
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rhodium catalyzed cycloisomerization and tandem Diels–Alder reaction for facile access to diverse bicyclic and tricyclic heterocycles†
Yirong
Zhou
ab,
Ali
Nikbakht
a,
Felix
Bauer
a and
Bernhard
Breit
 *a
*a
aInstitut für Organische Chemie und Biochemie, Albert-Ludwigs-Universität, Alberstr. 21, 79104 Freiburg, Germany. E-mail: bernhard.breit@chemie.uni-freiburg.de
bKey Laboratory of Functional Small Organic Molecules, Ministry of Education, College of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang, 330022, China
First published on 3rd April 2019
Abstract
A regioselective distal cycloisomerization of 1,6-allenenes was successfully developed to afford six-membered ring exocyclic 1,3-dienes employing a rhodium/diphosphine catalyst system. Deuterium labelling experiments and DFT calculations were performed to provide insights into the reaction mechanism of this unprecedented transformation. In addition, one-pot tandem Diels–Alder reactions with various dienophiles could readily construct diverse bicyclic and tricyclic nitrogen heterocycles, which are ubiquitous core scaffolds for a variety of natural products and bioactives. High efficiency and exclusive chemo and regioselectivities for a broad substrate scope were achieved under mild conditions using a low catalyst loading of 0.5 mol%.
Transition metal catalyzed cycloisomerization reactions have proved to be synthetically useful and elegant methods to construct structurally diverse all carbon and heterocyclic frameworks with high efficiency and an excellent atom and step economy.1 In particular, cycloisomerization reactions of linear di-unsaturated systems with a suitable linker chain, such as 1,6-diynes,2 1,6-enynes,3 allenynes,4 bisallenes,5 and allenedienes,6 have been intensely investigated. Furthermore, readily available 1,6-allenenes have attracted extensive synthetic interest and their cycloisomerization has been successfully realized to access diverse heterocycles by employing various transition metal catalysts, including nickel,7 palladium,8 ruthenium,9 rhodium,10 gold,11etc. To the best of our knowledge, all the previously reported examples involved a proximal allene π-bond activation and produced five-membered ring dienes as major products (Scheme 1a). In view of diversity-oriented synthesis,12 it is therefore highly desirable to develop a new versatile catalytic system to furnish alternative cycloisomerization products which enable subsequent multiple functionalizations.
 | ||
| Scheme 1 Transition metal catalyzed cycloisomerizations of allenenes. | ||
Recently, our group has made significant progress on the development of rhodium catalyzed atom-efficient addition reactions of various pronucleophiles to unactivated allenes and alkynes to provide enantioenriched branched allylic products.13 To further expand the synthetic potential, we envisioned that 1,6-allenenes would be interesting substrates for cycloisomerization to construct useful cyclic 1,3-dienes by using our rhodium/diphosphine catalytic system. Herein, we present an unprecedented rhodium catalyzed regioselective distal cycloisomerization of 1,6-allenenes to provide six-membered ring exocyclic 1,3-dienes exclusively (Scheme 1b). Based on deuterium labelling experiments and DFT calculations a plausible reaction mechanism could be elucidated. Moreover, a one-pot tandem Diels–Alder reaction with various dienophiles furnished diverse bicyclic and tricyclic nitrogen heterocycles with a high atom- and step-economy.14 Such fused bicyclic and tricyclic nitrogen containing heterocycles constitute privileged core skeletons of a variety of natural products and drugs as well as fluorescent probes (Fig. 1), which highlights the manifold potential applications of this new methodology in medicinal and materials chemistry.15
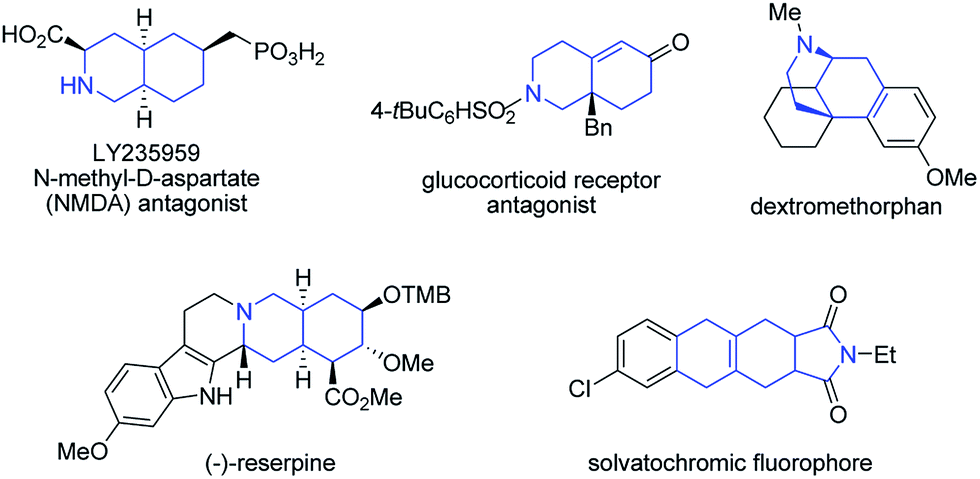 | ||
| Fig. 1 Selected examples of drugs and natural products containing bicyclic or tricyclic nitrogen heterocycles as core structures. | ||
To test our hypothesis, unactivated terminal 1,6-allenene 1a was chosen as a privileged model substrate for initial reactivity assays. Surprisingly, in contrast to the results reported in the literature, no proximal coupled five-membered ring 1,3-diene product (as shown in Scheme 1a) was detected. Conversely, a completely new kind of six-membered ring exocyclic 1,3-diene was obtained as the sole product with exclusive regioselectivity and in good yield. The molecular structure of product 2a was unambiguously confirmed by X-ray crystallography analysis. This unexpected preliminary result induced us to systematically optimize the reaction conditions (Table 1). First, several solvents were examined for the new cycloisomerization reaction (Table 1, entries 1–3) with DCE (1,2-dichloroethane) proving to be superior to THF (tetrahydrofuran) and toluene. Further investigations on the reaction temperature revealed that 60 °C was the most suitable temperature for this new transformation. The yield decreased with either higher or lower temperatures (Table 1, entry 5). Finally, the best result (80% yield) was obtained employing a lower substrate concentration of 0.1 M (Table 1, entry 6).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Temp./°C | Yieldb [%] |
| a The reactions were carried out on a 0.2 mmol scale of 1a in the presence of 2.5 mol% of [Rh(COD)Cl]2 and 5.0 mol% of DPEphos in DCE (1.0 mL) at different temperatures for 20 h. b Isolated yield. c 2.0 mL DCE was used (0.1 M). | |||
| 1 | DCE | 80 | 65 |
| 2 | THF | 80 | 58 |
| 3 | Toluene | 80 | 55 |
| 4 | DCE | 95 | 51 |
| 5 | DCE | 60 | 75 |
| 6c | DCE | 60 | 80 |
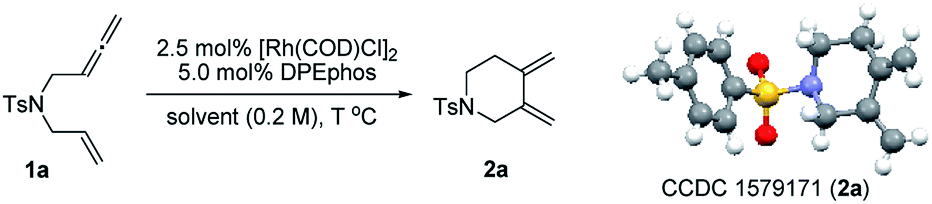
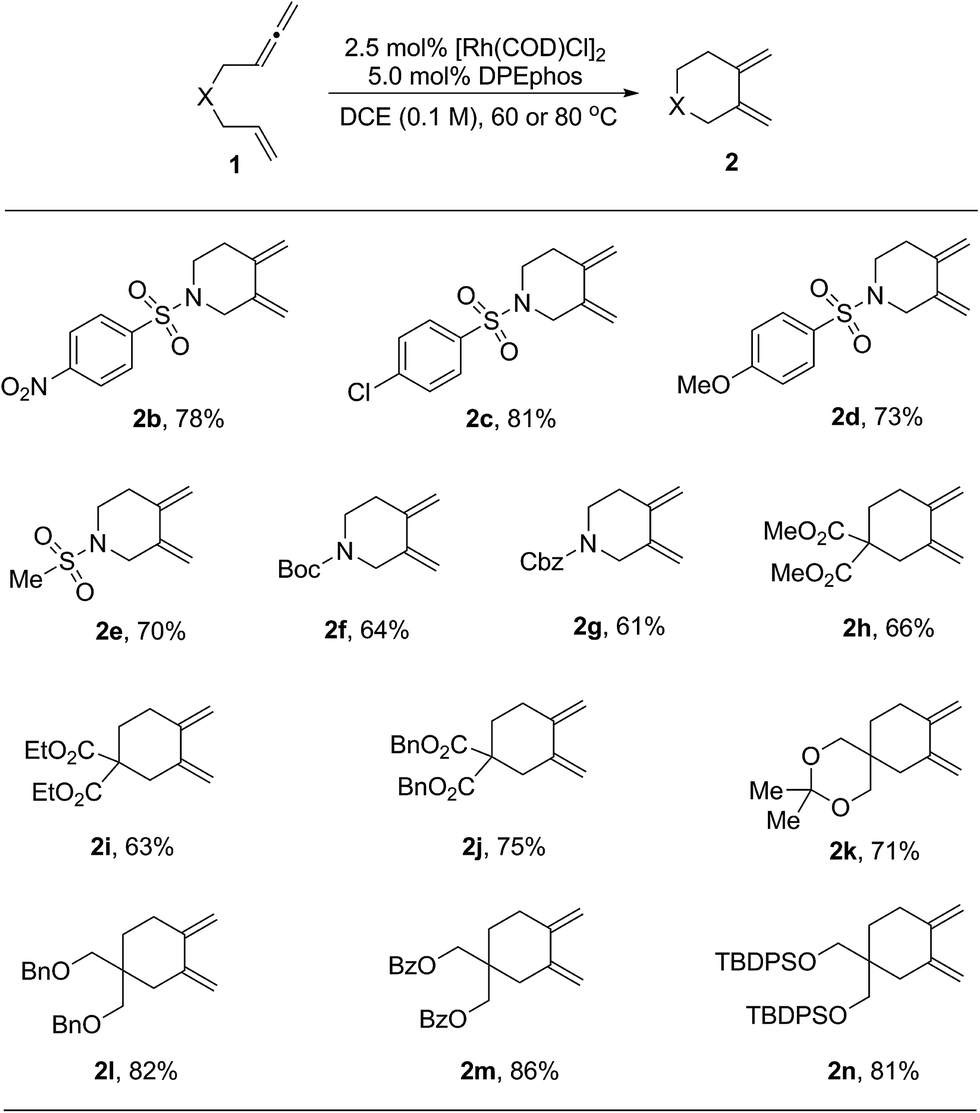
With the optimized reaction conditions in hand, the substrate scope was explored. The results are shown in Table 2. First, substrates having different protecting groups at the nitrogen linker atom were investigated (2a to 2g). Thus, in addition to the sulfonyl groups, easily removable Boc and Cbz carbamates were well tolerated. Second, a variety of allenenes with an all-carbon linker were examined (2h to 2n). In these cases, a slight increase of the reaction temperature to 80 °C was necessary in order to complete the cycloisomerization process. Different ester functions, such as methyl, ethyl and benzyl, were all compatible in this reaction (2h to 2j). Moreover, a group of masked hydroxyl functions were also suitable for the transformation (2l to 2n). It is noteworthy that an interesting diene product 2k with a spiro ketal structure was obtained in good yield.
| a The reactions were carried out on a 0.2 mmol scale of 1 in the presence of 2.5 mol% of [Rh(COD)Cl]2 and 5.0 mol% of DPEphos in DCE (2.0 mL) for 20 h. For products 2b to 2g, the reactions were performed at 60 °C. For products 2h to 2n, the reactions were performed at 80 °C. All the yields were isolated yields. |
|---|
|
To gain deeper insights into the reaction mechanism of this new cycloisomerization reaction, a series of deuterium labelled substrates were prepared and subjected to standard reaction conditions (Scheme 2). The results with substrates 1a-1 and 1a-2 indicated that the deuterium atoms at the terminal positions of alkene and allene completely remained in their original position. Hence, these carbon–hydrogen bonds should not change during the process (eqn (1) and (2)). However, the deuterium atom at the internal position of the alkene in 1a-3 completely shifted to the 5 position of the piperidine ring (2a-3), which indicated that an isomerization step might be involved to form the exocyclic carbon–carbon double bonds (eqn (3)).
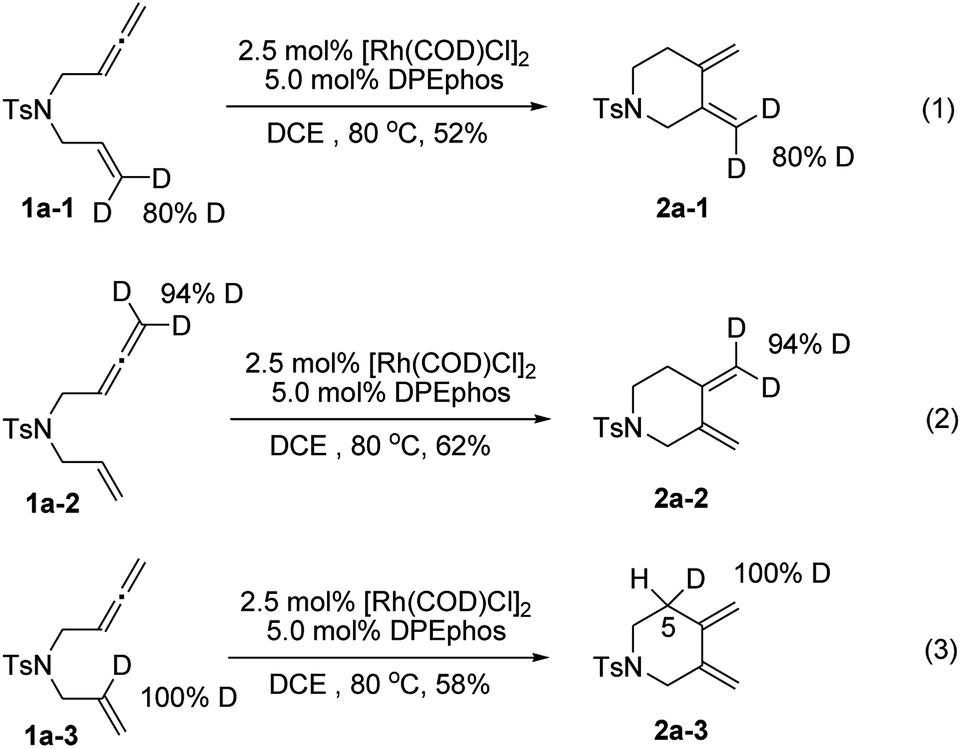 | ||
| Scheme 2 Deuterium labelling experiments. | ||
To shed further light on the mechanism of this new cycloisomerization reaction, DFT computations were performed.16 Based on the labeling experiments and the DFT calculations the mechanism depicted in Scheme 3 is proposed. Starting from a monomeric [ClRh(DPEphos)] complex ([Rh]) the substrate 1a could coordinate to form the chelate intermediate I1. An oxidative coupling viaTS1 furnishes the σ-allyl intermediate I2. This is followed by a β-hydride elimination (TS2) to give the σ-allyl hydride complex I3. Isomerization to the π-allyl intermediate I4 (viaTS3) and reductive elimination (viaTS4) deliver product 2a and regenerate the rhodium catalyst.
 | ||
| Scheme 3 Proposed mechanism for the rhodium-catalyzed cycloisomerization of 1,6-allenenes to six-membered exocyclic 1,3-dienes. | ||
Fig. 2 displays the energy profile of the reaction. The highest energetic barriers are the oxidative coupling to form a five-membered metallacycle (TS1) with a ΔG of 14.6 kcal mol−1 (M06/def2SVP) and the reductive elimination step (TS4) with a ΔG of 14.5 kcal mol−1 (M06/def2SVP). The calculated reaction mechanism is in accord with the results of the deuterium labelling experiments. Furthermore, calculations of the complete catalytic cycle for the traditional 5-membered ring cycloisomerization were performed. However, the energy barrier of the rate determining step was found to be significantly higher and is therefore unfavored.16
 | ||
| Fig. 2 Computed catalytic cycle of the Rh catalyzed cycloisomerization of 1,6-allenenes to six-membered exocyclic 1,3-dienes. All energies (PCM-M06/def2SVP//BP86/def2SVP) are given with respect to the energies of the [ClRh(DPEphos)] complex and the substrate. | ||
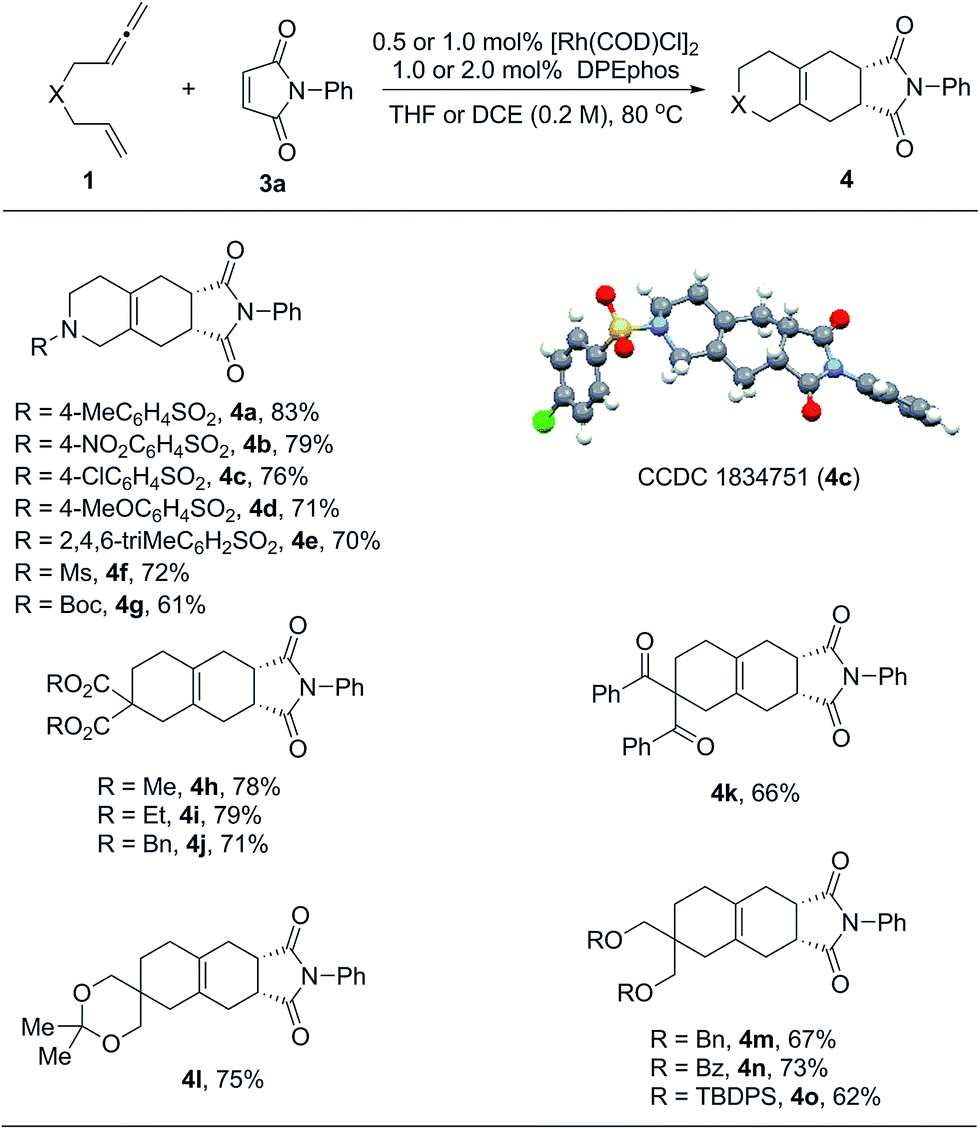
Considering that exocyclic 1,3-dienes 2 are potentially good reaction partners in Diels–Alder reactions, we anticipated that in the presence of suitable dienophiles, a one-pot tandem cycloisomerization/Diels–Alder reaction could be developed. This could become an efficient synthetic method to prepare diverse bicyclic and tricyclic nitrogen heterocycles.16 Indeed, as summarized in Table 3, we found that a wide range of 1,6-allenenes reacted smoothly in the presence of the rhodium catalyst and N-phenyl maleimide to furnish the desired tricyclic heterocycles 4 in good yields along with exclusive regio- and diastereoselectivities. The constitution and relative configuration of 4c were determined by X-ray crystallography analysis, while the others were assigned by analogy. For the protected amide allenenes (4a–4g), as low as 0.5 mol% of the rhodium catalyst was sufficient to achieve full conversion. Comparable yields showed that the protecting groups on the nitrogen atom exhibited negligible influences on the reaction. For carbon-linked allenene substrates (4h–4o) a slightly increased catalyst loading of 1 mol% was needed to allow for smooth and complete transformation. Various functional groups, such as ester, ketone, ketal and ethers, were all well tolerated.
| a The reactions were carried out on a 0.2 mmol scale of 1 with 1.0 equivalent of 3a at 80 °C for 20 h. For products 4b to 4g, the reactions were performed in THF (1.0 mL) using 0.5 mol% of [Rh(COD)Cl]2 and 1.0 mol% of DPEphos. For products 4h to 4o, the reactions were performed in DCE (1.0 mL) using 1.0 mol% of [Rh(COD)Cl]2 and 2.0 mol% of DPEphos. All the yields were isolated yields. |
|---|
|
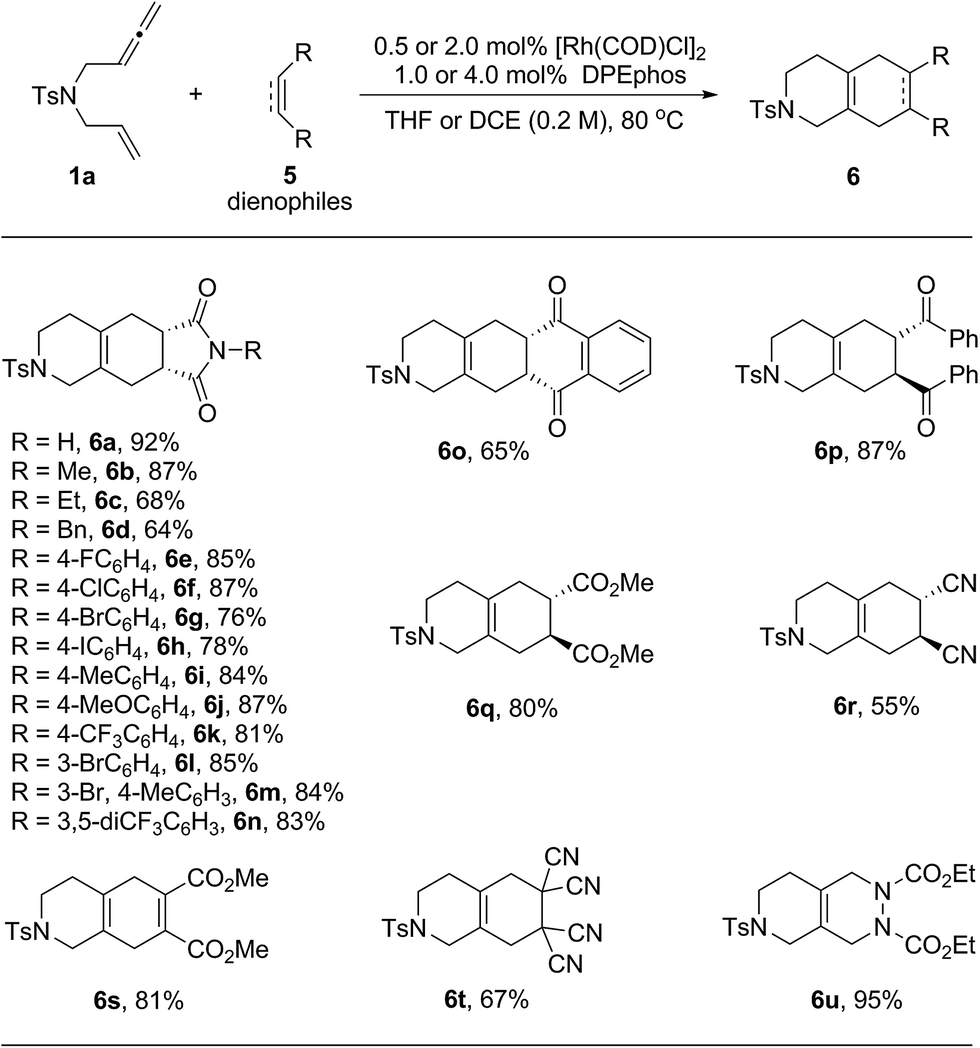
Next, the scope of dienophiles for the rhodium catalyzed domino cycloisomerization/Diels–Alder reaction was evaluated. As illustrated in Table 4, a wide range of symmetrical dienophiles proved suitable, providing the desired tricyclic and bicyclic heterocycles in good to high yields.
| a The reactions were carried out on a 0.2 mmol scale of 1a with 1.0 equivalent of 5 in the presence of 0.5 mol% of [Rh(COD)Cl]2 and 1.0 mol% of DPEphos in THF (1.0 mL) at 80 °C for 20 h. For products 6s to 6u, the reactions were performed in DCE (1.0 mL) using 2.0 mol% of [Rh(COD)Cl]2 and 4.0 mol% of DPEphos and the dienophiles were added after the first cycloisomerization step was completed. All the yields were isolated yields. |
|---|
|
Thus, a series of N-aryl maleimides with either electron poor or electron rich aryl substituents behaved well and provided the tandem products in good to high yields (6a–6n). A variety of aryl halides including F, Cl, Br and even I were well tolerated enabling subsequent derivatization through diverse cross-coupling methods (6e–6h). Other well behaving dienophiles were benzoquinone (6o), the diphenyl ketone derived from fumaric acid (6p), dimethyl fumarate (6q), trans-dicyano ethylene (6r), acetylene dicarboxylate (6s), tetra-cyano ethylene (6f) and azo dicarboxylate (6u).
The obtained fused tricyclic heterocyclic products contain an internal tetra-substituted alkene function, which permits a variety of functionalization reactions. Towards this goal the tricyclic products 4a and 4h were selected for preliminary studies (Scheme 4).
 | ||
| Scheme 4 Diverse synthetic transformations of 4a and 4h. | ||
First, the palladium catalyzed diastereoselective hydrogenation delivered the saturated azatricyclic 7 as a single diastereomer in high yield. By treatment with a suitable bromination reagent (PyH*Br3) in DCM, the dibrominated product 8 was obtained as a mixture of diastereomers. Epoxidation with m-CPBA furnished the epoxide 9 as a single diastereomer. A dihydroxylation yielded the vicinal diols 10a and 10h as single diastereomers from 4a and 4h, respectively. These diols could be oxidatively cleaved to give the ten-membered diketones 11a and 11h. Alternatively, the tricyclic product 4h could be directly transformed into 11hvia ozonolysis. It is noteworthy that such medium-sized fused bicyclic structures are difficult to access by other methods, highlighting the significance of this new method for potential application in the synthesis of natural products and medicinal chemistry.
Conclusions
In conclusion, a novel regioselective cycloisomerization of 1,6-allenenes was successfully developed to generate six-membered ring exocyclic 1,3-dienes by using a rhodium/diphosphine catalyst system. Based on labelling experiments corroborated by DFT computations a plausible reaction mechanism could be suggested. Moreover, one-pot tandem Diels–Alder reactions with various dienophiles led to the efficient and rapid construction of diverse bicyclic and tricyclic nitrogen heterocycles. The new method displays a high efficiency, broad substrate scope, complete atom and step economy, low catalyst loading of 0.5 mol%, and excellent chemo-, regio-, and diastereoselectivity. Further studies on the application of this new method are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DFG. Y. Z. is grateful for the Sino-German (CSC-DAAD) Postdoc Scholarship and thanks the National Natural Science Foundation of China (no. 21602089) for support. We thank Dr D. Kratzert for the X-ray diffraction analysis, Dr M. Keller for NMR analysis, and Solvias, Umicore, BASF and Wacker for generous gifts of chemicals.Notes and references
- Selected recent reviews: (a) Y. Lee, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2018, 57, 5212–5226 CrossRef CAS PubMed; (b) D. Zhang, J. Liu, A. Córdova and W. Liao, ACS Catal., 2017, 7, 7051–7063 CrossRef CAS; (c) C. I. Stathakis, P. L. Gkizis and A. L. Zografos, Nat. Prod. Rep., 2016, 33, 1093–1117 RSC; (d) D. Zhang, Z. Zhang and M. Shi, Chem. Commun., 2012, 48, 10271–10279 RSC; (e) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884–4908 RSC; (f) I. D. G. Watson and F. Dean Toste, Chem. Sci., 2012, 3, 2899–2919 RSC; (g) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed; (h) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed.
- Selected recent reviews and examples: (a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2915 CrossRef CAS PubMed; (b) P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS; (c) Y. Shibata and K. Tanaka, Synthesis, 2012, 44, 323–350 CrossRef CAS; (d) H. Jang and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 7875–7880 CrossRef CAS PubMed; (e) K. Tanaka, Y. Otake and M. Hirano, Org. Lett., 2007, 9, 3953–3956 CrossRef CAS PubMed.
- Selected recent reviews: (a) Y. Hu, M. Bai, Y. Yang and Q. Zhou, Org. Chem. Front., 2017, 4, 2256–2275 RSC; (b) R. Dorel and A. M. Echavarren, J. Org. Chem., 2015, 80, 7321–7332 CrossRef CAS PubMed; (c) S. I. Lee and N. Chatani, Chem. Commun., 2009, 371–384 RSC; (d) V. Michelet, P. Y. Toullec and J. Genét, Angew. Chem., Int. Ed., 2008, 47, 4268–4315 CrossRef CAS PubMed; (e) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef CAS; (f) C. Bruneau, Angew. Chem., Int. Ed., 2005, 44, 2328–2334 CrossRef CAS PubMed; (g) C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813–834 CrossRef CAS PubMed.
- (a) Y. Oonishi, Y. Kitano and Y. Sato, Angew. Chem., Int. Ed., 2012, 51, 7305–7308 CrossRef CAS PubMed; (b) S. I. Lee, S. H. Sim, S. M. Kim, K. Kim and Y. K. Chung, J. Org. Chem., 2006, 71, 7120–7123 CrossRef CAS PubMed; (c) K. M. Brummond, H. Chen, P. Sill and L. You, J. Am. Chem. Soc., 2002, 124, 15186–15187 CrossRef CAS PubMed.
- (a) S. Kitagaki, M. Kajita, S. Narita and C. Mukai, Org. Lett., 2012, 14, 1366–1369 CrossRef CAS PubMed; (b) S. M. Kim, J. H. Park, Y. K. Kang and Y. K. Chung, Angew. Chem., Int. Ed., 2009, 48, 4532–4535 CrossRef CAS PubMed; (c) P. Lu and S. Ma, Org. Lett., 2007, 9, 2095–2097 CrossRef CAS PubMed; (d) X. Jiang, X. Cheng and S. Ma, Angew. Chem., Int. Ed., 2006, 45, 8009–8013 CrossRef CAS PubMed.
- (a) I. Alonso, H. Faustino, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2011, 50, 11496–11500 CrossRef CAS PubMed; (b) M. Gulías, A. Collado, B. Trillo, F. López, E. Oñate, M. A. Esteruelas and J. L. Mascareñas, J. Am. Chem. Soc., 2011, 133, 7660–7663 CrossRef PubMed; (c) P. Mauleón, R. M. Zeldin, A. Z. González and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6348–6349 CrossRef PubMed; (d) P. A. Wender, T. E. Jenkins and S. Suzuki, J. Am. Chem. Soc., 1995, 117, 1843–1844 CrossRef CAS.
- (a) N. N. Noucti and E. J. Alexanian, Angew. Chem., Int. Ed., 2015, 54, 5447–5450 CrossRef CAS PubMed; (b) B. M. Trost and J. M. Tour, J. Am. Chem. Soc., 1988, 110, 5231–5233 CrossRef CAS.
- (a) A. K. Å. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2010, 49, 4624–4627 CrossRef CAS PubMed; (b) B. Trillo, M. Gulías, F. López, L. Castedo and J. L. Mascareñas, Adv. Synth. Catal., 2006, 348, 2381–2384 CrossRef CAS.
- (a) T. Nada, Y. Yoneshige, Y. Ii, T. Matsumoto, H. Fujioka, S. Shuto and M. Arisawa, ACS Catal., 2016, 6, 3168–3171 CrossRef CAS; (b) S. Kang, B. Ko and D. Lee, Tetrahedron Lett., 2002, 43, 6693–6696 CrossRef CAS.
- (a) K. Sugikubo, F. Omachi, Y. Miyanaga, F. Inagaki, C. Matsumoto and C. Mukai, Angew. Chem., Int. Ed., 2013, 52, 11369–11372 CrossRef CAS PubMed; (b) K. M. Brummond, H. Chen, B. Mitasev and A. D. Casarez, Org. Lett., 2004, 6, 2161–2163 CrossRef CAS PubMed; (c) T. Makino and K. Itoh, J. Org. Chem., 2004, 69, 395–405 CrossRef CAS PubMed; (d) T. Makino and K. Itoh, Tetrahedron Lett., 2003, 44, 6335–6338 CrossRef CAS; (e) P. A. Wender, F. Glorius, C. O. Husfeld, E. Langkopf and J. A. Love, J. Am. Chem. Soc., 1999, 121, 5348–5349 CrossRef CAS.
- (a) P. Aillard, P. Retailleau, A. Voituriez and A. Marinetti, Chem.–Eur. J., 2015, 21, 11989–11993 CrossRef CAS PubMed; (b) H. Teller, M. Corbet, L. Mantilli, G. Gopakumar, R. Goddard, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2012, 134, 15331–15342 CrossRef CAS PubMed; (c) A. Z. González, D. Benitez, E. Tkatchouk, W. A. Goddard and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 5500–5507 CrossRef PubMed; (d) H. Teller, S. Flügge, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 1949–1953 CrossRef CAS PubMed; (e) M. R. Luzung, P. Mauleón and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12402–12403 CrossRef CAS PubMed; (f) M. A. Tarselli, A. R. Chianese, S. J. Lee and M. R. Gagné, Angew. Chem., Int. Ed., 2007, 46, 6670–6673 CrossRef CAS PubMed.
- Selected recent reviews: (a) S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed; (b) C. J. O' Connor, H. S. G. Beckmann and D. R. Spring, Chem. Soc. Rev., 2012, 41, 4444–4456 RSC; (c) J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629–14638 CrossRef CAS PubMed; (d) M. Garcia-Castro, S. Zimmermann, M. G. Sankar and K. Kumar, Angew. Chem., Int. Ed., 2016, 55, 7586–7605 CrossRef CAS PubMed.
- (a) P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524–1536 CrossRef CAS PubMed; (b) A. M. Haydl, B. Breit, T. Liang and M. J. Krische, Angew. Chem., Int. Ed., 2017, 56, 11312–11325 CrossRef CAS PubMed; (c) Y. Zhou and B. Breit, Chem.–Eur. J., 2017, 23, 18156–18160 CrossRef CAS PubMed; (d) Z. Liu and B. Breit, Org. Lett., 2018, 20, 300–303 CrossRef CAS PubMed; (e) D. Berthold and B. Breit, Org. Lett., 2018, 20, 598–601 CrossRef CAS PubMed; (f) C. Grugel and B. Breit, Org. Lett., 2018, 20, 1066–1069 CrossRef CAS PubMed; (g) P. Steib and B. Breit, Angew. Chem., Int. Ed., 2018, 57, 6572–6576 CrossRef CAS PubMed.
- Selected recent reviews: (a) J. Funel and S. Abele, Angew. Chem., Int. Ed., 2013, 52, 3822–3863 CrossRef CAS PubMed; (b) S. Kotha, M. Meshram and A. Tiwari, Chem. Soc. Rev., 2009, 38, 2065–2092 RSC; (c) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650–1667 CrossRef CASDuring the preparation of this manuscript a related study using a cycloisomerization of 1,6-enynes and tandem Diels–Alder reaction strategy appeared: H. Zheng, Y. Wang, C. Xu, Q. Xiong, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 5327–5331 Search PubMed.
- (a) P. A. Wender, M. S. Jeffreys and A. G. Raub, J. Am. Chem. Soc., 2015, 137, 9088–9093 CrossRef CAS PubMed; (b) W. E. Childers Jr and R. B. Baudy, J. Med. Chem., 2007, 50, 2557–2562 CrossRef PubMed; (c) R. D. Clark, N. C. Ray, P. Blaney, P. H. Crackett, C. Hurley, K. Williams, H. J. Dyke, D. E. Clark, P. M. Lockey, R. Devos, M. Wong, A. White and J. K. Belanoff, Bioorg. Med. Chem. Lett., 2007, 17, 5704–5708 CrossRef CAS PubMed; (d) F. Chen and J. Huang, Chem. Rev., 2005, 105, 4671–4706 CrossRef CAS PubMed.
- For detailed information, see the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1579171 (2a) and 1834751 (4c). Experimental procedures and detailed characterization data of all new compounds. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00980a |
| This journal is © The Royal Society of Chemistry 2019 |