ir]-6 (0.075 mol%) | 140 °C, PBS (pH = 2.5), 60 bar H2, 1 h | 100 | 71a | 42 |
| [Ru]-3 (5 mol%) | 120 °C, H2O, 12 eq. HCOOH, 12 h | 96 | 69 (42)a | 43 |
3 Applications of 1-hydroxyhexane-2,5-dione
There are a limited number of reports on the utilisation of HHD for the synthesis of useful products. In most cases, these were transformations of HHD formed in situ. This is a rapidly growing field.3.1 Preparation of cyclopentanone derivatives and follow-up products
The intramolecular aldol condensation of HHD leads to the formation of five-membered ring compounds. It can occur via two different routes depending on the reaction conditions as described in Scheme 7. The group of Satsuma performed extensive research on the conversion of HMF to 3-hydroxycyclopentanone (HCPN, 14) under acidic conditions and H2 pressure.34,48,49 HCPN is an important chemical intermediate in the synthesis of polymers, pharmaceuticals and fuels.50,51 The first approach to the conversion of HMF to HCPN via HHD was based on the use of supported gold nanoparticles.34 Various metal oxides were examined as the supports under 80 bar of H2; they acted as a Lewis acids and catalysed the ring closure of HHD as it was formed. The highest reported selectivity towards HCPN was 86% after 12 hours using 1 wt% Au supported on niobium oxide at 140 °C. Moreover, the activity of the catalyst did not decrease significantly after the second catalytic cycle (84% selectivity). Following the reaction over time revealed the formation of HHD in 20% yield for the first 6 hours and after this time HCPN was produced exclusively. Based on this result, a plausible mechanism was proposed for the conversion of HMF to HCPN (Scheme 8). Hydrogenation/hydrolytic ring opening of HMF leads to the formation of HHD, which is consistent with the generally proposed mechanism. Subsequently, intramolecular aldol condensation of HHD occurs under acidic conditions resulting in the formation of 3-(hydroxymethyl)cyclopent-2-en-1-one (HCPEN, 13), which is next hydrogenated to 14. The same group investigated the effect of acid–base catalysis on the ring rearrangement of HMF by using a combination of a hydrogenation catalyst (Pt/SiO2) with acidic and basic metal oxides.47 The results of the experiments showed that basic metal oxides such as La2O3 and CeO2 produce HCPN in low yields (<25% yield), however, addition of acidic metal oxides (e.g. Ta2O5, ZrO2, Nb2O5) enhanced the yield of HCPN to around 80%. The best catalytic performance was observed by using 1 wt% Pt/SiO2 together with Ta2O5 in water at 140 °C under 30 bar of H2 resulting in 82% yield of HCPN after 12 hours.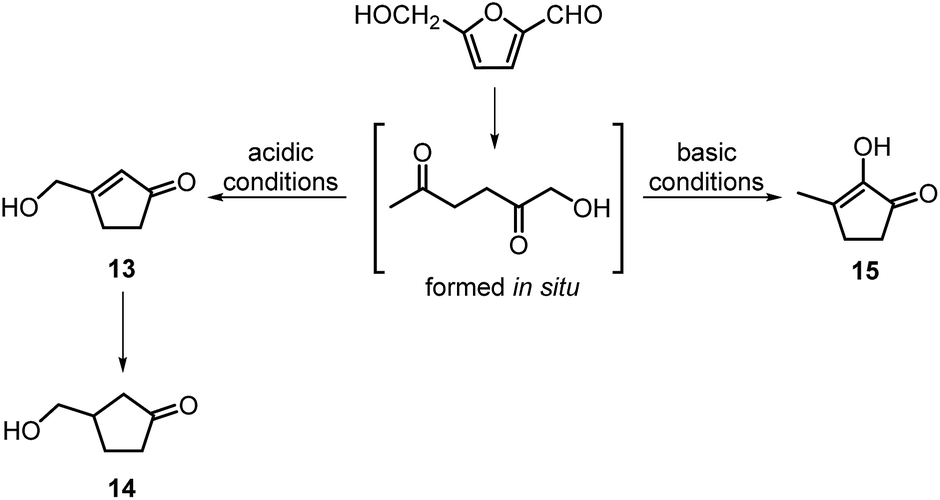 | ||
| Scheme 7 Aldol condensation products starting from HHD. | ||
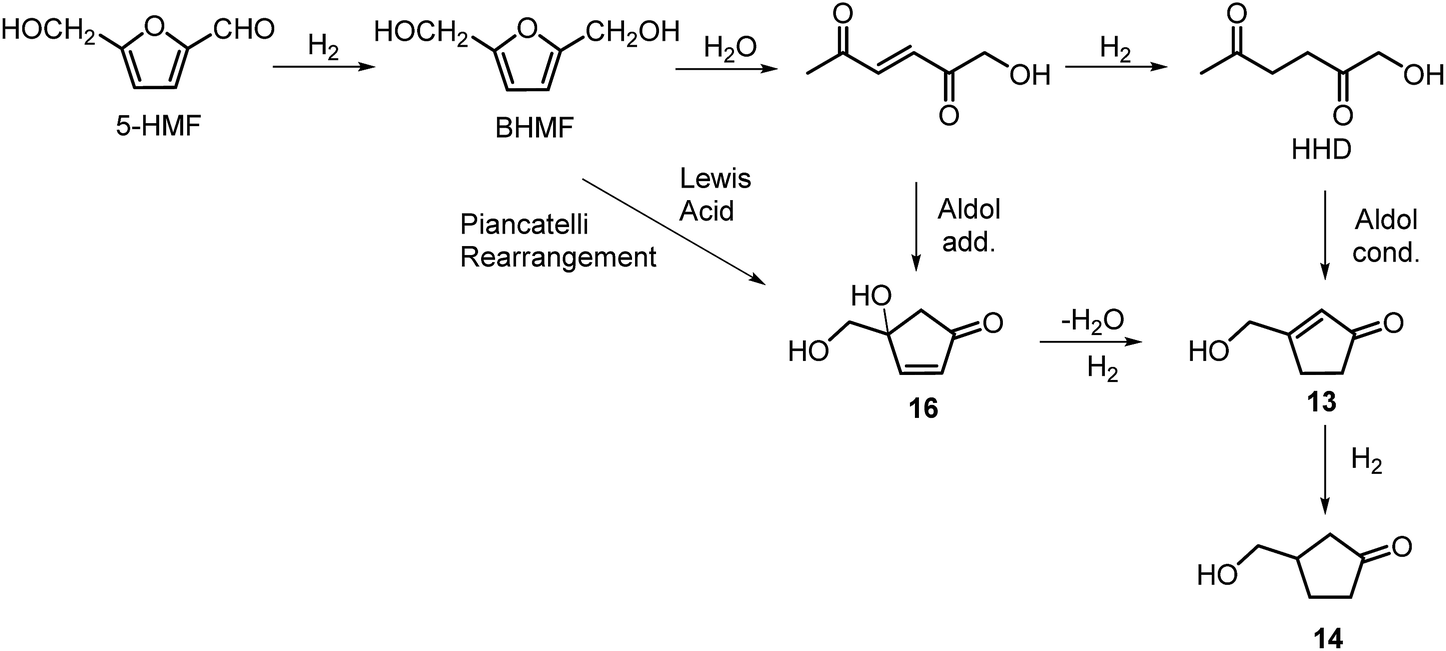 | ||
| Scheme 8 Plausible mechanism for the conversion of HMF to HCPN via HHD. | ||
The formation of HCPN via HHD was also proposed by Perret et al.52 To avoid the use of noble metal complexes, the catalytic activity of nickel supported on alumina was tested. A comparable yield of HCPN (81%) was obtained after 6 hours at 140 °C and 20 bar of H2. Further research on the selective conversion of HMF to cyclopentanone derivatives was performed using Cu/Al2O3 and Co/Al2O3 catalysts.53 When 26 wt% of Cu/Al2O3 was used as a catalyst at 180 °C and 20 bar of H2, the HCPN yield was 86% at full conversion after 6 hours. However, hydrogenative ring-rearrangement of HMF catalysed by 9 wt% Co/Al2O3 revealed the formation of 3-hydroxymethylcyclopentanol (HCPL), a hydrogenation product of HCPN. The highest HCPL yield of 94% was achieved at 140 °C and 20 bar of H2 after 48 hours. Ohyama et al. also reported the direct conversion of HMF to HCPL using a combination of Pt/SiO2 and lanthanide oxides.49 High product yields (ca. 88%) were obtained by applying 5 wt% Pt/Nd2O3 in water at 140 °C and 30 bar of H2 after 30 hours.
Deng, Fu and co-workers proposed a different mechanism for the formation of 14.43 They were able to show that BHMF undergoes a Piancatelli rearrangement under the influence of the Lewis acid to form 16, which subsequently is dehydrated and hydrogenated to obtain 14 (Scheme 8). They were able to obtain evidence for all the steps of the mechanism.
In contrast to acidic systems, intramolecular aldol condensation of HHD under basic conditions led to another cyclopentanone derivative: 2-hydroxy-3-methylcyclopent-2-enone (MCP, 15).38,43,45 MCP is a known naturally occurring flavour ingredient in roasted coffee54 and maple syrup.55 On the basis of these properties it has found commercial use in the food industry as a flavouring agent. Apart from its natural occurrence, MCP is commonly produced from adipic acid in a multistep reaction; however, the use of toxic reagents such as Cl2 and potentially carcinogenic CH3I is required for this process.56 Therefore, the development of a bio-based route to MCP is highly desired.
Deng, Fu and co-workers discovered the preparation of MCP from HHD under basic conditions.43 A typical experiment was performed with 25 mol% KOH in refluxing water. An MCP yield of 45% was obtained after stirring the reaction mixture for 40 minutes. Yang and co-workers examined the conversion of HHD to MCP in the presence of different bases.38 The addition of solid base Ca–Al (200 mg) to a refluxing aqueous solution of HHD (0.1 M) led to 88% selectivity toward MCP after 6 hours. In addition, neutralisation was not required, since the pH value of the aqueous solution after reaction was neutral. Other solvents, such as n-butanol, ethanol and MIBK were also tested but did not lead to any improvements in MCP selectivities. A reaction mechanism was proposed as described in Scheme 9. Deprotonation of HHD under basic conditions occurs at the α-hydroxy position. In the next step, the enolate reacts intramolecular with the other ketone group to form the α,β-bishydroxy-cyclopentanone derivative, followed by dehydration to give MCP. In addition, the direct synthesis of MCP from HMF under optimised conditions using Pd/Nb2O5 and Ca–Al was reported. The product was isolated in 58% yield.
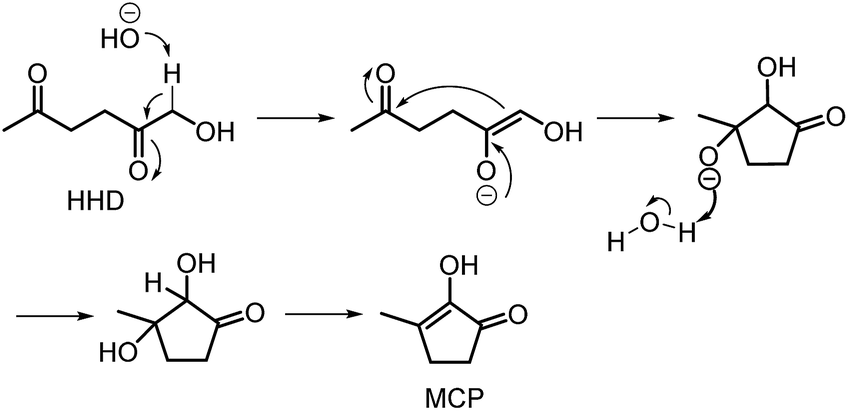 | ||
| Scheme 9 Base-promoted intramolecular aldol condensation of HHD. | ||
Wozniak et al. examined the role of the base and solvent in the aldol condensation reaction of HHD at 60 °C.44 An isolated MCP yield of 72% was achieved in aqueous KOH solution after 15 minutes. Alternatively, an 80% yield at full conversion of the starting material was obtained after 5 min in a THF solution of KOtBu. The authors also established a one-pot synthesis of MCP directly from HMF via HHD as an intermediate (Scheme 10). The application of the optimised conditions afforded 55% isolated yield of MCP in a one-pot reaction. In addition, they reported the X-ray structure of MCP (Fig. 5).
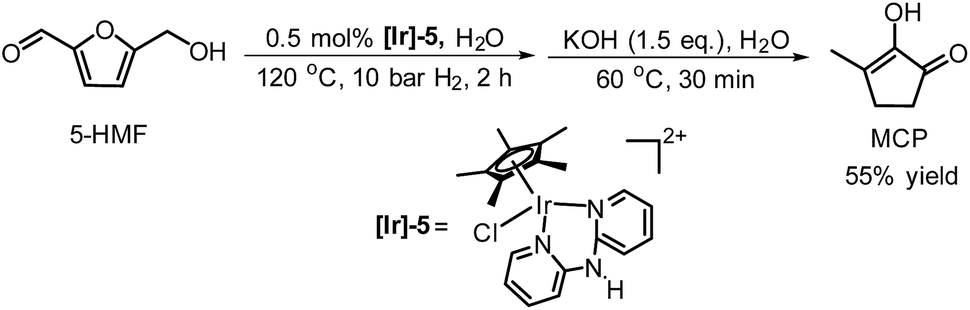 | ||
| Scheme 10 A one-pot synthesis of MCP from HMF. | ||
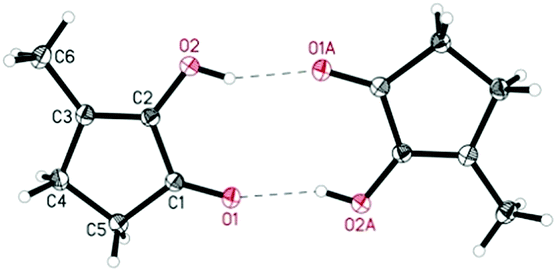 | ||
| Fig. 5 ORTEP representation of MCP showing intermolecular O–H⋯O hydrogen bonds. | ||
Furthermore, MCP was converted to a number of valuable products as described in Scheme 11. For example, the fully hydrogenated form of MCP, 3-methyl-1,2-cyclopentanediol (17) was obtained in near quantitative yield as a mixture of isomers by using the commercially available Ru-MACHO-BH catalyst (Scheme 11, eqn (a)). The oxidative carbonylation of diol 17 has been reported to lead to the formation of the cyclic carbonates, which are important intermediates in the synthesis of pharmaceuticals, polymers and agrochemicals.57 The application of aliphatic and aromatic diamines in the reaction with MCP led to excellent yields of the corresponding α,β-unsaturated heterocyclic imines and a quinoxaline derivate (Scheme 11, eqn (b), 18–20). Quinoxalines are known intermediates for the manufacturing of pharmaceuticals due to their antiviral, anticancer and antidepressant properties.58 Another possible useful synthetic approach is the transformation of MCP to the corresponding enol acetate (21, Scheme 11, eqn (c)), which is a starting material for the synthesis of dihydrojasmone, a fragrance used in perfumes.59
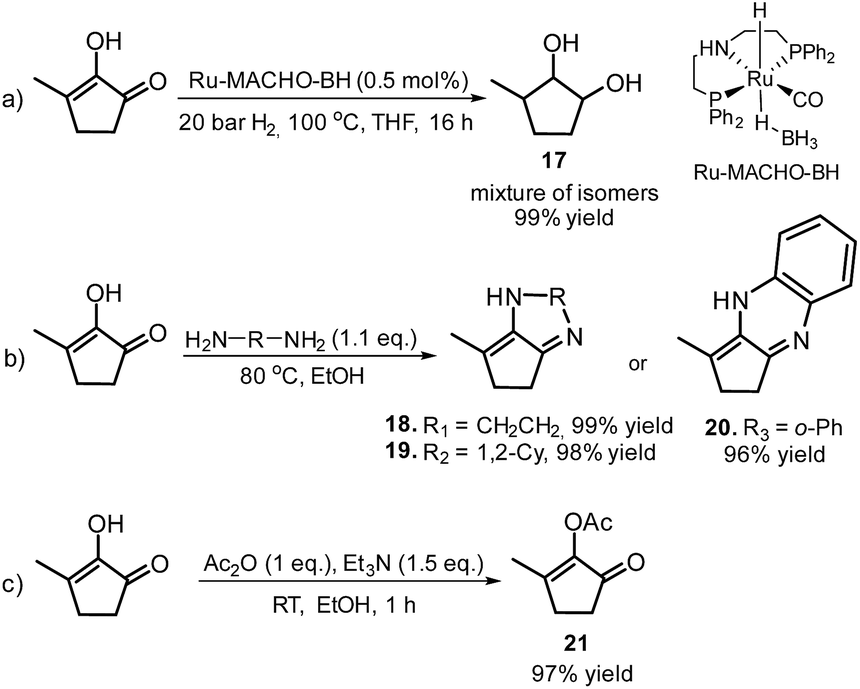 | ||
| Scheme 11 Conversion of MCP into valuable biomass-based products. | ||
3.2 Synthesis of pyrroles
In another publication by de Vries and co-workers, the main focus was on the utilisation of the diketone moiety of HHD.46 1,4-diketones are common substrates in the Paal–Knorr synthesis for the production of pyrroles in reaction with primary amines. Usually, weakly acidic conditions are required for this transformation. However, the authors achieved the synthesis of N-substituted 2-hydroxymethyl-5-methyl-pyrroles from HHD and a variety of primary amines in ethanol as solvent at room temperature, without catalyst (Scheme 12).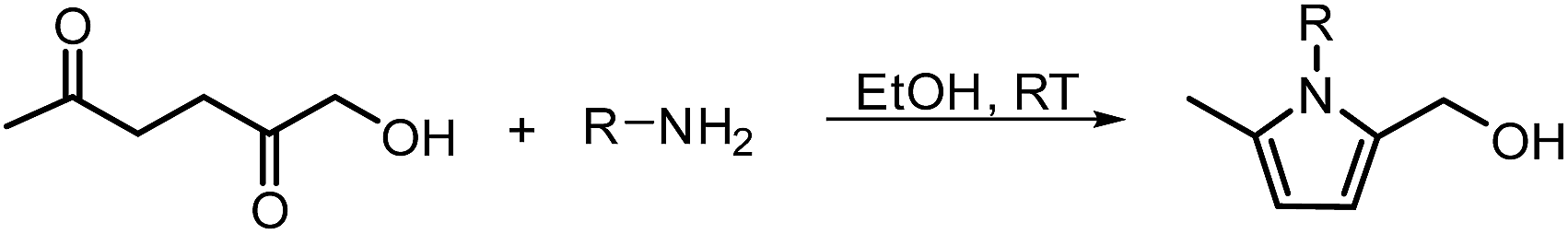 | ||
| Scheme 12 The Paal–Knorr synthesis of N-substituted pyrroles starting from HHD. | ||
In general, most of the N-alkyl and N-benzyl substituted pyrroles were isolated with excellent yields after 5–25 minutes (Fig. 6). In case of more sterically hindered amines, like cyclopentylamine and aniline derivatives prolonged reaction times (up to 48 h) were required. However the utilisation of anilines bearing electron-donating groups led to the full conversion of HHD in shorter reaction times (up to 20 h). In addition, a series of pyrroles containing heteroatoms such as sulfur, oxygen and nitrogen was synthesised in up to 99% isolated yields. On the basis of these results, the described Paal–Knorr synthesis meets the Click Reaction60 requirements as it provides the products in near quantitative yields in very short reaction times and proceeds under mild conditions in a benign solvent. Moreover, in most cases, the only by-product of the reaction is water and the pure product was simply isolated by evaporation of the solvent.
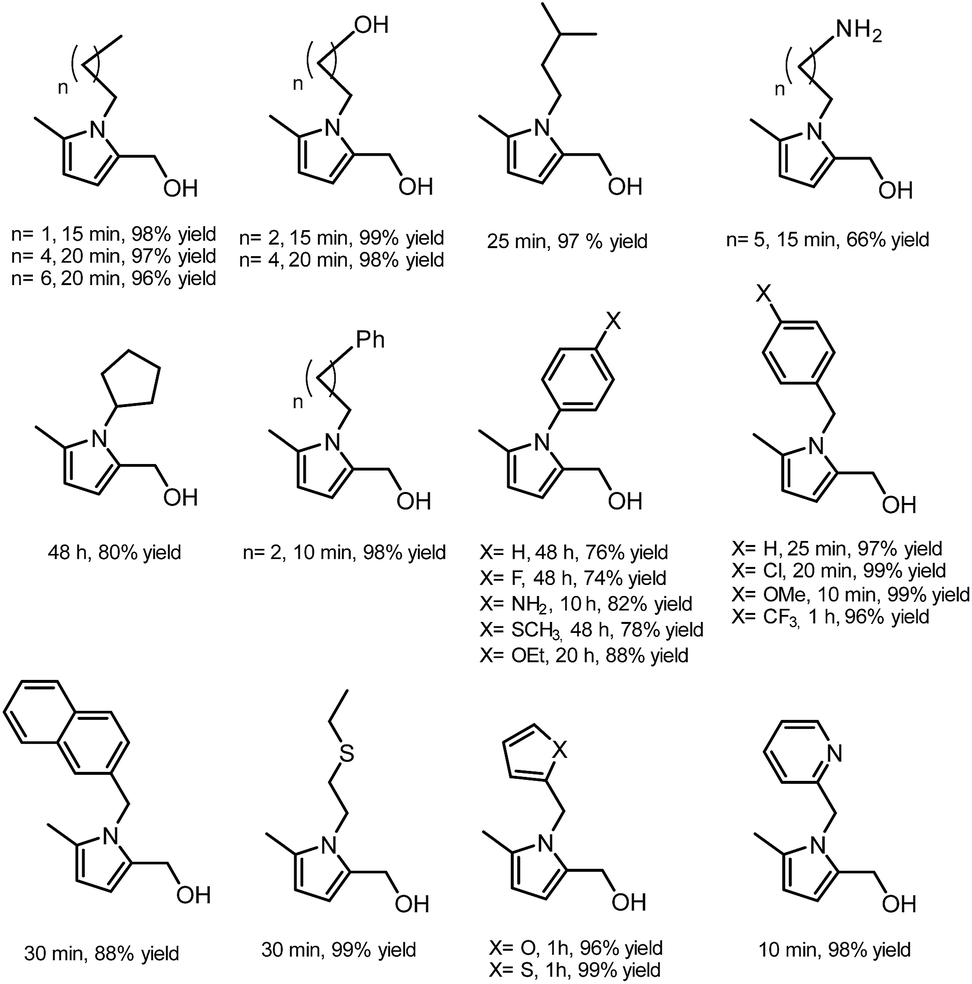 | ||
| Fig. 6 N-substituted pyrroles derived from HHD. | ||
3.3 Others
The hydrogenation of HMF often resulted not only in the formation of HHD, but also in its fully hydrogenated form: 1,2,5-hexanetriol (1,2,5-HT). In 1991, Descotes was the first who reported the synthesis of 1,2,5-HT directly from HMF using heterogeneous Ru/C as a catalyst.31 The same compound has been described as one of the by-products in prolonged hydrogenation of HMF catalysed by supported metals.61,62 Melián-Cabrera and co-workers obtained 1,2,5-HT by hydrogenating BHMF using Rh–Re/SiO2 as catalyst with 92% selectivity.63 Xu and co-workers described the hydrogenation of HHD in aqueous solution, which provided isolated 1,2,5-HT yield of 95% after 4 hours.37 The reaction was carried out in the presence of 5 wt% Ru/C under 40 bar H2 at 120 °C. Wozniak et al. developed a method for synthesis of 1,2,5-HT starting from HHD using homogeneous catalysis.64 A near quantitative yield of triol was achieved after 18 hours at 100 °C with 30 bar of H2 using 0.5 mol% Ru-MACHO-BH and isopropanol as the solvent (Scheme 13). Moreover, the authors showed that 1,2,5-HT can be further converted into useful products in the presence of rhenium(VII) oxide. Rhenium has received increasing interest caused by the high activity in many transformations, especially the deoxydehydration reaction (DODH) of vicinal diols to produce alkenes in the presence of a reductant.65–67 This is a very promising approach for oxygen removal from biomass-derived platform chemicals without losing any carbon atoms. De Vries and co-workers described DODH of 1,2,5-HT under neat conditions at 165 °C in the presence of 3 mol% Re2O7 and 1.1 eq. PPh3 as a reductant.64 A mixture of cis- and trans-4-hexen-2-ol as the main products was obtained (Scheme 14). The same Re2O7 catalysed reaction in the absence of the reductant resulted in the selective formation of the cyclic product 5-methyltetrahydrofurfuryl alcohol (5-MTHFA). This compound has been described as a potentially useful fuel additive and has been reported as the main product of 5-methylfurfural hydrogenation.68,69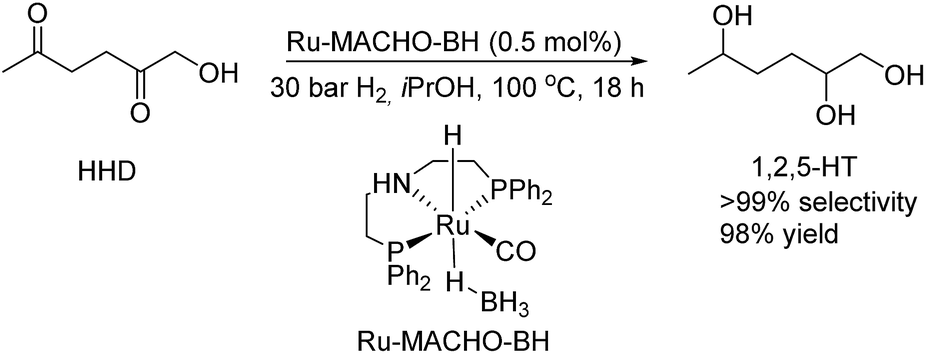 | ||
| Scheme 13 Homogeneous hydrogenation of HHD. | ||
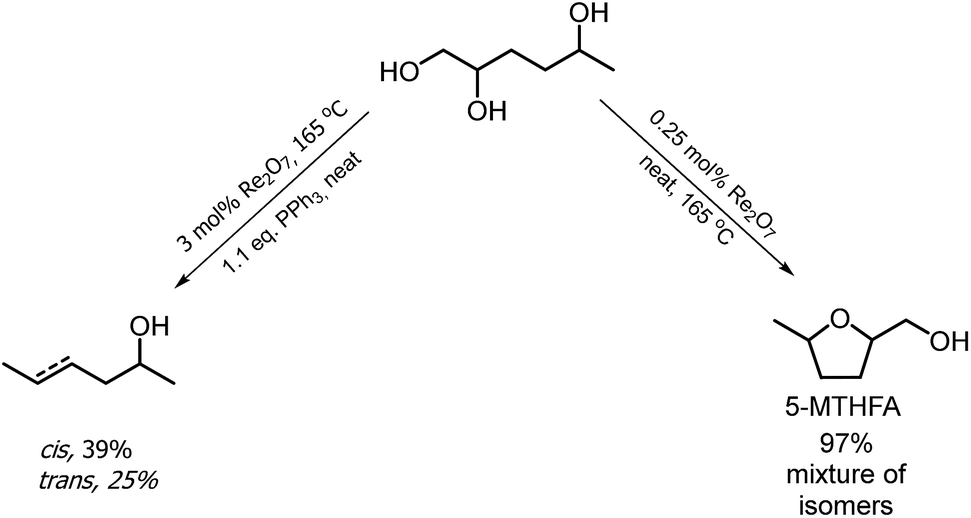 | ||
| Scheme 14 Rhenium catalysed transformations of 1,2,5-hexanetriol. | ||
4 Conclusions
In conclusion, this review summarises both the state of art for HHD synthesis including the mechanistic aspects of its formation, as well as the recent progress in the application of HHD as a building block for useful chemicals. A broad range of compounds was synthesised from it, which can serve in the production of consumer products of the chemical industry including pharmaceuticals, food additives, agrochemicals and transportation fuels. However, the key to the success of HHD will be the development of an production method at larger (commercial) scale for HMF. This would further boost the interest in the chemistry of HHD, which has the potential to become one of a new generation of Platform Chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leibniz Foundation for financial support (SAW-LIKAT-1 523). We thank Martyna Wozniak for the TOC illustration.References
- M. A. Rubio Rodriguez, J. De Ruyck, P. Roque Diaz, V. K. Verma and S. Bram, Appl. Energy, 2011, 88, 630–635 CrossRef.
- W. P. Nel and C. J. Cooper, Energy Policy, 2009, 37, 166–180 CrossRef.
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- B. Kamm, P. R. Gruber and M. Kamm, Biorefineries: Industrial Processes and Products, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- T. Werpy and G. Petersen, Top Value-Added Chemicals from Biomass, Vol. I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U.S. Department of Energy (DOE) by the National Renewable Energy Laboratory, Golden, CO, 2004 Search PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- P. J. Deuss, K. Barta and J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 RSC.
- J. G. de Vries, Chem. Rec., 2016, 16, 2787–2800 CrossRef PubMed.
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as a feedstock for a bioenergy and bioproducts industry: the technical feasibility of a billion-ton annual supply, US Department of Agriculture, Washington, DC, 2005 Search PubMed.
- C.-H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- L. T. Mika, E. Cséfalvay and A. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- G. Düll, Chem.-Ztg., 1895, 19, 216–217 Search PubMed.
- J. Kiermayer, Chem.-Ztg., 1895, 19, 1003–1006 CAS.
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS.
- S. W. Teong, G. Yi and Y. Zhang, Green Chem., 2014, 16, 2015–2026 RSC.
- T. Buntara, S. Noel, P. H. Phua, I. Melián-Cabrera, J. G. de Vries and H. J. Heeres, Angew. Chem., Int. Ed., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- J. Shen, H. Chen, K. Chen, Y. Qin, X. Lu, P. Ouyang and J. Fu, Ind. Eng. Chem. Res., 2018, 57, 2811–2818 CrossRef CAS.
- F. Jie, S. Jinshan, C. Hao, L. Xiuyang and O. Pingkai, CN106749130, 2017.
- T. R. Boussie, E. L. Dias, Z. M. Fresco and V. J. Murphy, Production of adipic acid and derivatives from carbohydrate-containing materials, WO2010144873, 2010to Rennovia inc Search PubMed.
- T. R. Boussie, G. M. Diamond, E. Dias and V. Murphy, in Chemicals and Fuels from Bio-Based Building Blocks, ed. F. Cavani, S. Albonetti, F. Basile and A. Gandini, Wiley-VCH, Weinheim, 2016, pp. 152–171 Search PubMed.
- S. Krawielitzki and T. M. Kläusli, Ind. Biotechnol., 2015, 11(1), 6–8 CrossRef.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CrossRef CAS PubMed.
- (a) G. J. M. Gruter and F. Dautzenberg, WO2007104514, 2007, to Avantium International BV; (b) A. S. Dias, R.-J. van Putten and G. J. Gruter, WO2012091570, 2012, to Furanix Technologies; (c) J. C. van der Waal, E. Mazoyer, H. J. Baars and G. J. M. Gruter, in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives, ed. S. S. Stahl and P. L. Alsters, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, pp. 311–329 Search PubMed.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS PubMed.
- V. Schiavo, G. Descotes and J. Mentech, Bull. Soc. Chim. Fr., 1991, 128, 704–711 Search PubMed.
- G. C. A. Luijkx, N. P. M. Huck, F. van Rantwijk, L. Maat and H. van Bekkum, Heterocycles, 2009, 77, 1037–1044 CrossRef CAS.
- J. Ohyama, A. Esaki, Y. Yamamoto, S. Arai and A. Satsuma, RSC Adv., 2013, 3, 1033–1036 RSC.
- J. Ohyama, R. Kanao, A. Esaki and A. Satsuma, Chem. Commun., 2014, 50, 5633–5636 RSC.
- F. Liu, M. Audemar, K. De Oliveira Vigier, J.-M. Clacens, F. De Campo and F. Jérôme, ChemSusChem, 2014, 7, 2089–2095 CrossRef CAS PubMed.
- F. Liu, M. Audemar, K. De Oliveira Vigier, J.-M. Clacens, F. De Campo and F. Jérôme, Green Chem., 2014, 16, 4110–4114 RSC.
- Y. Yang, Z. Du, J. Ma, F. Lu, J. Zhang and J. Xu, ChemSusChem, 2014, 7, 1352–1356 CrossRef CAS PubMed.
- Y. Duan, M. Zheng, D. Li, D. Deng, L.-F. Ma and Y. Yang, Green Chem., 2017, 19, 5103–5113 RSC.
- Z. Xu, P. Yan, W. Xu, X. Liu, Z. Xia, B. Chung, S. Jia and Z. C. Zhang, ACS Catal., 2015, 5, 788–792 CrossRef CAS.
- K. Gupta, D. Tyagi, A. D. Dwivedi, S. M. Mobin and S. K. Singh, Green Chem., 2015, 17, 4618–4627 RSC.
- A. D. Dwivedi, K. Gupta, D. Tyagi, R. K. Rai, S. M. Mobin and S. K. Singh, ChemCatChem, 2015, 7, 4050–4058 CrossRef CAS.
- W.-P. Wu, Y.-J. Xu, R. Zhu, M.-S. Cui, X.-L. Li, J. Deng and Y. Fu, ChemSusChem, 2016, 9, 1209–1215 CrossRef CAS PubMed.
- Y.-J. Xu, J. Shi, W.-P. Wu, R. Zhu, X.-L. Li, J. Deng and Y. Fu, Appl. Catal., A, 2017, 543, 266–273 CrossRef CAS.
- Z. Xu, P. Yan, H. Li, K. Liu, X. Liu, S. Jia and Z. C. Zhang, ACS Catal., 2016, 6, 3784–3788 CrossRef CAS.
- B. Wozniak, A. Spannenberg, Y. Li, S. Hinze and J. G. de Vries, ChemSusChem, 2018, 11, 356–359 CrossRef CAS PubMed.
- B. Wozniak, Y. Li, S. Hinze, S. Tin and J. G. de Vries, Eur. J. Org. Chem., 2018, 2009–2012 CrossRef CAS.
- A. D. Dwivedi, V. K. Sahu, S. M. Mobin and S. K. Singh, Inorg. Chem., 2018, 57, 4777–4787 CrossRef CAS PubMed.
- J. Ohyama, R. Kanao, Y. Ohira and A. Satsuma, Green Chem., 2016, 18, 676–680 RSC.
- J. Ohyama, Y. Ohira and A. Satsuma, Catal. Sci. Technol., 2017, 7, 2947–2953 RSC.
- T. Kitahara, Y. Warita, M. Abe, M. Seya, Y. Takagi and K. Mori, Agric. Biol. Chem., 1991, 55, 1013–1017 CAS.
- P. Marcé, Y. Díaz, M. I. Matheu and S. Castillón, Org. Lett., 2008, 10, 4735–4738 CrossRef PubMed.
- N. Perret, A. Grigoropoulos, M. Zanella, D. T. Manning, J. B. Claridge and M. J. Rosseinsky, ChemSusChem, 2016, 9, 521–531 CrossRef CAS PubMed.
- R. Ramos, A. Grigoropoulos, N. Perret, M. Zanella, A. P. Katsoulidis, T. D. Manning, J. B. Claridge and M. J. Rosseinsky, Green Chem., 2017, 19, 1701–1713 RSC.
- M. A. Gianturco, A. S. Giammarino and R. G. Pitcher, Tetrahedron, 1963, 19, 2051–2059 CrossRef CAS PubMed.
- V. J. Filipic, J. C. Underwood and C. O. Willits, J. Food Sci., 1965, 30, 1008–1015 CrossRef CAS.
- C. M. Leir, J. Org. Chem., 1970, 35, 3203–3205 CrossRef CAS.
- S. P. Chavan and B. M. Bhanage, Tetrahedron Lett., 2014, 55, 1199–1202 CrossRef CAS.
- R. Sarges, H. R. Howard, R. C. Browne, L. A. Label and P. A. Seymour, J. Med. Chem., 1990, 33, 2240–2254 CrossRef CAS PubMed.
- J. L. E. Erickson and F. E. Collins Jr, J. Org. Chem., 1965, 30, 1050–1052 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 ( Angew. Chem. , 2001 , 113 , 2056–2075 ) CrossRef CAS PubMed.
- R. Alamillo, M. Tucker, M. Chia, Y. Pagán-Torres and J. Dumesic, Green Chem., 2012, 14, 1413–1419 RSC.
- Y. Duan, J. Zhang, D. Li, D. Deng, L.-F. Ma and Y. Yang, RSC Adv., 2017, 7, 26487–26493 RSC.
- T. Buntara, I. Melián-Cabrera, Q. Tan, J. L. G. Fierro, M. Neurock, J. G. de Vries and H. J. Heeres, Catal. Today, 2013, 210, 106–116 CrossRef CAS.
- B. Wozniak, Y. Li, S. Tin and J. G. de Vries, Green Chem., 2018, 20, 4433–4437 RSC.
- S. Raju, M.-E. Moret and R. J. M. Klein Gebbink, ACS Catal., 2015, 5, 281–300 CrossRef CAS.
- J. R. Dethlefsen and P. Fristrup, ChemSusChem, 2015, 8, 767–775 CrossRef CAS PubMed.
- S. Tin and J. G. de Vries, in Science of Synthesis: Catalytic Reduction in Organic Synthesis, ed. J. G. de Vries, Georg Thieme Verlag KG, Stuttgart, 2018, vol. 1, pp. 229–243 Search PubMed.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 1543–1551 RSC.
- C. Li, G. Xu, X. Liu, Y. Yhang and Y. Fu, Ind. Eng. Chem. Res., 2017, 56, 8843–8849 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |