
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of two stereocenters to one or two chiral axes: atroposelective synthesis of 2,3-diarylbenzoindoles†
Yu-Long
Hu‡
,
Zhe
Wang‡
,
Hui
Yang‡
,
Jie
Chen
 ,
Zi-Bo
Wu
,
Yibo
Lei
and
Ling
Zhou
*
,
Zi-Bo
Wu
,
Yibo
Lei
and
Ling
Zhou
*
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, Department of Chemistry & Materials Science, National Demonstration Center for Experimental Chemistry Education, Northwest University, Xi'an 710127, P. R. China. E-mail: zhoul@nwu.edu.cn
First published on 20th May 2019
Abstract
Central-to-axial chirality conversion provides efficient access to axially chiral compounds, and several examples regarding the conversion of one, two or four stereocenters to one axis have been reported. Herein, we report the conversion of two stereocenters to one or two chiral axes for the first time. In this study, a new class of enantiomerically enriched 2,3-diarylbenzoindoles was efficiently synthesized using a chiral phosphoric acid-catalyzed [3 + 2] formal cycloaddition and a mild DDQ oxidation strategy. Moreover, a speculative model of the central-to-axial chirality conversion outcome was proposed based on preliminary mechanistic studies and DFT calculations. Potentially, using this strategy, useful chiral phosphine ligand can be synthesized smoothly (99% ee).
Introduction
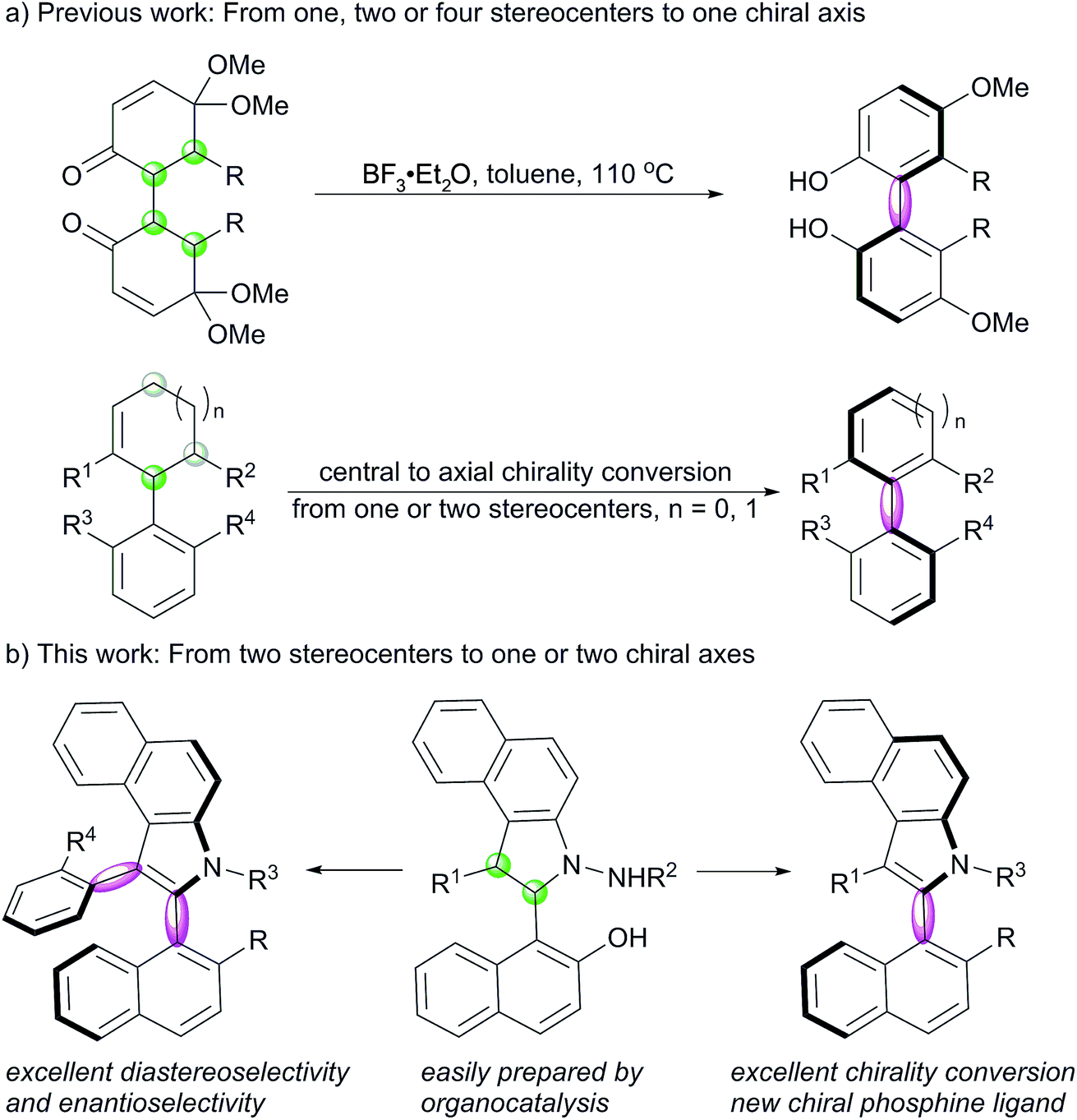
Axially chiral compounds are widely spread in natural products and bioactive molecules,1 which are recognized as a type of privileged scaffolds for catalyst development, ligand design, drug discovery and materials science.2,3 Many remarkable endeavors have been made to develop strategies, such as dynamic-kinetic resolution,5 atroposelective coupling,6 cycloaddition,7 chirality conversion,8 and so on,9 for the construction of axial chirality.4 Central-to-axial chirality conversion often provides access to axially chiral compounds that are difficult to obtain otherwise, which has been used in the asymmetric synthesis of allenes,10 biaryl atropisomers8 and natural products.11 However, although it is an attractive strategy, only limited studies have been reported on it ever since Berson proposed this concept in 1955.8,12 In 2011, Thomson reported BF3–Et2O-triggered aromatization of 1,4-diketones for the preparation of biphenols with excellent enantioselectivities.13a Recently, Rodriguez and Bugaut have successfully disclosed the atroposelective synthesis of 4-aryl pyridines and 3-arylfurans with excellent enantioselectivities by this strategy.13b,c Sparr developed a remote central to axial chirality conversion to prepare biaryl silanes.13d However, the previously reported examples focus on the construction of a single stereogenic axis, and no studies have been reported on the synthesis of five-six-membered N-heterocyclic atropisomers by this strategy. Moreover, the preparation of compounds with multiple configurational axes is a challenge in asymmetric synthetic chemistry,7b,9c,14 and to the best of our knowledge, the conversion of stereocenters to two axes is unprecedented.N-Heterocyclic atropisomers are of particular interest for chemists due to the presence of a nitrogen atom that would exert steric and electronic effects during their bonding to substrates or metals; this makes these atropisomers ideal candidates for application as catalysts or ligands.3,15 Compared to the well-documented preparation of six-six-membered biaryl atropisomers, the construction of five-six-membered species is still a challenge in modern asymmetric synthesis.16 This is due to the fact that the increased distance between ortho-substituents adjacent to the axis dramatically reduces the conformational stability; this makes racemization easy to take place. Indole-based biaryl atropisomers including N-aryl, C(2)-aryl, and C(3)-aryl axial chirality are potential ligands and found in some natural products.17 In 2010, Kitagawa reported Pd-catalyzed hydroaminocyclization to construct indole N-aryl axial chirality.18 Later, Kamikawa, Takahashi and Ogasawara reported an enantioselective desymmetrising ring-closing metathesis strategy to prepare axially chiral indole N-aryl compounds.19 Very recently, Shi and Tan independently reported a well-designed chiral phosphoric acid-catalyzed asymmetric synthesis of indol C(3)-aryl axial chirality;20 Gu reported Pd-catalyzed dynamic kinetic intramolecular C–H cyclization to construct indol C(3)-aryl atropisomers.21 However, no studies have been reported on the synthesis of indol C(2)-aryl axial chirality. On the other hand, nonchiral indol C(2)-aryl phosphine has been utilized as a unique ligand for Pd-catalyzed cross-coupling reaction of aryl mesylates.22 Herein, we report an efficient method for the construction of benzoindole-based C(2)-aryl and C(2,3)-aryls atropisomers via a [3 + 2] formal cycloaddition and an oxidative central-to-axial chirality conversion reaction (Scheme 1).
 | ||
| Scheme 1 Central-to-axial chirality conversion in the synthesis of atropisomers. | ||
Results and discussion
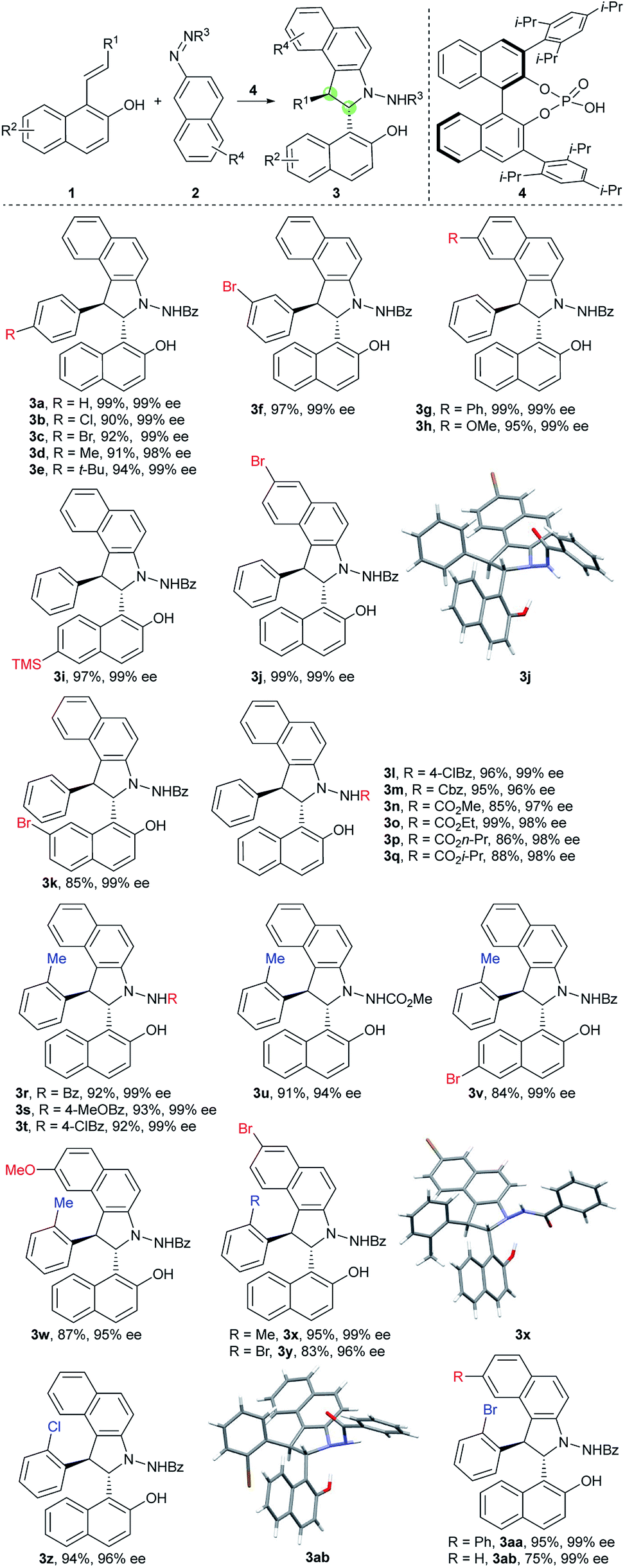
Inspired by the abovementioned elegant studies on central-to-axial chirality conversion and our previous study on the asymmetric [3 + 2] formal cycloaddition reaction,23 we envisioned that an enantioselective [3 + 2] annulation of 1-styrylnaphthols with azonaphthalenes followed by oxidative aromatization would generate benzoindole (C(2) position) naphthyl atropisomers. To achieve this goal, we needed to address the following issues: (1) accomplishment of a highly diastereoselective and enantioselective [3 + 2] annulation; (2) installation of suitable groups around the C–C axis to avoid free rotation; and (3) realization of efficient chirality conversion with enantioselective and diastereoselective control via the development of mild oxidation conditions.With these challenges in mind, we initially evaluated the reaction between 1-styryl naphthol (1a) and azonaphthalene (2a) in the presence of a chiral phosphoric acid. After systematic optimization of the reaction conditions,24 the desired cycloaddition product 3a was obtained in a 99% yield with exclusive diastereoselectivity and 99% ee using the chiral phosphoric acid 4 as a catalyst in CH2Cl2 at −30 °C for 36 h. We then examined the substrate scope of this [3 + 2] cycloannulation. As shown in Table 1, substrates with the substituents Cl, Br, Me and t-Bu at the para-position or Br at the meta-position of the phenyl ring resulted in the desired products with excellent yields and enantioselectivities (3a–f, 98–99% ee). Substrates with substituents at the 6 or 7-position of azonaphthalene were well tolerated, and the corresponding trans-2,3-diarylbenzoindolines were obtained in excellent yields and enantioselectivities (3g, h, and j 99%ee). Substrates with substituents at the 6 or 7-position of the naphthol led to excellent enantioselectivities, diastereoselectivities and yields (3i and k, 99% ee). In addition, the reaction was applicable to azonaphthalene derivatives with different esters including benzyl (3m), methyl (3n), ethyl (3o), n-propyl (3p) and i-propyl (3q) esters. Not surprisingly, substrates with the substituents Me, Cl and Br at the ortho-position of the phenyl ring provided the desired products with excellent yields, diastereoselectivities and enantioselectivities (3r–ab, 93 ≥ 99% ee). The exact structures of 3j, 3x and 3ab were also confirmed by X-ray crystallography, and the absolute configuration was identified as (2S, 3S)-3j, x, and ab.25
a Reactions were carried out with 1a (0.15 mmol), 2 (0.1 mmol), 4 (0.01 mmol) and 3 Å molecular sieve (100 mg) in CH2Cl2 (2.0 mL) at −30 °C for 36 h. All dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. :1.
|
|---|
|
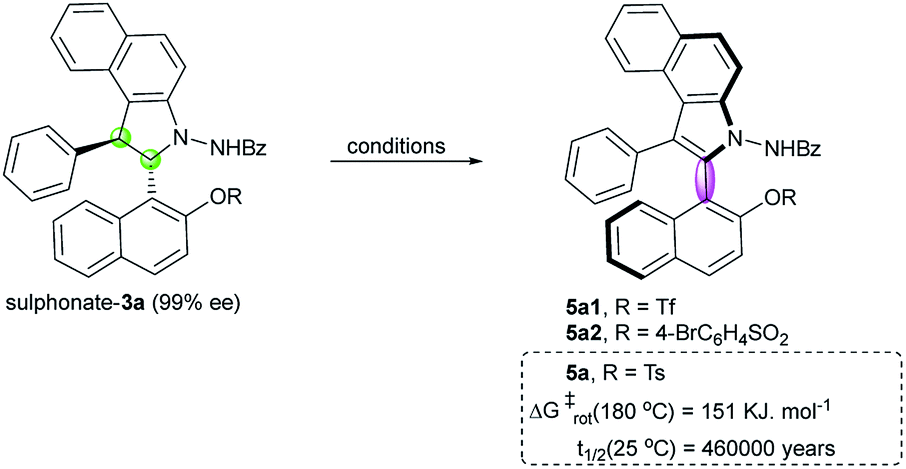
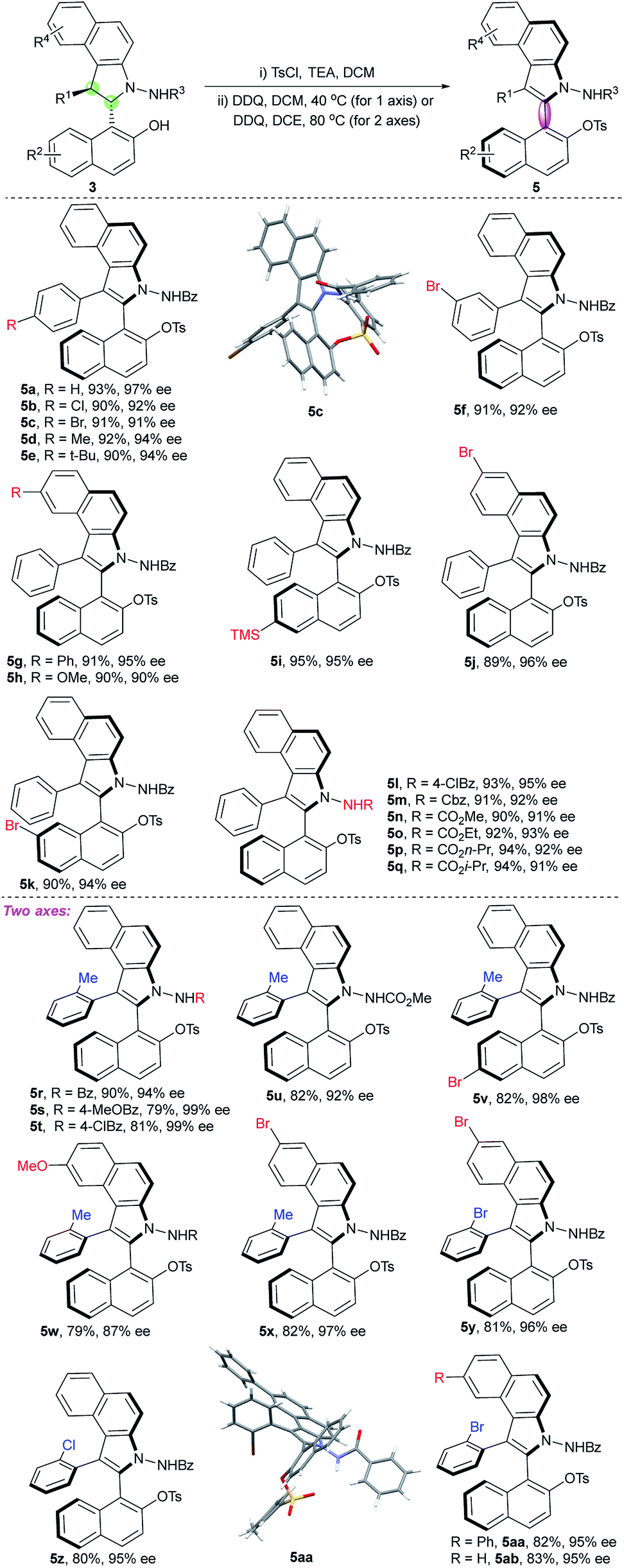
Then, we emphasized on the construction of an axially chiral biaryl system by the chirality conversion strategy. To avoid the dearomatization side reactions, sulphonate derivatives were prepared from compound 3a. Oxidation of triflate 3a using DDQ in CH2Cl2 at room temperature provided the desired atropisomer 5a1 in quantitative yield but with poor enantioselectivity (Table 1, entry 1). We proposed that a bulky substituent adjacent to the C–C axis may be helpful in chirality conversion and stabilizing the axial chirality. Thus, tosylate 3a was prepared; indeed, the corresponding atropisomer 5a was obtained with a significant increase in enantioselectivity (85% ee, entry 2) under the same reaction conditions. Satisfyingly, a measurable increase in enantioselectivity was observed by the slow addition of DDQ to CH2Cl2 (entry 3 vs. entry 2). Interestingly, the reaction temperature also plays an important role in this chirality conversion: higher reaction temperature leads to higher enantioselectivity (entries 3–5). The best result (97% ee) was obtained at 40 °C by the slow addition of DDQ (entry 6). Moreover, other solvents, such as DCE and toluene, were tested. DCE provided similar result at 60 °C (entry 7), whereas toluene led to only 77% ee (entry 8). Another oxidant i.e. MnO2 led to poor enantioselectivity (entry 9). In addition to the tosyl group, 4-bromobenzenesulfonyl is a suitable group for the construction of a stable atropisomer (entry 10). To investigate the configurational stability of these new atropisomers, thermal racemization was conducted.24 Compound 5a displays excellent stability with a barrier to rotation of 151 kJ mol−1, and the half-life time of racemization was determined to be 460000 years at 25 °C.
Once the optimized conditions were identified (Table 2, entry 6), other substrates were examined. As shown in Table 3, the substrates 3a–q were well tolerated in this central-to-axial chirality conversion process, and the corresponding five-six-membered atropisomers were obtained with excellent yields and enantioselectivities (5a–q). The absolute configuration of 5a–q was assigned as aR on the basis of the X-ray crystallographic structure of 5c.25 The substrates 3r–ab were also used to obtain vicinal biaryl benzoindole atropisomers bearing two stereogenic axes. Gratifyingly, the desired products 5r–x containing the ortho-methyl group were obtained with excellent diastereoselectivities (all > 20:1) and enantioselectivities (87–99% ee) under the newly developed optimal reaction conditions with higher reaction temperature in DCE (Table 3). Not only methyl substituent at the ortho-position of the phenyl ring, but also chloride and bromide were suitable groups for the reaction and produced axially chiral products (5y, 5z, 5aa, and 5ab) with excellent diastereoselectivities and enantioselectivities. The second chiral axis of compound 5x also displayed good stability with a barrier to rotation of 126.6 kJ mol−1, and the half-life time of racemization was determined to be 28 years at 25 °C.24 Note that all halogen-substituted substrates were suitable for the conversion, exhibiting highly impressive performance (5b, c, f, i, k, v, and x–ab), which are appropriate substrates for synthetic transformations through cross-coupling reactions. The absolute configuration of 5r–ab was assigned as (2aR, 3aS) on the basis of the X-ray crystallographic structure of 5aa.25
|
|
|||||
|---|---|---|---|---|---|
| Entry | 5 | Solvent | T (°C) | Yieldb (%) | ee (%) |
| a Reactions were carried out with sulphonate-3a (0.2 mmol) in CH2Cl2, DDQ (0.4 mmol in CH2Cl2, 0.01 M) was added over 1 h by a syringe pump. b Isolated yields. c DDQ was added in one batch. d DDQ (0.01 M) was added dropwise over 30 min. e MnO2 (1.0 mmol) was used. | |||||
| 1c | 5a1 | CH2Cl2 | r.t. | 98 | 7 |
| 2c | 5a | CH2Cl2 | r.t. | 98 | 85 |
| 3d | 5a | CH2Cl2 | r.t. | 98 | 90 |
| 4d | 5a | CH2Cl2 | 0 | 96 | 70 |
| 5d | 5a | CH2Cl2 | 40 | 97 | 94 |
| 6 | 5a | CH2Cl2 | 40 | 99 | 97 |
| 7 | 5a | DCE | 60 | 97 | 96 |
| 8 | 5a | Toluene | 60 | 97 | 77 |
| 9e | 5a | CH2Cl2 | r.t. | 95 | 45 |
| 10 | 5a2 | CH2Cl2 | 50 | 97 | 91 |
We next conducted DFT studies to gain insight into the reaction mechanism and the origin of the high levels of chirality conversion observed. Tosylation of 3a provided the compound Ts-3a as a 73:27 mixture of the conformational diastereoisomers (sp)-Ts-3a and (ap)-Ts-3a at 25 °C. There exists an equilibrium between two rotamers because different ratios of them can be observed by 1H NMR at various temperatures.24,26 The rotational barrier was determined to be 80.8 kJ mol−1via the 1D-EXSY experiment in C2D2Cl4 at 40 °C.24 Selective 1D NOE and 2D NOESY of Ts-3a have indicated that ap-Ts-3a is the minor conformer.24 Hence, the fundamental step of the process would be the deprotonation of the central chiral substrate (ap)-Ts-3a((sp)-Ts-3a), leading to the cationic intermediate IM-A (IM-B), wherein a central-to-axial chirality conversion simultaneously occurred. Upon in situ elimination of the other proton, the product 5a (ent-5a) was generated. All calculations were performed with Gaussian 09. The geometries of all intermediates and transition states were optimized at the B3LYP-D3/6-1G(d) level, and energies were calculated at the M06-2X-D3/6-311++G(d,p) level with the solvation effect.24 Indeed, the potential energy of (sp)-Ts-3a was found to be lower than that of (ap)-Ts-3a by 0.3 kcal mol−1; thus, DDQ first coordinated with (sp)-Ts-3a to form a charge-transfer complex (Fig. 1).24 The steric effect between DDQ and Ts group in (sp)-Ts-3a is observable (Fig. 2). The charge transfer in the hydride transfer transition states TS-A was favored by 5.3 kcal mol−1 over TS-B. Moreover, the potential energy of the intermediate IM-A was found to be clearly lower than that of the intermediate IM-B by 10.3 kcal mol−1. These results indicate that the hydride transfer is the rate-determining step of the overall transformation, in agreement with the studies reported in the literature.27
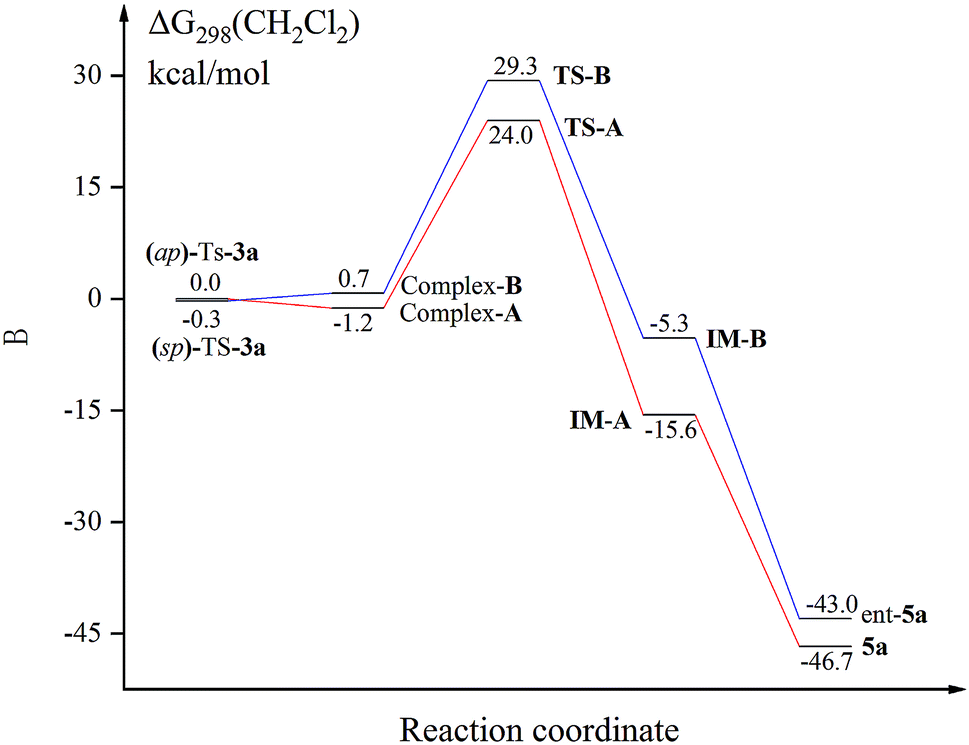 | ||
| Fig. 1 Energy profile of the DDQ-mediated central-to-axial chirality conversion. | ||
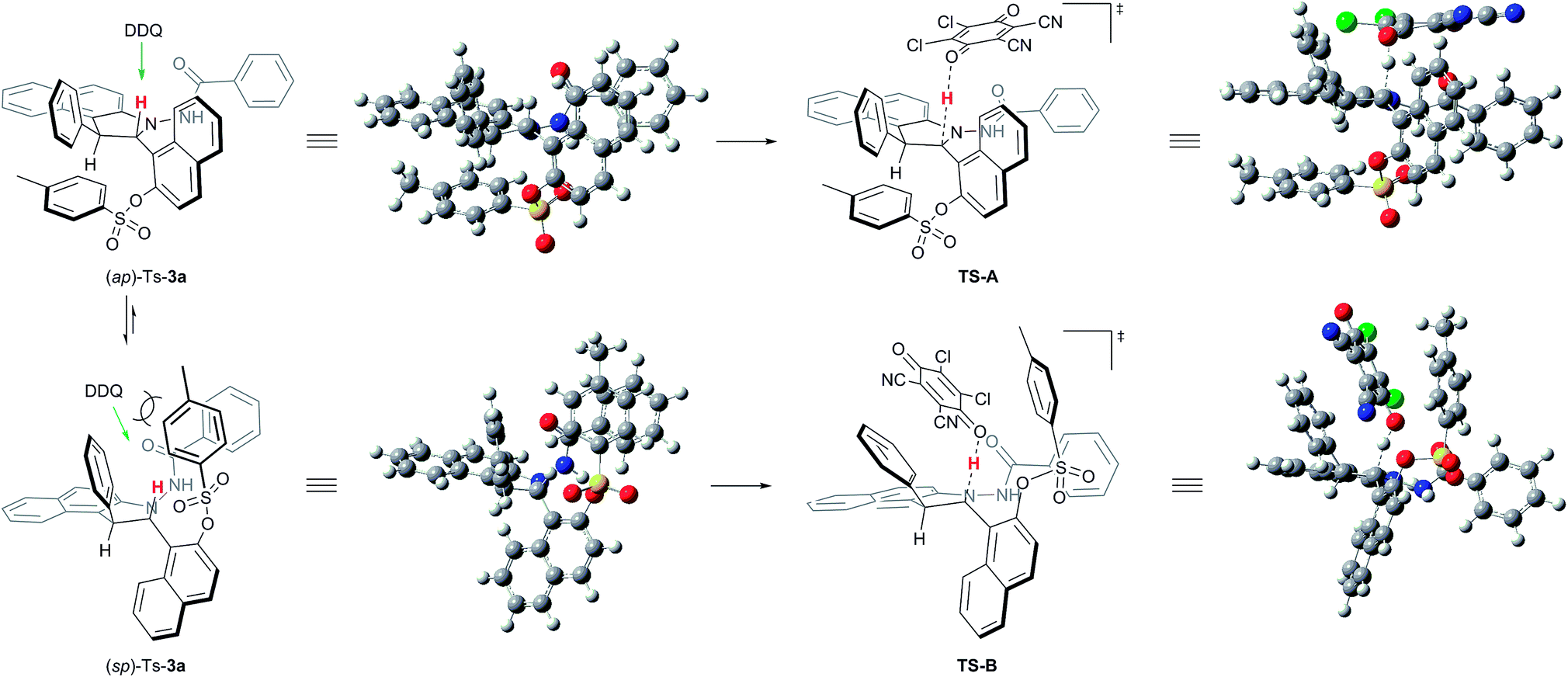 | ||
| Fig. 2 3D structures of the substrates and transition states. | ||
To demonstrate the utility of this method, a scale-up experiment was carried out. The desired product 5a (2 mmol scale) was successfully prepared without losing any efficiency and enantioselectivity. The replacement of the Ts group with the more reactive Tf group was achieved by conventional transformation, and then, Pd-catalyzed coupling of this modified compound with Ph2P(O)H provided a diphenyl phosphine oxide product (Scheme S2†).24 Unfortunately, a measurable decrease in the ee value (from 99% ee to 92% ee) has been observed when the Ts group was removed, suggesting that a bulky group in the naphthyl moiety also plays an important role in the configurational stability of this scaffold.
Based on the central-to-axial chirality conversion strategy, we designed a new synthetic route for the preparation of benzoindole-based C(2)-naphthyl phosphine ligands. As shown in Scheme 2, compound 7, bearing a bulky diphenyl phosphine oxide group, was prepared smoothly from 3a, and the enantioselectivity was retained (99% ee). Direct oxidation of 7 provided a nearly racemic benzoindolenaphthyl atropisomer (<5% ee); this indicated that the N-substituent was also crucial for this axial chirality. Thus, benzylation of 7 was conducted, and then, oxidation under our abovementioned optimized conditions resulted in the desired product 8 in a 90% yield with 99% ee. Finally, the reduction of 8 with HSiCl3 resulted in the formation of the new benzoindolenaphthyl phosphine ligand 9 (99% ee). The utility of this ligand was primarily demonstrated by an asymmetric allylation reaction between (E)-1,3-diphenylallyl acetate and dimethyl malonate. The desired product 11 was obtained in an 80% yield with 68% ee.
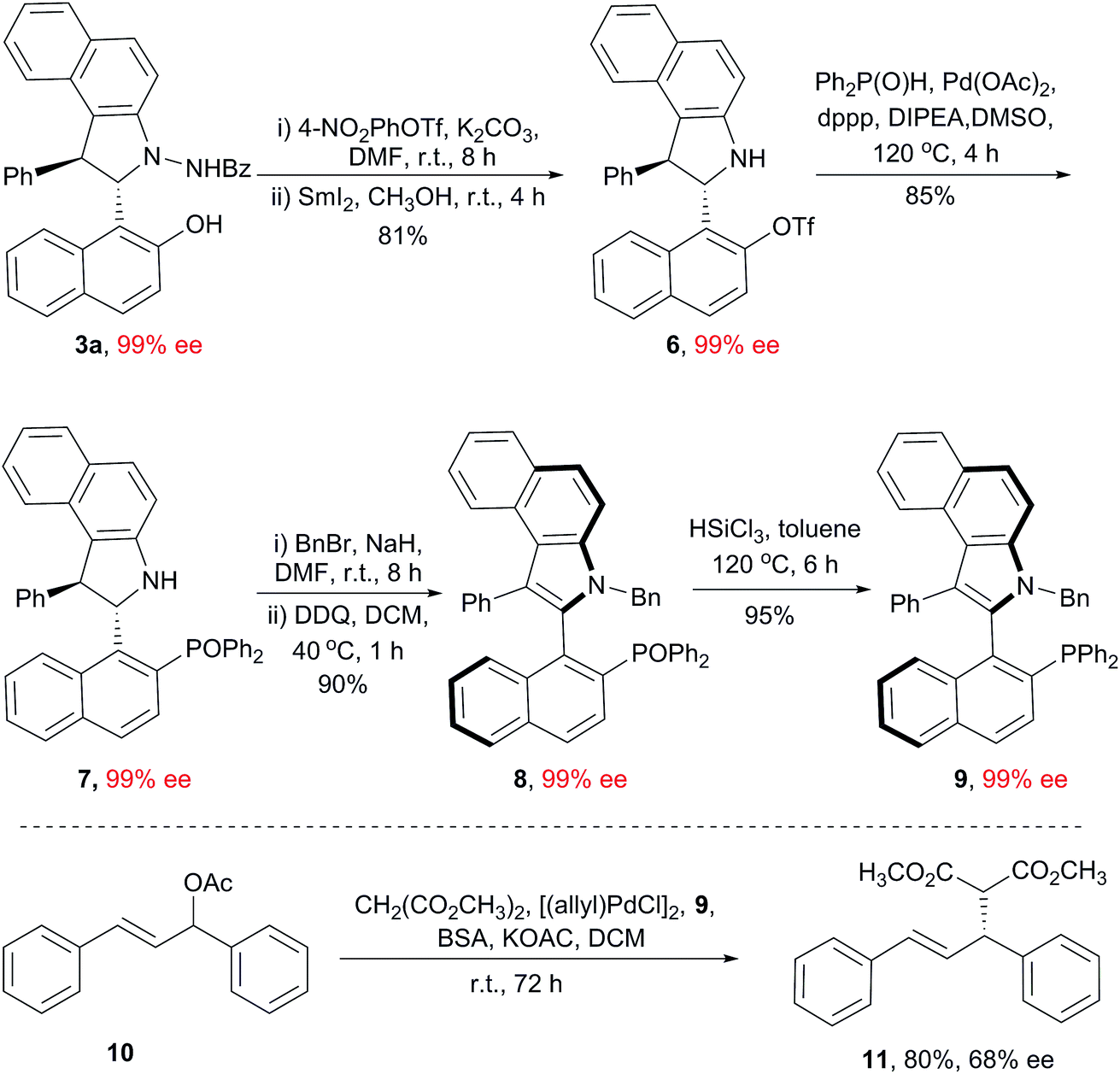 | ||
| Scheme 2 Synthesis and application of the phosphine ligand 9. | ||
Conclusions
In summary, we developed the first construction of newly 2,3-diarylbenzoindoles in high yields and enantioselectivities using an efficient stepwise strategy, including chiral phosphoric acid-catalyzed [3 + 2] formal cycloaddition of 1-styrylnaphthols with azonaphthalenes and a DDQ-oxidized central-to-axial chirality conversion reaction. This constitutes the first stereoselective conversion of two stereocenters to two axes. Preliminary, mechanistic studies and DFT calculations indicate that the hydride transfer is the rate-determining step, and the more stable conformational atropdiastereomer is prone to undergo oxidation to form the more stable iminium cation during the central-to-axial chirality conversion reaction. Moreover, new chiral phosphine compounds can be prepared efficiently by this strategy, which is attractive for the development of novel catalysts or ligands. However, further investigations on the development of this new chiral phosphine ligand and its application in some asymmetric reactions are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We warmly thank Prof. Yanxia Zhao and Guoping Yang for X-ray diffraction analysis and Prof. Wenhua Xu for the DFT calculations. The calculations were performed at chemical HPC center of NWU. We thank the National Natural Science Foundation of China (NSFC-21672170), Natural Science Basic Research Plan in Shaanxi Province of China (2018JC-020, 2018JM2029), China Postdoctoral Science Foundation (2018M643705), and the Key Science and Technology Innovation Team of Shaanxi Province (2017KCT-37) for providing financial support.Notes and references
- (a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC; (c) B. Chen, X. Liu, Y.-J. Hu, D.-M. Zhang, L. Deng, J. Lu, L. Min, W.-C. Ye and C.-C. Li, Chem. Sci., 2017, 8, 4961 RSC; (d) R. W. Hoffmann, H.-G. Schmalz, U. Koert and G. K. Pierens, Chem. Sci., 2019, 10, 943 RSC; (e) J. Hu, F. Sarrami, H. Li, G. Zhang, K. A. Stubbs, E. Lacey, S. G. Stewart, A. Karton, A. M. Piggott and Y.-H. Chooi, Chem. Sci., 2019, 10, 1457 RSC.
- (a) Y. Wang, A. Urbasand and Q. Li, J. Am. Chem. Soc., 2012, 134, 3342 CrossRef CAS PubMed; (b) Y.-N. Ma, S.-X. Liand and S.-D. Yang, Acc. Chem. Res., 2017, 50, 1480 CrossRef CAS PubMed; (c) P. W. Glunz, Bioorg. Med. Chem. Lett., 2018, 28, 53 CrossRef CAS PubMed.
- (a) W. Fuand and W. Tang, ACS Catal., 2016, 6, 4814 CrossRef; (b) P. Kočovský, Š. Vyskočiland and M. Smrčina, Chem. Rev., 2003, 103, 3213 CrossRef PubMed; (c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed.
- For reviews, see: (a) J. Wencel-Delord, A. Panossian, F. R. Lerouxand and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418 RSC; (b) G. Bencivenni, Synlett, 2015, 26, 1915 CrossRef CAS; (c) E. Kumarasamy, R. Raghunathan, M. P. Sibiand and J. Sivaguru, Chem. Rev., 2015, 115, 11239 CrossRef CAS PubMed; (d) D. Zhang and Q. Wang, Coord. Chem. Rev., 2015, 286, 1 CrossRef CAS; (e) P. Loxq, E. Manoury, R. Poli, E. Deydierand and A. Labande, Coord. Chem. Rev., 2016, 308, 131 CrossRef CAS; (f) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534 CrossRef CAS PubMed; (g) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981 CrossRef CAS.
- (a) J. Clayden, S. P. Fletcher, J. J. W. McDoualland and S. J. M. Rowbottom, J. Am. Chem. Soc., 2009, 131, 5331 CrossRef CAS PubMed; (b) J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS PubMed; (c) K. Mori, Y. Ichikawa, M. Kobayashi, Y. Shibata, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2013, 135, 3964 CrossRef CAS PubMed; (d) A. Ros, B. Estepa, P. Ramirez-Lopez, E. Alvarez, R. Fernandez and J. M. Lassaletta, J. Am. Chem. Soc., 2013, 135, 15730 CrossRef CAS PubMed; (e) V. Bhat, S. Wang, B. M. Stoltz and S. C. Virgil, J. Am. Chem. Soc., 2013, 135, 16829 CrossRef CAS PubMed; (f) S. Lu, S. B. Poh and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 11041 CrossRef CAS PubMed; (g) J. Wang, M.-W. Chen, Y. Ji, S.-B. Hu and Y.-G. Zhou, J. Am. Chem. Soc., 2016, 138, 10413 CrossRef CAS PubMed; (h) C. Yu, H. Huang, X. Li, Y. Zhang and W. Wang, J. Am. Chem. Soc., 2016, 138, 6956 CrossRef CAS PubMed; (i) P. Ramirez-Lopez, A. Ros, A. Romero-Arenas, J. Iglesias-Sigüenza, R. Fernandez and J. M. Lassaletta, J. Am. Chem. Soc., 2016, 138, 12053 CrossRef CAS PubMed; (j) Y. Liu, Y.-L. S. Tse, F. Y. Kwong and Y.-Y. Yeung, ACS Catal., 2017, 7, 4435 CrossRef CAS; (k) J. D. Jolliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558 CrossRef CAS PubMed.
- (a) T. Shimada, Y.-H. Cho and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 13396 CrossRef CAS PubMed; (b) G.-Q. Li, H. Gao, C. Keene, M. Devonas, D. H. Ess and L. Kürti, J. Am. Chem. Soc., 2013, 135, 7414 CrossRef CAS PubMed; (c) J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244 CrossRef CAS PubMed; (d) Y.-H. Chen, D.-J. Cheng, J. Zhang, Y. Wang, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 15062 CrossRef CAS PubMed; (e) S. Narute, R. Parnes, F. D. Toste and D. Pappo, J. Am. Chem. Soc., 2016, 138, 16553 CrossRef CAS PubMed; (f) M. Moliterno, R. Cari, A. Puglisi, A. Antenucci, C. Sperandio, E. Moretti, A. Di Sabato, R. Salvio and M. Bella, Angew. Chem., Int. Ed., 2016, 55, 6525 CrossRef CAS PubMed; (g) J.-Z. Wang, J. Zhou, C. Xu, H. Sun, L. Kürti and Q.-L. Xu, J. Am. Chem. Soc., 2016, 138, 5202 CrossRef CAS PubMed; (h) H. Gao, Q.-L. Xu, C. Keene, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2016, 55, 566 CrossRef CAS PubMed; (i) J. Feng, B. Li, Y. Heand and Z. Gu, Angew. Chem., Int. Ed., 2016, 55, 2186 CrossRef CAS PubMed; (j) Y.-H. Chen, L.-W. Qi, F. Fang and B. Tan, Angew. Chem., Int. Ed., 2017, 56, 16308 CrossRef CAS PubMed; (k) Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS PubMed.
- (a) A. Gutnov, B. Heller, C. Fischer, H. J. Drexler, A. Spannenberg, B. Sundermann and C. Sundermann, Angew. Chem., Int. Ed., 2004, 43, 3795 CrossRef CAS PubMed; (b) T. Shibata, T. Fujimoto, K. Yokota and K. Takagi, J. Am. Chem. Soc., 2004, 126, 8382 CrossRef CAS PubMed; (c) K. Tanaka, G. Nishida, A. Wada and K. Noguchi, Angew. Chem., Int. Ed., 2004, 43, 6510 CrossRef CAS PubMed; (d) G. Nishida, K. Noguchi, M. Hirano and K. Tanaka, Angew. Chem., Int. Ed., 2007, 46, 3951 CrossRef CAS PubMed; (e) M. Auge, A. Feraldi-Xypolia, M. Barbazanges, C. Aubert, L. Fensterbank, V. Gandon, E. Kolodziej and C. Ollivier, Org. Lett., 2015, 17, 3754 CrossRef CAS PubMed; (f) Y. Kwon, A. J. Chinn, B. Kim and J. S. Miller, Angew. Chem., Int. Ed., 2018, 57, 6251 CrossRef CAS PubMed; (g) Y. Liu, X. Wu, S. Li, L. Xue, C. Shan and Z. Z. H. Yan, Angew. Chem., Int. Ed., 2018, 57, 6491 CrossRef CAS PubMed; (h) S.-C. Zheng, Q. Wangand and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 1494 CrossRef CAS PubMed.
- (a) A. I. Meyers and D. G. Wettlaufer, J. Am. Chem. Soc., 1984, 106, 1135 CrossRef CAS; (b) A. Straub and A. Goehrt, Angew. Chem., Int. Ed., 1996, 35, 2662 CrossRef CAS; (c) Y. Nishii, K. Wakasugi, K. Koga and Y. Tanabe, J. Am. Chem. Soc., 2004, 126, 5358 CrossRef CAS PubMed; (d) A. V. Vorogushin, W. D. Wulff and H.-J. Hansen, J. Am. Chem. Soc., 2002, 124, 6512 CrossRef CAS PubMed.
- (a) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458 CrossRef CAS PubMed; (b) R. J. Armstrong and M. D. Smith, Angew. Chem., Int. Ed., 2014, 53, 12822 CrossRef CAS PubMed; (c) D. Lotter, M. Neuburger, M. Rickhaus, D. Häussinger and C. Sparr, Angew. Chem., Int. Ed., 2016, 55, 2920 CrossRef CAS PubMed; (d) J.-W. Zhang, J.-H. Xu, D.-J. Cheng, C. Shi, X.-Y. Liu and B. Tan, Nat. Commun., 2016, 7, 10677 CrossRef PubMed; (e) L. Zhang, J. Zhang, J. Ma, D.-J. Cheng and B. Tan, J. Am. Chem. Soc., 2017, 139, 1714 CrossRef CAS PubMed; (f) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617 CrossRef CAS PubMed; (g) K. Zhao, L. Duan, S. Xu, J. Jiang and Y. F. Z. Gu, Chem, 2018, 4, 599 CrossRef CAS; (h) T. Yang, X. Guo, Q. Yin and X. Zhang, Chem. Sci., 2019, 10, 2473 RSC.
- (a) T. Konoike and Y. Araki, Tetrahedron Lett., 1992, 33, 5093 CrossRef CAS; (b) C. Schultz-Fademrecht, B. Wibbeling, R. Fröhlich and D. Hoppe, Org. Lett., 2001, 3, 1221 CrossRef CAS PubMed; (c) S. Wu, X. Huang, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2015, 6, 7946 CrossRef PubMed.
- (a) R. A. Gibbs and W. H. Okamura, J. Am. Chem. Soc., 1988, 110, 4062 CrossRef CAS; (b) T. Qin, S. L. Skraba-Joiner, Z. G. Khalil, R. P. Johnson, R. J. Capon and J. A. Porco, Nat. Chem., 2015, 7, 234 CrossRef CAS PubMed.
- J. A. Berson and E. Brown, J. Am. Chem. Soc., 1955, 77, 450 CrossRef CAS.
- (a) F. Guo, L. C. Konkol and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 18 CrossRef CAS PubMed; (b) O. Quinonero, M. Jean, N. Vanthuyne, C. Roussel, D. Bonne, T. Constantieux, C. Bressy, X. Bugaut and J. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 1401 CrossRef CAS PubMed; (c) V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140 CrossRef CAS PubMed; (d) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2018, 57, 7136 CrossRef CAS PubMed.
- (a) K. T. Barrett, A. J. Metrano, P. R. Rablen and S. J. Miller, Nature, 2014, 509, 71 CrossRef CAS PubMed; (b) K. Tanaka, A. Kamisawa, T. Suda, K. Noguchi and M. Hirano, J. Am. Chem. Soc., 2007, 129, 12078 CrossRef CAS PubMed; (c) D. Lotter, A. Castrogiovanni, M. Neuburger and C. Sparr, ACS Cent. Sci., 2018, 4, 656 CrossRef CAS PubMed; (d) Y. Tan, S. Jia, F. Hu, Y. Liu, L. Peng, D. Li and H. Yan, J. Am. Chem. Soc., 2018, 140, 16893 CrossRef CAS PubMed.
- (a) S. Mishra, J. Liu and A. Aponick, J. Am. Chem. Soc., 2017, 139, 3352 CrossRef CAS PubMed; (b) M. Pappoppula and A. Aponick, Angew. Chem., Int. Ed., 2015, 54, 15827 CrossRef CAS PubMed; (c) M. Pappoppula, F. S. P. Cardoso, B. O. Garrett and A. Aponick, Angew. Chem., Int. Ed., 2015, 54, 15202 CrossRef CAS PubMed; (d) B. Song, T. Knauber and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 2954 CrossRef CAS PubMed.
- (a) D. Bonne and J. Rodriguez, Eur. J. Org. Chem., 2018, 2018, 2417 CrossRef CAS; (b) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385 RSC.
- (a) R. S. Nortonand and R. J. Wells, J. Am. Chem. Soc., 1982, 104, 3628 CrossRef; (b) T. Benincori, O. Piccolo, S. Rizzo and F. Sannicolò, J. Org. Chem., 2000, 65, 8340 CrossRef CAS PubMed; (c) G. Bringmann, S. Tasler, H. Endress, J. Kraus, K. Messer, M. Wohlfarth and W. Lobin, J. Am. Chem. Soc., 2001, 123, 2703 CrossRef CAS.
- (a) N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem.–Eur. J., 2010, 16, 6752 CrossRef CAS PubMed; (b) Y. Morimoto, S. Shimizu, A. Mokuya, N. Ototake, A. Saito and O. Kitagawa, Tetrahedron, 2016, 72, 5221 CrossRef CAS.
- K. Kamikawa, S. Arae, W.-Y. Wu, C. Nakamura, T. Takahashi and M. Ogasawara, Chem.–Eur. J., 2015, 21, 4954 CrossRef CAS PubMed.
- (a) L.-W. Qi, J.-H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58 CrossRef CAS PubMed; (b) H.-H. Zhang, C.-S. Wang, C. Li, G.-J. Mei, Y. Li and F. Shi, Angew. Chem., Int. Ed., 2017, 56, 116 CrossRef CAS PubMed; (c) C. Ma, F. Jiang, F.-T. Sheng, Y. Jiao, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014 CrossRef CAS PubMed.
- C. He, M. Hou, Z. Zhu and Z. Gu, ACS Catal., 2017, 7, 5316 CrossRef CAS.
- (a) C. M. So, C. P. Lau, A. S. C. Chan and F. Y. Kwong, J. Org. Chem., 2008, 73, 7731 CrossRef CAS PubMed; (b) C. M. So, z. Zhou, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 6402 CrossRef CAS PubMed; (c) C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2008, 47, 8059 CrossRef CAS PubMed.
- W. Feng, H. Yang, Z. Wang, B.-B. Gou, J. Chen and L. Zhou, Org. Lett., 2018, 20, 2929 CrossRef CAS PubMed.
- See ESI for details.†.
- CCDC 1884267 (3j), 1891943 (3x), 1905908 (3ab), 1882145 (5c) and 1905131 (5aa) contain the supplementary crystallographic data for this paper.†.
- N. Di Iorio, G. Filippini, A. Mazzanti, P. Righi and G. Bencivenni, Org. Lett., 2017, 19, 6692 CrossRef CAS PubMed.
- (a) H. H. Jung and P. E. Floreancig, Tetrahedron, 2009, 65, 10830 CrossRef CAS PubMed; (b) O. R. Luca, T. Wang, S. J. Konezny, V. S. Batista and R. H. Crabtree, New J. Chem., 2011, 35, 998 RSC; (c) X. Guo, H. Zipse and H. Mayr, J. Am. Chem. Soc., 2014, 136, 13863 CrossRef CAS PubMed; (d) C. A. Morales-Rivera, P. E. Floreancig and P. Liu, J. Am. Chem. Soc., 2017, 139, 17935 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1884267, 1891943, 1882145, 1905908 and 1905131. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00810a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |