
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA reactive oxygen species-generating, cancer stem cell-potent manganese(II) complex and its encapsulation into polymeric nanoparticles†
Arvin
Eskandari
a and
Kogularamanan
Suntharalingam
 *b
*b
aDepartment of Chemistry, King's College London, London, SE1 1DB, UK
bDepartment of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: k.suntharalingam@leicester.ac.uk
First published on 2nd July 2019
Abstract
Intracellular redox modulation offers a viable approach to effectively remove cancer stem cells (CSCs), a subpopulation of tumour cells thought to be responsible for cancer recurrence and metastasis. Here we report the breast CSC potency of reactive oxygen species (ROS)-generating manganese(II)- and copper(II)-4,7-diphenyl-1,10-phenanthroline complexes bearing diclofenac, a nonsteriodial anti-inflammatory drug (NSAID), 1 and 3. Notably, the manganese(II) complex, 1, exhibits 9-fold, 31-fold, and 40-fold greater potency towards breast CSCs than 3, salinomycin (an established breast CSC-potent agent), and cisplatin (a clinically approved anticancer drug) respectively. Encouragingly, 1 displays 61-fold higher potency toward breast CSCs than normal skin fibroblast cells. Clinically relevant epithelial spheroid studies show that 1 is able to selectively inhibit breast CSC-enriched HMLER-shEcad mammosphere formation and viability (one order of magnitude) over non-tumorigenic breast MCF10A spheroids. Mechanistic studies show that 1 prompts breast CSC death by generating intracellular ROS and inhibiting cyclooxygenase-2 (COX-2) activity. The manganese(II) complex, 1, induces a greater degree of intracellular ROS in CSCs than the corresponding copper(II) complex, 3, highlighting the ROS-generating superiority of manganese(II)- over copper(II)-phenanthroline complexes. Encapsulation of 1 by biodegradable methoxy poly(ethylene glycol)-b-poly(D,L-lactic-co-glycolic) acid (PEG-PLGA) copolymers at the appropriate feed (5%, 1 NP5) enhances breast CSC uptake and greatly reduces overall toxicity. The nanoparticle formulation 1 NP5 indiscriminately kills breast CSCs and bulk breast cancer cells, and evokes a similar cellular response to the payload, 1. To the best of our knowledge, this is the first study to investigate the anti-CSC properties of managense complexes and to demonstrate that polymeric nanoparticles can be used to effectively deliver managense complexes into CSCs.
Introduction
A small subset of tumour cells, termed cancer stem cells (CSCs), are thought to be one of the underlying causes of metastasis and relapse.1,2 CSCs, akin to normal stem cells, replicate slowly and thus evade conventional cancer treatments (such as platinum chemotherapy and radiotherapy), which primarily target proliferating cells.3–5 Having bypassed conventional treatments, CSCs can instigate metastasis and relapse due to their cell motility proficiency and high secondary tumour seeding frequency, respectively.6,7 Therefore it is imperative for new generations of anticancer agents to remove entire populations of tumour cells, including CSCs, in order to provide a durable clinical response. Several CSC functional targets have been identified, however, there is still no clinically approved chemical or biological agent that can remove both bulk cancer cells and CSCs at non-lethal doses.8,9 All of the chemical agents currently undergoing clinical trials as prospective anti-CSC agents are completely organic in nature.8 Recently, we and others have shown that metal complexes can exhibit promising anti-CSC activities and deserve further investigation in this context.10Copper- and iron-containing complexes have emerged as promising anti-CSC agents owing to their ability to partake in Fenton-type reactions and produce lethal levels of reactive oxygen species (ROS) inside CSCs.11–13 Notably, copper(II)-phenanthroline and -Schiff base complexes bearing non-steroidal anti-inflammatory drugs (NSAIDs) kill breast CSCs and bulk breast cancer cells, both selectivity and indiscriminately at micromolar and sub-micromolar concentrations.12,14,15 Mechanistic studies show that these complexes induce CSC death by elevating intracellular ROS levels and inhibiting cyclooxygenase isoenzyme-2 (COX-2). This approach exploits the vulnerability of CSCs and bulk cancer cells to changes in their intracellular redox state and the overexpression of COX-2, a major player in regulation, in certain CSCs and bulk cancer cells.16,17 Here we have sought to improve CSC potency by developing related manganese(II)-phenanthroline complexes bearing diclofenac (a potent COX-2 inhibitor with anti-metastatic potential)18,19 with higher ROS-generating power. Analogous copper(II)-phenanthroline complexes bearing diclofenac were also prepared and used as controls to showcase the superiority of manganese(II) over copper(II) (within this system) to elevate intracellular ROS levels.
Manganese is an essential trace element for animals and plants, and plays an important role in many biological processes including cell protection, metabolism, bone mineralisation, and reproductive function.20 Although the anticancer properties of manganese complexes are underexplored, a number of bulk cancer cell-potent manganese(II/III) complexes containing Schiff base, porphyrin, flavonoids, and polypyridyl ligands have been identified.21–24 The cellular mechanism of action of the manganese(II/III) complexes is highly dependent on the nature of the coordinated ligand.25 Notably, a number of studies have shown that manganese metal and its coordination complexes increase markers of oxidative stress in various cell types due to intracellular ROS generation.20,26–29 By utilising manganese over other more common endogenous metals such as copper and iron, we hope to take advantage of the many physiologically accessible oxidation states of manganese and its ability to undergo Fenton-type reactions (whereby the manganese(II) form can effectively generate hydroxide radicals from endogenous hydrogen peroxide), to potently kill CSCs via efficient ROS production. Indeed, a series of di-nuclear manganese(II)-phenanthroline complexes were previously reported to significantly increase intracellular ROS levels and thus cytotoxicity in human-derived colorectal cancer lines, relative to equivalent copper(II) complexes.27 The impact of manganese-mediated ROS generation in CSCs, however, is completely unknown.
Therapeutic agents (especially metal complexes) can suffer from systematic toxicity due to non-specificity, resulting from their degradation in biological solutions. Nanoscale technologies offer a method to precisely deliver anticancer therapeutics to tumour microenvironments.30 Further, nano-systems increase drug solubility, bioavailability, and extend drug half-life.31,32 Nano-sized particles passively target certain tumours by taking advantage of the enhanced permeability and retention (EPR) effect in tumour tissues.33,34 From recent clinical studies, it is clear that the EPR effect is highly dependent on tumour histology.35,36 For instance, human carcinomas are very vascular and have many porous blood vessels and thus more susceptible to targeting by the EPR effect.37 Slow-growing human sarcomas on the other hand are not extremely vascular and thus less able to accumulate nano-sized entities via the EPR effect. The work presented in this manuscript involves breast carcinomas (bulk and CSCs). Several nanoparticle formulations exist, including those based on iron oxide, carbon, gold, hydrogels, liposomes, and polymers.38 A number of these formulations are currently used in the clinic to deliver chemotherapies to tumours.39 Polymeric nanoparticles are of particular interest due to their biocompatibility, synthetic versatility, and tuneable properties.40 We recently reported a proof-of-concept study where the biodegradable, amphiphilic copolymer, methoxy poly(ethylene glycol)-b-poly(D,L-lactic-co-glycolic) acid (PEG-PLGA) was used to encapsulate and deliver a copper(II)-NSAID complex into (carcinoma-derived) breast CSCs.41 In addition to improved cellular uptake, cytotoxicity toward breast CSCs over bulk breast cancer cells, was enhanced compared to the payload. Here we use similar polymeric nanoparticles to encapsulate and deliver the most CSC-potent manganese(II)-phenanthroline complex identified in this study into breast CSCs.
Results and discussion
Synthesis and characterisation
The manganese(II)- and copper(II)-phenanthroline complexes, 1–4 investigated in this study are shown in Fig. 1. The manganese(II) complexes, 1 and 2 were prepared by reacting MnCl2·4H2O with 3,4,7,8-tetramethyl-1,10-phenanthroline or 4,7-diphenyl-1,10-phenanthroline and two equivalents of diclofenac sodium salt in methanol, under basic conditions. The corresponding copper(II) complexes, 3 and 4 were synthesised via the same procedure, with CuCl2·2H2O used as the metal source. The manganese(II) complexes, 1 and 2 were isolated as yellow solids whereas the copper(II) complexes, 3 and 4 were obtained as green solids. All of the complexes, 1–4 were characterised by infrared spectroscopy and elemental analysis (Fig. S1†). The difference between the vibrational stretching frequencies between the asymmetric, νasym(CO2) and symmetric, νsym(CO2) carbonyl peaks gives an indication into the binding mode of the associated carboxylic acid group to a given metal centre.42,43 According to the IR spectra, the difference (Δ) between νasym(CO2) and νsym(CO2) stretching bands for 1 and 2 varied between 125–128 cm−1 (Fig. S1A and B†), suggestive of a bidentate binding mode for the carboxylate group on diclofenac to the manganese(II) centre (as depicted in Fig. 1). Similarly, the difference (Δ) between νasym(CO2) and νsym(CO2) stretching bands for 3 and 4 was 107–125 cm−1 (Fig. S1C and D†), indicative of a bidentate binding mode between diclofenac and the copper(II) centre (as depicted in Fig. 1). The purity of 1–4 was established by elemental analysis (see Experimental details section in the ESI†). Mn(3,4,7,8-tetramethyl-1,10-phenanthroline)Cl2 was also prepared to serve as a control compound – a manganese(II)-phenanthroline complex without diclofenac. The synthetic protocol and characterisation of Mn(3,4,7,8-tetramethyl-1,10-phenanthroline)Cl2 is reported in the Experimental details section in the ESI.†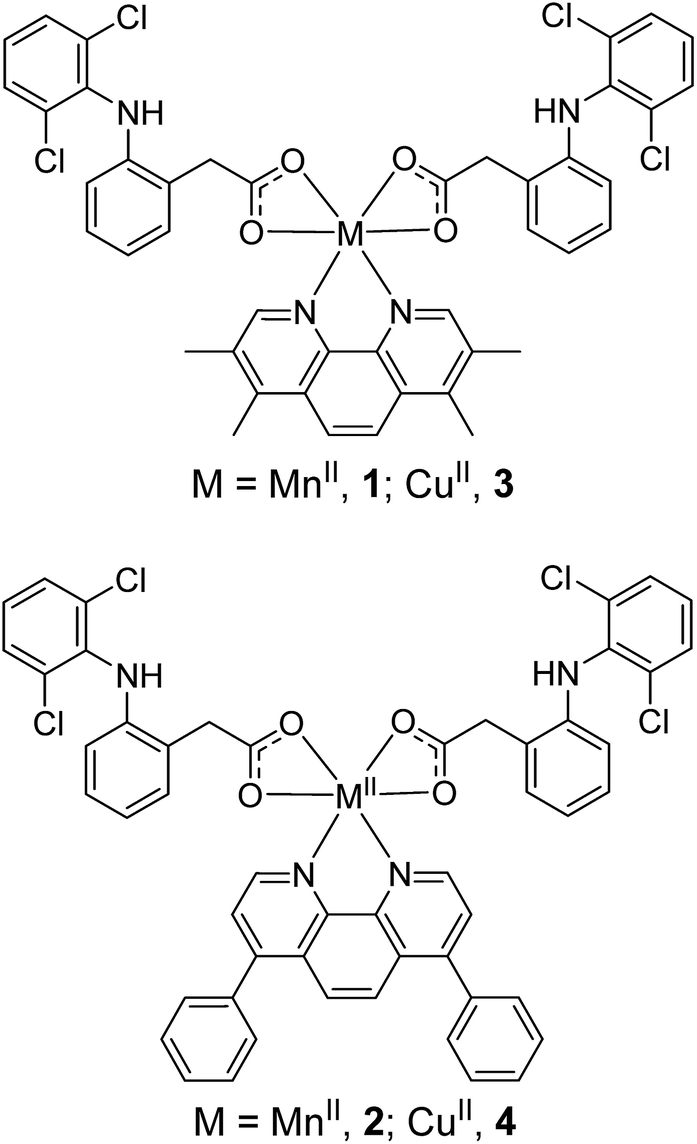 | ||
| Fig. 1 Proposed chemical structures of the manganese(II)- and copper(II)-phenanthroline complexes bearing two diclofenac molecules, 1–4 under investigation in this study. | ||
Lipophilicity and stability in biological solutions
The lipophilicity of 1–4 was determined by measuring the extent to which it partitioned between octanol and water, P. The experimentally determined log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values varied from 0.9 to 1.1 for the manganese(II) complexes, 1 and 2, and 0.2 to 0.8 for the copper(II) complexes, 3 and 4 (Table S1†). The hydrophobic nature of 1–4 suggests that the manganese(II) and copper(II) complexes should be readily internalized by cells. UV-vis spectroscopy studies were carried out to assess the stability of 1–4, in biological relevant solutions. The UV-vis metal-perturbed π–π* absorption band of 1 (25 μM) in PBS:DMSO (200:1) with and without 10 equivalents of glutathione (a cellular reductant) remained constant over the course of 24 h at 37 °C (Fig. S2 and S3†), indicative of stability. In contrast, the 4,7-diphenyl-1,10-phenanthroline-containing manganese(II) complex, 2 was relatively unstable in PBS:DMSO (200:1) with and without 10 equivalents of glutathione (Fig. S4 and S5†). Like the trend observed for the manganese(II) complexes, the copper(II) complex bearing 3,4,7,8-tetramethyl-1,10-phenanthroline, 3 was comparatively more stable in PBS:DMSO (200:1) with and without 10 equivalents of glutathione than the 4,7-diphenyl-1,10-phenanthroline-bearing complex, 4 (Fig. S6–S9†). The UV-vis bands associated to 1 and 3 (25 μM) in mammary epithelial cell growth medium (MEGM)/DMSO (200:1) remained unaltered over the course of 24 h at 37 °C, suggestive of stability in conditions required for cellular studies (Fig. S10 and S11†). Under the same conditions, 2 and 4 were relatively unstable (Fig. S12 and S13†). Collectively, the UV-vis spectroscopy studies suggest that the stability of the complexes in biologically relevant solutions is dependent on the metal and the polypyridyl ligand, with the manganese(II)-3,4,7,8-tetramethyl-1,10-phenanthroline complex, 1 being the most stable. High-resolution ESI mass spectrometry studies were also performed to confirm the stability of 1 in biologically-relevant conditions. Upon incubation of 1 (100 μM) in PBS:DMSO (200:1) for 72 h at 37 °C, a peak corresponding to [1-2H + 2Na]+ (925.1902 m/z) with the appropriate isotopic distribution was observed in the positive mode of the ESI mass spectrum (Fig. S14†), further suggesting that 1 is stable under these conditions.
P values varied from 0.9 to 1.1 for the manganese(II) complexes, 1 and 2, and 0.2 to 0.8 for the copper(II) complexes, 3 and 4 (Table S1†). The hydrophobic nature of 1–4 suggests that the manganese(II) and copper(II) complexes should be readily internalized by cells. UV-vis spectroscopy studies were carried out to assess the stability of 1–4, in biological relevant solutions. The UV-vis metal-perturbed π–π* absorption band of 1 (25 μM) in PBS:DMSO (200:1) with and without 10 equivalents of glutathione (a cellular reductant) remained constant over the course of 24 h at 37 °C (Fig. S2 and S3†), indicative of stability. In contrast, the 4,7-diphenyl-1,10-phenanthroline-containing manganese(II) complex, 2 was relatively unstable in PBS:DMSO (200:1) with and without 10 equivalents of glutathione (Fig. S4 and S5†). Like the trend observed for the manganese(II) complexes, the copper(II) complex bearing 3,4,7,8-tetramethyl-1,10-phenanthroline, 3 was comparatively more stable in PBS:DMSO (200:1) with and without 10 equivalents of glutathione than the 4,7-diphenyl-1,10-phenanthroline-bearing complex, 4 (Fig. S6–S9†). The UV-vis bands associated to 1 and 3 (25 μM) in mammary epithelial cell growth medium (MEGM)/DMSO (200:1) remained unaltered over the course of 24 h at 37 °C, suggestive of stability in conditions required for cellular studies (Fig. S10 and S11†). Under the same conditions, 2 and 4 were relatively unstable (Fig. S12 and S13†). Collectively, the UV-vis spectroscopy studies suggest that the stability of the complexes in biologically relevant solutions is dependent on the metal and the polypyridyl ligand, with the manganese(II)-3,4,7,8-tetramethyl-1,10-phenanthroline complex, 1 being the most stable. High-resolution ESI mass spectrometry studies were also performed to confirm the stability of 1 in biologically-relevant conditions. Upon incubation of 1 (100 μM) in PBS:DMSO (200:1) for 72 h at 37 °C, a peak corresponding to [1-2H + 2Na]+ (925.1902 m/z) with the appropriate isotopic distribution was observed in the positive mode of the ESI mass spectrum (Fig. S14†), further suggesting that 1 is stable under these conditions.
Cytotoxicity toward CSCs, bulk cancer cells, and normal cells in monolayer systems
Given the instability of the 4,7-diphenyl-1,10-phenanthroline-containing complexes, 2 and 4 in MEGM (Fig. S12 and S13†), cellular studies were not performed with these complexes. The cytotoxicity of 1 and 3 against bulk bone (U2OS), liver (HepG2), and breast (HMLER) cancer cells, and breast CSC-enriched cells (HMLER-shEcad) was determined using the MTT assay. The IC50 values were determined from dose–response curves (Fig. S15 and S16†) and are summarised in Tables 1 and S2.† Both complexes displayed submicro- or micro-molar potency towards the cell lines tested. The manganese(II) complex, 1 killed CSC-enriched HMLER-shEcad cells and CSC-depleted HMLER cells similarly whereas the copper(II) complex, 3 killed CSC-depleted HMLER cells preferentially (9-fold selectivity) (Table 1). Notably, 1 displayed 9-fold, 31-fold, and 40-fold greater potency (p < 0.05, n = 18) for HMLER-shEcad cells than 3, salinomycin (an established breast CSC-potent agent),12 and cisplatin (a clinically approved anticancer drug) respectively (Fig. S17,†Table 1). As a measure of therapeutic potential, the cytotoxicity of 1 and 3 towards normal skin fibroblast GM07575 cells was determined. The complexes, 1 and 3 were 61-fold and 11-fold less potent toward GM07575 cells (IC50 value = 8.5 ± 1.5 μM for 1 and 13.9 ± 0.1 μM for 3, Fig. S18 and S19†) than HMLER-shEcad cells respectively, indicating selective toxicity for breast CSCs over non-tumorigenic cells. Taken together, the cytotoxicity studies showed that the manganese(II) complex, 1 is significantly (p < 0.05) more cytotoxic towards breast CSCs than its copper(II) analogous, 3, and displays reduced toxicity towards normal cells.| Compound | HMLER IC50/nM | HMLER-shEcad IC50/nM |
|---|---|---|
| a Taken from ref. 12. | ||
| 1 | 186 ± 26 | 137 ± 15 |
| 3 | 133 ± 8 | 1250 ± 282 |
| Cisplatin | 2565 ± 21 | 5645 ± 304 |
| Salinomycina | 11400 ± 400 |
4200 ± 400 |
| MnCl2·4H2O | 10385 ± 1491 |
7765 ± 318 |
| CuCl2·2H2O | 47545 ± 176 |
47920 ± 1803 |
| Diclofenac | 69495 ± 8704 |
54735 ± 233 |
| 3,4,7,8-Tetramethyl-1,10-phenanthroline | 785 ± 10 | 720 ± 84 |
| Mn(3,4,7,8-tetramethyl-1,10-phenanthroline)Cl2 | 163 ± 21 | 170 ± 14 |
Control cytotoxicity studies showed that the potency of MnCl2·4H2O, CuCl2·2H2O, diclofenac, and the polypyridyl ligand, 3,4,7,8-tetramethyl-1,10-phenanthroline, towards HMLER and HMLER-shEcad cells was significantly lower (p < 0.05, n = 18) than 1 and 3 (Table 1 and Fig. S20–S23†). This suggests that the cytotoxicity of 1 and 3 towards bulk breast cancer cells and breast CSCs is likely to result from the intact manganese(II) and copper(II) complexes rather than their individual components (manganese, copper, diclofenac, or the free polypyridyl ligand). Interestingly the potency of Mn(3,4,7,8-tetramethyl-1,10-phenanthroline)Cl2 towards HMLER and HMLER-shEcad cells was similar to 1 (Table 1 and Fig. S24†). This suggests that the Mn-polypyridyl unit is largely responsible for the bulk breast cancer cell and breast CSC toxicity of 1.
Epithelial spheroid inhibitory studies
Epithelial breast cells (cancer and non-tumorigenic), when grown in serum-free media under low-attachment conditions, are capable of forming three-dimensional spheroids called mammospheres.44 The ability of a given compound to inhibit mammosphere formation from single cell suspensions (with respect to number, size, and viability) is often used as a marker for in vivo potency, given that three-dimensional systems are more representative of organs and tumours than monolayer cell cultures. The ability of 1 and 3 to inhibit breast CSC-enriched HMLER-shEcad and non-tumorigenic breast epithelial MCF10A mammosphere formation (at a non-lethal dose, IC20 value for 5 days) was assessed using an inverted microscope. The addition of 1 (IC20 value for 5 days) to single cell suspensions of HMLER-shEcad cells significantly (p < 0.05) decreased the number and size of HMLER-shEcad mammospheres formed (Fig. 2A and B). Notably, the HMLER-shEcad mammosphere inhibitory effect of 1 (51% decrease in number of HMLER-shEcad mammospheres formed compared to the untreated control) was comparable to that of salinomycin under identical conditions (53% decrease in number of HMLER-shEcad mammospheres formed compared to the untreated control, Fig. 2A and B). The copper(II) complex, 3 (IC20 value for 5 days) did not significantly alter the number or size of HMLER-shEcad mammospheres formed (Fig. S25 and S26†). We have previously shown that diclofenac does not significantly affect the number or size of HMLER-shEcad mammospheres formed.15 This suggests the HMLER-shEcad mammosphere inhibitory effect of 1 is a due to the manganese(II)-polypyridyl component. To gauge the ability of 1 to decrease HMLER-shEcad mammosphere viability, the colorimetric resazurin-based reagent, TOX8 was employed. Encouragingly, the IC50 value of 1 (3.0 ± 0.1 μM) (Fig. S27†) was 6-fold lower than that reported for salinomycin (18.5 ± 1.5 μM)15 under identical conditions. As diclofenac is non-toxic towards HMLER-shEcad mammospheres (IC50 > 133 μM),15 we believe that the manganese(II)-polypyridyl component in 1 is the major contributor for HMLER-shEcad mammosphere toxicity.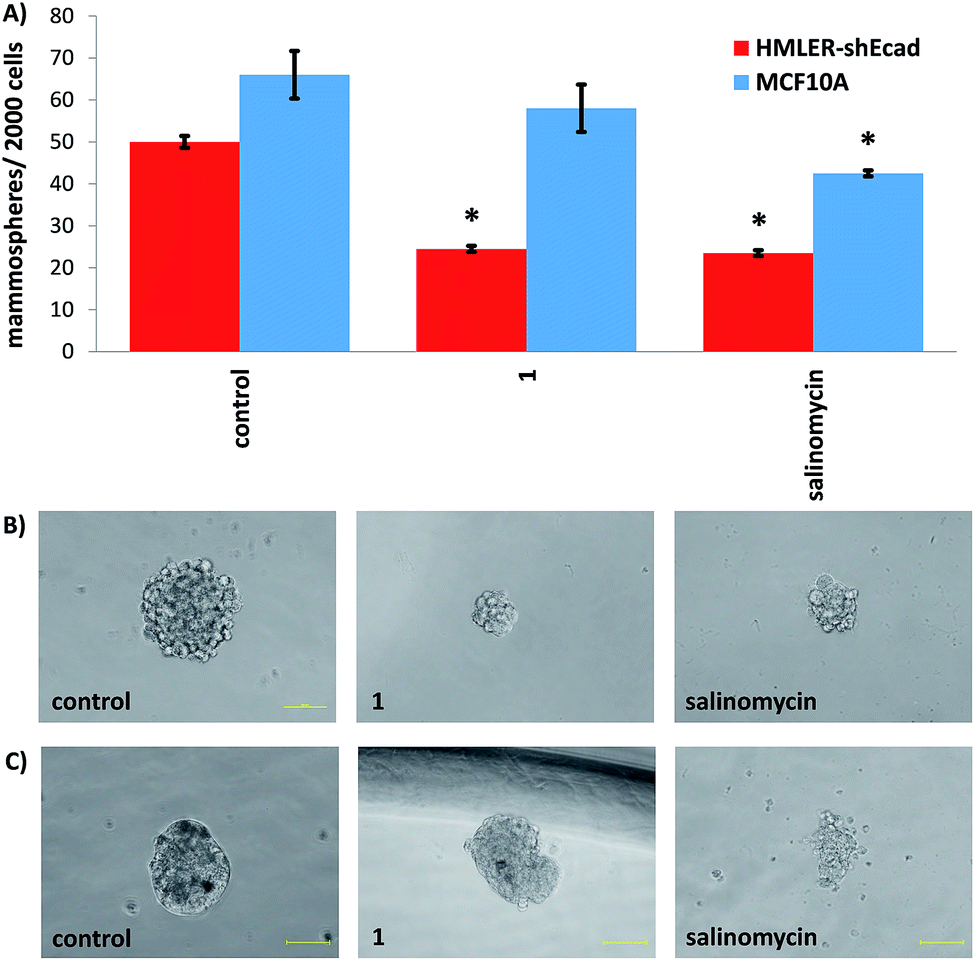 | ||
| Fig. 2 (A) Quantification of spheroid formation with HMLER-shEcad and MCF10A cells untreated and treated with 1 and salinomycin at their respective IC20 values for 5 days. Error bars = SD and Student t-test, * = p < 0.05. (B and C) Representative bright-field images (×20) of HMLER-shEcad and MCF10A spheroids in the absence and presence of 1 and salinomycin at their respective IC20 values for 5 days. | ||
The addition of 1 (IC20 value for 5 days) to single cell suspensions of non-tumorigenic MCF10A cells did not significantly (p = 0.25) change the number or size of MCF10A spheroids formed (Fig. 2A and B). In contrast, treatment with salinomycin under the same conditions resulted in a significant (p < 0.05) decrease in the number (40% decrease) and size of MCF10A spheroids formed (Fig. 2A and B). This is similar to the result obtained with HMLER-shEcad mammospheres (Fig. 2A and B). Spheroid viability studies showed that 1 killed non-tumorigenic MCF10A spheroids (IC50 = 36.7 ± 1.9 μM) with 12-fold lower potency than CSC-enriched HMLER-shEcad mammospheres (Fig. S28†). Salinomycin on the other hand killed MCF10A (IC50 = 14.9 ± 0.5 μM) and HMLER-shEcad mammospheres equipotently (Fig. S29†). Overall the epithelial spheroid studies, show that 1 is able to selectively inhibit CSC-enriched HMLER-shEcad mammospheres formation and viability over non-tumorigenic MCF10A spheroids. These properties are highly desirable for selecting CSC drug candidates in preclinical studies.
Cellular uptake and fractionation in breast CSCs
Cellular uptake studies were carried out to determine the breast CSC permeability of 1 and 3. CSC-enriched HMLER-shEcad cells were incubated with 1 or 3 (0.25 μM for 12 h) and the intracellular manganese or copper concentration was determined by inductively coupled plasma mass spectrometry (ICP-MS). The complex, 1 was modestly taken up by HMLER-shEcad cells, with whole cell uptake totalling 5.4 ± 0.1 ppb of Mn/million cells (Fig. S30†). Under identical conditions, 3 (0.25 μM for 12 h) entered HMLER-shEcad cells to a higher extent (17.5 ± 0.5 ppb of Cu/million cells) than 1. Although the amount of manganese and copper found inside HMLER-shEcad and HMLER cells is relatively low, it is reasonable considering the low administration concentration (0.25 μM). Fractionation studies were conducted to determine the cell localisation of 1 in HMLER-shEcad cells (Fig. S30†). In terms of the cell distribution pattern, 6% of internalised 1 was found in the nucleus, 39% in the cytoplasm, and 10% in the membrane (Fig. S30†). The ability of 1 to access the nucleus could be attributed to the presence of the diclofenac ligand, which is known to interact with COX-2 located on the nuclear envelope.45 Since a relatively large amount of internalised 1 was also found in the cytoplasm and membrane, the fractionation studies suggest that 1-induced cell toxicity could be related to a mixture of genomic DNA-dependent or -independent mechanisms.Mechanism of CSC toxicity of 1
The manganese(II) complex, 1 was envisaged to kill CSCs by increasing intracellular ROS levels to lethal doses. To test this hypothesis, the ability of 1 to produce ROS in HMLER-shEcad cells was determined using 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), a well-established ROS indicator. HMLER-shEcad cells treated with 1 (0.25 μM) exhibited a substantial increase in intracellular ROS levels over the course of 48 h, peaking after 6 h exposure (3.4-fold increase) (Fig. 3A). Under identical conditions, the corresponding copper(II) complex, 3 (0.25 μM) displayed a general increase in ROS levels in HMLER-shEcad cells over the course of 48 h, however, the extent was significantly lower than 1 (Fig. S31†). Cytotoxicity studies in the presence of N-acetylcysteine (NAC, 2 mM, 72 h), a ROS scavenger showed that the potency of 1 and 3 towards HMLER-shEcad cells decreased significantly (IC50 value increased from 137 ± 15 nM to 1100 ± 89 nM, 8-fold, p < 0.05 for 1 and 1.3 ± 0.3 μM to 3.6 ± 0.1 μM, 3-fold, p < 0.05 for 3) (Fig. S32 and S33†) suggesting that 1 and 3-induced CSC death is related to intracellular ROS generation. The differential increases in IC50 values of 1 and 3 in the presence of NAC indicates that 1-induced CSC death is more reliant on intracellular ROS production than 3-induced CSC death. Therefore, the greater intracellular ROS-generating power of 1 over 3 could explain the 9-fold greater potency of 1 over 3 against HMLER-shEcad cells (Table 1).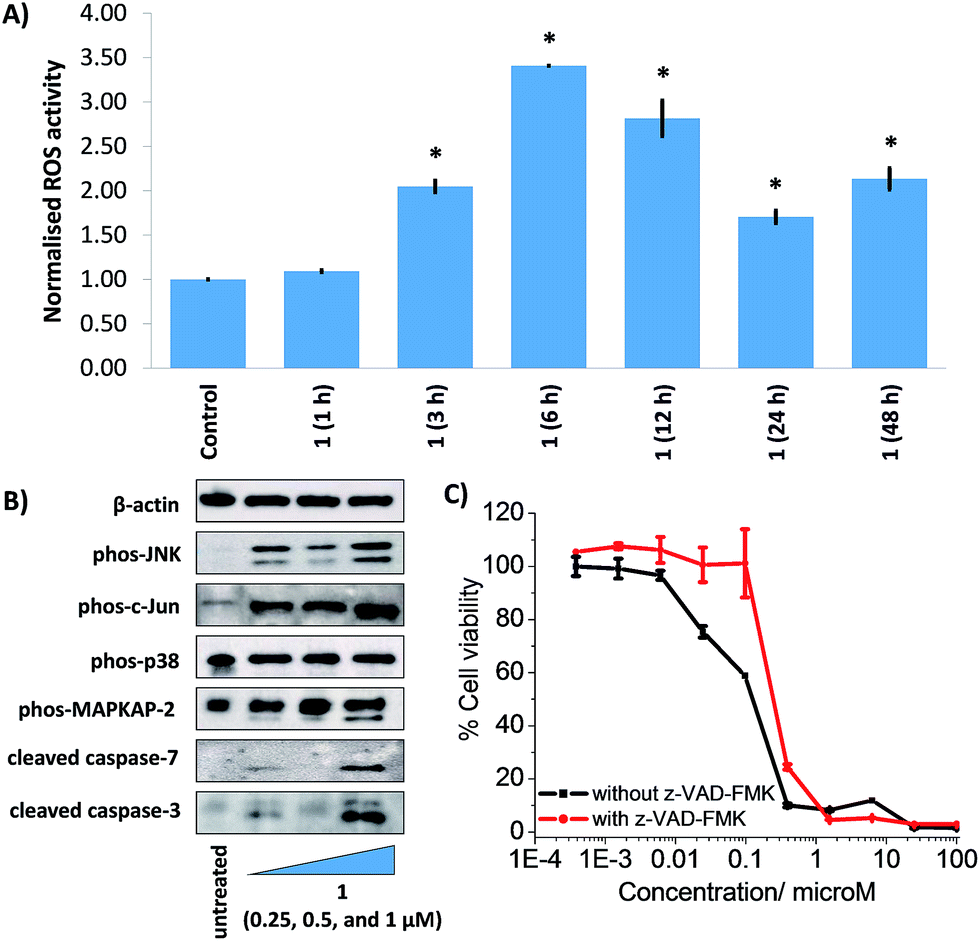 | ||
| Fig. 3 (A) Normalised ROS activity in untreated HMLER-shEcad cells (control) and HMLER-shEcad cells treated with 1 (0.25 μM for 1, 3, 6, 12, 24, and 48 h). Error bars represent SD and Student t-test, * = p < 0.05. (B) Immunoblotting analysis of proteins related to the JNK, p38, and apoptosis pathways. Protein expression in HMLER-shEcad cells following treatment with 1 (0.25, 0.5, and 1 μM for 72 h) (C) representative dose–response curves of 1 against HMLER-shEcad cells in the absence and presence of z-VAD-FMK (5 μM) after 72 h incubation. | ||
Intracellular ROS production can activate Jun-amino-terminal kinase (JNK) and/or p38 MAP kinase (MAPK) pathways.46 To provide further insight into the mechanism of cytotoxicity of the manganese(II) complex, 1 we conducted immunoblotting studies to monitor changes in the expression of biomarkers related to these pathways. HMLER-shEcad cells dosed with 1 (0.25–1 μM for 72 h) exhibited enhanced phosphorylation of JNK and its downstream effector, c-Jun indicative of JNK pathway activation (Fig. 3B). Interestingly, p38 phosphorylation was not markedly effected although its downstream effector, MAP kinase-activated protein kinase 2 (MAPKAPK-2) was greatly phosphorylated in the presence of 1 (Fig. 3B). Constant activation of JNK or p38 pathways can induce apoptosis.47 Immunoblotting studies showed that HMLER-shEcad cells treated with 1 (0.25–1 μM for 72 h) displayed increased expression of cleaved caspase-3 and -7 compared to untreated cells, indicative of caspase-dependent apoptosis (Fig. 3B). Interestingly, 1 induced a greater level of caspase-3 and -7 cleavage upon treatment of HMLER-shEcad cells with 0.25 μM of 1 than 0.5 μM of 1. Cytotoxicity studies in the presence of z-VAD-FMK (5 μM), a peptide-based caspase-dependent apoptosis inhibitor showed that the potency of 1 towards HMLER-shEcad cells decreased significantly (p < 0.05, IC50 value = 220 ± 14 nM) (Fig. 3C). This confirms that 1 induces caspase-dependent CSC death. As expected the potency of cisplatin, a well-known apoptosis-inducer, towards HMLER-shEcad cells decreased significantly (p < 0.05, IC50 value = 10.21 ± 0.78 μM) in the presence of z-VAD-FMK (5 μM) (Fig. S34†). Collectively, the ROS, immunoblotting, and cytotoxicity studies show that 1 elevates intracellular ROS levels, activates the JNK pathway, and triggers apoptotic CSC death.
COX-2 is overexpressed in certain bulk breast cancer cells and breast CSCs and is thought to play a meaningful role in CSC maintenance and regulation, as well as chemotherapeutic resistance.17,48,49 COX-2 is widely regarded as a molecular target for CSC-directed therapy. Given that the manganese(II) complex, 1 contains two diclofenac ligands, flow cytometric studies were performed to determine if the mechanism of action of 1 involved COX-2 downregulation. HMLER-shEcad cells were pre-treated with lipopolysaccharide (LPS) (2.5 μg L−1 for 24 h), to increase basal COX-2 levels, and then treated with 1 (0.25 μM), diclofenac (20 μM) or Mn(3,4,7,8-tetramethyl-1,10-phenanthroline)Cl2 (0.25 μM) for 48 h and the COX-2 expression was determined. A noticeable decrease in COX-2 expression was also observed in HMLER-shEcad cells treated with 1 and diclofenac compared to untreated cells (Fig. S35†). No appreciable change in COX-2 expression was detected in the presence of Mn(3,4,7,8-tetramethyl-1,10-phenanthroline)Cl2 suggesting that the COX-2 downregulation observed in the presence of the 1 is most likely mediated by the diclofenac moieties (Fig. S36†). Overall this suggests that the cytotoxic mechanism of action of 1 may involve COX-2 downregulation. To probe this further, cytotoxicity studies were carried out with HMLER-shEcad cells in the presence of prostaglandin E2 (PGE2) (20 μM, 72 h), the functional product of COX-2-catalysed arachidonic acid metabolism. The IC50 value for 1 against HMLER-shEcad cells in the presence of PGE2 was significantly (p < 0.05, IC50 value = 255 ± 50 nM) higher than for 1 alone (Fig. S37†), implying that 1 induces COX-2-dependent CSC death.
Encapsulation of 1 into polymeric nanoparticles
In order to improve the anti-CSC properties of 1 further (especially the cellular uptake), biodegradable PEG-PLGA polymeric nanoparticles were employed. PEG-PLGA copolymers are amphiphilic and tend to self-assemble in water to yield sphere-like nanoparticles with a hydrophobic PLGA inner core and a hydrophilic PEG outer shell.50 The hydrophobic nature of 1 (logP = 1.1) enabled encapsulation into the lipophilic core of PEG-PLGA (5000:30000 Da, 1:1 LA:GA) nanoparticles using the nanoprecipitation method. Various nanoparticle formulations (1 NP2.5–60) were prepared using a range of feeds (2.5–60%), where feed refers to the percentage (w/w) of 1 to polymer. To determine the most suitable formulation for biological studies, the loading and encapsulation efficiency of 1 was calculated by measuring the manganese concentration using ICP-MS. The loading and encapsulation efficiency was highly dependent on the feed (Fig. 4). Optimal encapsulation conditions were achieved at 5% feed (1 NP5), where the encapsulation efficiency = 5.1% and loading efficiency = 0.25%. The diameter of 1 NP5 was 114.7 nm and the polydispersity was 0.116, according to dynamic light scattering (DLS) studies (Fig. S38†). These values are comparable to those reported for other polymeric nanoparticles with internalised metal complexes.51,52 For reference, the diameter of empty PEG-PLGA nanoparticles was 94.5 nm and the polydispersity was 0.086 (Fig. S39†). As expected the zeta-potential of 1 NP5 was negative (−14.4 mV) owing to the PEG outer shell (Fig. S40†). The zeta-potential of empty PEG-PLGA nanoparticles was −33.3 mV (Fig. S41†). The surface morphology and size distribution of 1 NP5 was examined further by scanning electron microscopy (SEM). SEM images showed that 1 NP5 does indeed adopt relatively uniform spherical structures with an average size of 93 ± 6 nm (Fig. S42†). The latter is in good agreement with the DLS measurements (Fig. S38†). The nanoparticle formulation, 1 NP5 was stable in water, PBS, and PBS with 10% FBS (all at pH 7.4) over the course of 72 h at 37 °C (Fig. S43†). Furthermore, 1 NP5 effectively releases reasonable amounts of its payload over the course of 72 h at 37 °C in PBS (pH 7.4) (Fig. S44†). The stability of 1 NP5 and its ability to release sufficient amount of the payload bode well for the effective delivery of 1 into breast CSCs.
 | ||
| Fig. 4 The effect of feed variation on loading and encapsulation efficiency of 1 incorporated into PEG-PLGA nanoparticles. | ||
Delivery of 1 into CSCs using polymeric nanoparticles
To gauge the ability of the nanoparticle formulation, 1 NP5 to deliver its payload, 1, into CSCs, the manganese content in HMLER-shEcad cells treated with 1 NP5 (1 μM, 12 h) was measured by ICP-MS. The intracellular manganese concentration (297.1 ± 1.8 ppb of Mn/million cells) was 15-fold higher than that observed for HMLER-shEcad cells treated with 1 under identical conditions (18.6 ± 0.7 ppb of Mn/million cells, Fig. 5A). This shows that 1 NP5 is able to markedly improve the internalisation of 1 into breast CSCs. Polymeric nanoparticles such as PEG-PLGA tend to undergo cell uptake via energy-dependent endocytosis.53 To probe the mechanism of cellular uptake of 1 NP5, studies were conducted in the presence of endocytosis inhibitors. HMLER-shEcad cells treated with 1 NP5 (1 μM, 12 h) in the presence of chloroquine (100 μM) or ammonium chloride (50 mM) displayed significantly (p < 0.01) lower levels of manganese than HMLER-shEcad cells treated with 1 NP5 alone (Fig. 5A). This suggests that the mechanism of CSC uptake of 1 NP5 is likely to be endocytosis. Nanoparticles undergoing endocytosis often end up in acidic endosomes. Therefore we investigated the ability of 1 NP5 to release its payload under acidic conditions (sodium acetate buffer, pH 5.2 at 37 °C over 72 h). The nanoparticle formulation, 1 NP5 released 1 to a better extent than in PBS (pH 7.4), under similar conditions (Fig. S44†), indicating that it is capable of releasing its payload in CSCs upon endocytic uptake. | ||
| Fig. 5 (A) Manganese content in HMLER-shEcad cells treated with 1 NP5 (1 μM for 12 h) at 37 °C, 1 (1 μM for 12 h) at 37 °C, 1 NP5 (1 μM for 12 h) co-incubated with ammonium chloride (50 mM) or chloroquine (100 μM) at 37 °C. Error bars represent standard deviations and Student t-test, ** = p < 0.01. (B) Representative dose–response curves for the treatment of HMLER-shEcad and HMLER cells with 1 NP5 after 72 h incubation. (C) Representative bright-field images (×20) of HMLER-shEcad mammospheres in the absence and presence of 1 NP5 and salinomycin at their respective IC20 values for 5 days. | ||
CSC potency and mechanism of action of the nanoparticle formation, 1 NP5
The cytotoxicity of 1 NP5 against breast CSC-enriched (HMLER-shEcad) and breast CSC-depleted (HMLER) cells was determined using the MTT assay (Fig. 5B). The nanoparticle formulation displayed micromolar toxicity toward both cell lines (IC50 value = 1.3 ± 0.1 μM for HMLER-shEcad cells and IC50 value = 1.8 ± 0.1 μM for HMLER cells, Fig. 5B). The nanoparticle formulation, 1 NP5 was 10-fold less toxic than the payload, 1 for both HMLER-shEcad and HMLER cells. This result was somewhat expected, as polymeric nanoparticles are well known to reduce the toxicities of incorporated compounds.54 Importantly, 1 NP5 killed HMLER-shEcad and HMLER cells with equal potency (like 1), showing that the encapsulation of 1 by PEG-PLGA polymeric nanoparticles does not affect its spectrum of activity. The empty PEG-PLGA nanoparticles were non-toxic towards CSC-enriched HMLER-shEcad cells (IC50 value > 5 mg L−1) (Fig. S45†). This implies that the CSC-potency of the nanoparticle formation, 1 NP5 is due to its payload, 1 rather than the PEG-PLGA components. Spheroid formation studies showed that 1 NP5 (when treated at the IC20 value for 5 days) was able to significantly reduce the number and size of HMLER-shEcad mammospheres formed from single cell suspensions of HMLER-shEcad cells (Fig. 5C and S46†). This result was comparable to the payload, 1 (Fig. 2A and B). Notably, 1 NP5 was able to reasonably recapitulate the potency of the payload, 1 towards HMLER-shEcad mammospheres (IC50 value = 3.6 ± 0.1 μM, Fig. S47†). Interestingly, 1 NP5 did not significantly (p = 0.18) change the number or size of non-tumorigenic MCF10A spheroids formed (Fig. S48 and S49†). Viability studies showed that 1 NP5 killed non-tumorigenic MCF10A spheroids (IC50 = 9.5 ± 0.1 μM) with significantly (p < 0.05) lower potency than CSC-enriched HMLER-shEcad mammospheres (Fig. S50†). Collectively, the monolayer and three-dimensional toxicity studies show that encapsulation of 1 into PEG-PLGA polymeric nanoparticles (1 NP5) does not detrimentally affect its potency towards CSCs.Further cell studies were conducted to determine the mechanism of action of the nanoparticle formulation, 1 NP5. Given that the payload, 1 induced CSC death by elevating intracellular ROS levels and inhibiting COX-2, studies were carried to determine if 1 NP5 was able to retain the mechanism of action of the payload. HMLER-shEcad cells treated with 1 NP5 (1.3 μM) displayed a massive increase in intracellular ROS levels over the course of 48 h, peaking after 3 h exposure (9-fold increase) (Fig. S51†). Under identical conditions, after 3 h exposure, the payload, 1 increased intracellular ROS levels by only 2-fold (Fig. 3A). The markedly different ROS generating power of the nanoparticle formulation, 1 NP5 and the payload, 1 is likely to be due to the better internalisation of the nanoparticle formulation (Fig. 5A). Cell viability studies in the presence of NAC (2 mM, 72 h) showed that the potency of 1 NP5 towards HMLER-shEcad cells decreased significantly (IC50 value increased from 1.3 ± 0.1 μM to 4.8 ± 0.1 μM, 3.7-fold, p < 0.05) (Fig. S52†) suggesting that 1 NP5-induced CSC death, like 1-induced CSC death, is related to intracellular ROS generation. Immunoblotting studies showed that like 1, 1 NP5 is able to activate the JNK and p38 pathways (probably due to ROS formation) and induce caspase-dependent apoptosis (Fig. S53†). Cytotoxicity studies in the presence of z-VAD-FMK (5 μM), showed that the potency of 1 NP5 towards HMLER-shEcad cells decreased significantly (p < 0.05, IC50 value = 4.4 ± 0.1 μM) (Fig. S54†), confirming that 1 NP5 induces caspase-dependent CSC apoptosis.
HMLER-shEcad cells pre-treated with LPS (2.5 μg L−1 for 24 h) and incubated with 1 NP5 (0.6–1.3 μM for 48 h) displayed a noticeable decrease in COX-2 levels according to flow cytometric studies (Fig. S55†). This is comparable to the COX-2 downregulation induced by the payload, 1 (Fig. S35†). Cytotoxicity studies in the presence of PGE2 (20 μM, 72 h) showed that the potency of 1 NP5 towards HMLER-shEcad cells decreased significantly (IC50 value increased from 1.3 ± 0.1 μM to 2.7 ± 0.02 μM, 2.1-fold, p < 0.05) (Fig. S56†) suggesting that 1 NP5, like 1, induces COX-2-dependent CSC death. Collectively, the mechanistic studies shows that encapsulation of 1 by PEG-PLGA polymeric nanoparticles (1 NP5) does not alter its cellular properties, which augers well for further pre-clinical development.
Conclusions
In summary we report the synthesis, characterisation, anti-CSC properties, and encapsulation of a manganese(II)-3,4,7,8-tetramethyl-1,10-phenanthroline complex, 1 bearing a NSAID moiety, namely diclofenac. The manganese(II) complex, 1 is stable in biologically relevant solutions, including PBS with and without glutathione (a cellular reductant) and cell culture media, under physiological conditions. The manganese(II) complex, 1 displays equally toxicity against bulk breast cancer cells (HMLER) and breast CSCs (HMLER-shEcad), in the sub-micromolar range, suggesting that it has the potential to eliminate heterogeneous breast tumour populations (made up of bulk cancer cells and CSCs) with a single dose. The analogous copper(II) complex, 3 exhibited preferential potency towards bulk breast cancer cells (HMLER) over breast CSCs (HMLER-shEcad), implying that the metal (within this system) plays a determinant role in bulk cancer versus CSC toxicity. Extraordinarily, the manganese(II) complex, 1 inhibited HMLER-shEcad mammospheres formation and viability favourably over non-tumorigenic MCF10A spheroids (12-fold selectivity), indicating that 1 can potentially remove breast CSCs with reduced toxicity towards normal breast epithelial cells. In contrast, salinomycin, one of the leading anti-breast CSC agents identified thus far, killed HMLER-shEcad mammospheres and MCF10A spheroids equipotently. Detailed mechanistic studies showed that the cytotoxic mechanism of action of 1 involved intracellular ROS generation and COX-2 inhibition. The manganese(II) complex, 1 generated significantly higher levels of ROS inside CSCs than the analogous copper(II) complex, 3 proving that manganese(II)-phenanthroline complexes are, in general, better intracellular ROS generators than their copper(II) counterparts.In an attempt to improve CSC delivery, the manganese(II) complex, 1 was encapsulated into PEG-PLGA nanoparticles. The optimal loading conditions were obtained when the feed was set at 5%, 1 NP5. This yielded predominantly spherical nanoparticles with a diameter of ca. 100 nm. Importantly, 1 NP5 was stable and able to release the payload, 1 in biologically relevant solutions over the course of 72 h. The nanoparticle formulation delivered 15-fold higher levels of manganese into CSCs (via an endocytotic pathway) than the free payload. Encapsulation of 1 into PEG-PLGA nanoparticles significantly reduced overall toxicity towards bulk breast cancer cells (HMLER) and breast CSCs (HMLER-shEcad). The nanoparticle formulation retained the ability of the payload to indiscriminately kill bulk breast cancer cells and breast CSCs and thus its potential to remove whole breast cancer cell populations (including bulk cancer cells and CSCs) with a single dose. Mechanistic studies proved that 1 NP5 exhibited a similar mechanism of action as 1 – CSC death by intracellular ROS elevation and COX-2 inhibition. This is a highly desirable trait, as one of the major drawbacks associated to nanoparticle encapsulation as a strategy for drug delivery is the potential discrepancy in cellular mechanism of action of the nanoparticle formulation and its payload.
Overall this study highlights the expanding potential of redox modulating metal complexes as anti-CSC agents and opens the door for the development of other manganese complexes as CSC-potent agents. Furthermore, in light of the findings reported in this manuscript, the anti-CSC potential of ROS-generating manganese complexes previously reported in the literature should be determined as they could provide promising anti-CSC leads. As well as presenting the first manganese complex to show therapeutically relevant CSC potency, we demonstrate that polymeric nanoparticles can be used to effectively deliver manganese complexes into CSCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. thanks the Leverhulme Trust (ECF-2014-178) for funding to start this project. A. E. is supported by a King's College London Faculty Graduate School International Studentship. We thank Prof. Robert Weinberg for providing the HMLER and HMLER-shEcad cell lines used in this study.Notes and references
- L. V. Nguyen, R. Vanner, P. Dirks and C. J. Eaves, Nat. Rev. Cancer, 2012, 12, 133–143 CrossRef CAS
.
- V. Plaks, N. Kong and Z. Werb, Cell Stem Cell, 2015, 16, 225–238 CrossRef CAS
- M. Dean, T. Fojo and S. Bates, Nat. Rev. Cancer, 2005, 5, 275–284 CrossRef CAS
- L. N. Abdullah and E. K. Chow, Clin. Transl. Med., 2013, 2, 3 CrossRef
- K. Rycaj and D. G. Tang, Int. J. Radiat. Biol., 2014, 90, 615–621 CrossRef CAS
- J. Marx, Science, 2007, 317, 1029–1031 CrossRef CAS
- D. R. Pattabiraman and R. A. Weinberg, Nat. Rev. Drug Discovery, 2014, 13, 497–512 CrossRef CAS
- J. Kaiser, Science, 2015, 347, 226–229 CrossRef CAS
- X. Ning, J. Shu, Y. Du, Q. Ben and Z. Li, Cancer Biol. Ther., 2013, 14, 295–303 CrossRef CAS
- K. Laws and K. Suntharalingam, ChemBioChem, 2018, 19, 2246–2253 CrossRef CAS
- M. Gonzalez-Bartulos, C. Aceves-Luquero, J. Qualai, O. Cusso, M. A. Martinez, S. Fernandez de Mattos, J. A. Menendez, P. Villalonga, M. Costas, X. Ribas and A. Massaguer, PLoS One, 2015, 10, e0137800 CrossRef
- J. N. Boodram, I. J. McGregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Angew. Chem., Int. Ed., 2016, 55, 2845–2850 CrossRef CAS
- P. Liu, S. Brown, T. Goktug, P. Channathodiyil, V. Kannappan, J. P. Hugnot, P. O. Guichet, X. Bian, A. L. Armesilla, J. L. Darling and W. Wang, Br. J. Cancer, 2012, 107, 1488–1497 CrossRef CAS PubMed
- C. Lu, A. Eskandari, P. B. Cressey and K. Suntharalingam, Chem.–Eur. J., 2017, 23, 11366–11374 CrossRef CAS
- C. Lu, K. Laws, A. Eskandari and K. Suntharalingam, Dalton Trans., 2017, 46, 12785–12789 RSC
- M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman and M. F. Clarke, Nature, 2009, 458, 780–783 CrossRef CAS
- L. Y. Pang, E. A. Hurst and D. J. Argyle, Stem Cells Int., 2016, 2016, 11 Search PubMed
- J. A. Mitchell, P. Akarasereenont, C. Thiemermann, R. J. Flower and J. R. Vane, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11693–11697 CrossRef CAS PubMed
- P. Pantziarka, V. Sukhatme, G. Bouche, L. Meheus and V. P. Sukhatme, Ecancermedicalscience, 2016, 10, 610 Search PubMed
- L. Li and X. Yang, Oxid. Med. Cell. Longevity, 2018, 2018, 1–11 Search PubMed
- K. I. Ansari, J. D. Grant, S. Kasiri, G. Woldemariam, B. Shrestha and S. S. Mandal, J. Inorg. Biochem., 2009, 103, 818–826 CrossRef CAS
- K. I. Ansari, S. Kasiri, J. D. Grant and S. S. Mandal, Dalton Trans., 2009, 8525–8531 RSC
- Z. Zhang, R. He, K. Yan, Q. N. Guo, Y. G. Lu, X. X. Wang, H. Lei and Z. Y. Li, Bioorg. Med. Chem. Lett., 2009, 19, 6675–6678 CrossRef CAS PubMed
- X. Chen, L. J. Tang, Y. N. Sun, P. H. Qiu and G. Liang, J. Inorg. Biochem., 2010, 104, 379–384 CrossRef CAS
- M. Prudhomme, Anti-Cancer Agents Med. Chem., 2013, 2 Search PubMed
- C. Slator, Z. Molphy, V. McKee and A. Kellett, Redox Biol., 2017, 12, 150–161 CrossRef CAS
- A. Kellett, M. O'Connor, M. McCann, O. Howe, A. Casey, P. McCarron, K. Kavanagh, M. McNamara, S. Kennedy, D. D. May, P. S. Skell, D. O'Shea and M. Devereux, MedChemComm, 2011, 2, 579 RSC
- J. I. A. Liu, W. Guo, J. Li, X. Li, J. I. Geng, Q. Chen and J. Gao, Int. J. Mol. Med., 2015, 35, 607–616 CrossRef CAS PubMed
- R. Farghadani, J. Rajarajeswaran, N. B. Mohd Hashim, M. A. Abdulla and S. Muniandy, RSC Adv., 2017, 7, 24387–24398 RSC
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS
- N. P. Barry and P. J. Sadler, ACS Nano, 2013, 7, 5654–5659 CrossRef CAS
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CrossRef CAS
- J. W. Nichols and Y. H. Bae, J. Controlled Release, 2014, 190, 451–464 CrossRef CAS
- F. Danhier, J. Controlled Release, 2016, 244, 108–121 CrossRef CAS
- A. Z. Wang, Sci. Transl. Med., 2015, 7, 294ec112 CrossRef
-
F. Alexis, E. M. Pridgen, R. Langer and O. C. Farokhzad, Handbook of experimental pharmacology, 2010, pp. 55–86 Search PubMed
- R. Wang, P. S. Billone and W. M. Mullett, J. Nanomater., 2013, 2013, 12 Search PubMed
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS
- A. Eskandari, J. N. Boodram, P. B. Cressey, C. Lu, P. M. Bruno, M. T. Hemann and K. Suntharalingam, Dalton Trans., 2016, 45, 17867–17873 RSC
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS
- D. Martinez, M. Motevalli and M. Watkinson, Dalton Trans., 2010, 39, 446–455 RSC
- G. Dontu, W. M. Abdallah, J. M. Foley, K. W. Jackson, M. F. Clarke, M. J. Kawamura and M. S. Wicha, Genes Dev., 2003, 17, 1253–1270 CrossRef CAS
- I. Morita, M. Schindler, M. K. Regier, J. C. Otto, T. Hori, D. L. DeWitt and W. L. Smith, J. Biol. Chem., 1995, 270, 10902–10908 CrossRef CAS
- J. A. McCubrey, M. M. Lahair and R. A. Franklin, Antioxid. Redox Signaling, 2006, 8, 1775–1789 CrossRef CAS
- M. R. Junttila, S. P. Li and J. Westermarck, FASEB J., 2008, 22, 954–965 CrossRef CAS
- B. Singh, K. R. Cook, L. Vincent, C. S. Hall, C. Martin and A. Lucci, J. Surg. Res., 2011, 168, e39–e49 CrossRef CAS
- C. Bocca, M. Ievolella, R. Autelli, M. Motta, L. Mosso, B. Torchio, F. Bozzo, S. Cannito, C. Paternostro, S. Colombatto, M. Parola and A. Miglietta, Expert Opin. Ther. Targets, 2014, 18, 121–135 CrossRef CAS PubMed
- E. Locatelli and M. Comes Franchini, J. Nanopart. Res., 2012, 14, 1–17 CrossRef
- M. A. Miller, Y. R. Zheng, S. Gadde, C. Pfirschke, H. Zope, C. Engblom, R. H. Kohler, Y. Iwamoto, K. S. Yang, B. Askevold, N. Kolishetti, M. Pittet, S. J. Lippard, O. C. Farokhzad and R. Weissleder, Nat. Commun., 2015, 6, 8692 CrossRef CAS
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01275c |
| This journal is © The Royal Society of Chemistry 2019 |