DOI:
10.1039/C9SC00793H
(Edge Article)
Chem. Sci., 2019,
10, 5405-5422
Tuning the π-bridge of quadrupolar triarylborane chromophores for one- and two-photon excited fluorescence imaging of lysosomes in live cells†‡
Received
15th February 2019
, Accepted 20th April 2019
First published on 23rd April 2019
Abstract
A series of tetracationic quadrupolar chromophores containing three-coordinate boron π-acceptors linked by different π-bridges, namely 4,4′-biphenyl, 2,7-pyrene, 2,7-fluorene, 3,6-carbazole and 5,5′-di(thien-2-yl)-3,6-diketopyrrolopyrrole, were synthesized. While their neutral precursors 1–5 displayed highly solvatochromic fluorescence, the water-soluble tetracationic target molecules 1M–5M, did not, but their emission colour could be tuned from blue to pink by changing the π-bridge. Compound 5M, containing the diketopyrrolopyrrole bridge, exhibits the most red-shifted absorption and emission maxima and the largest two-photon absorption cross-section (4560 GM at 740 nm in MeCN). Confocal laser scanning fluorescence microscopy studies in live cells confirm localization of the dye at the lysosome. Moreover, the low cytotoxicity, and high photostability of 5M combined with two-photon excited fluorescence imaging studies demonstrate its excellent potential for lysosomal imaging in live cells.
Introduction
The synthesis and applications of triarylboranes have increased tremendously in the last few decades.1–9 In order for the three-coordinate boron moiety to function as a strong π-electron acceptor (A) due to its vacant pz-orbital, the trigonal-planar geometry of the boron atom must be maintained, as its strong Lewis acidity can otherwise lead to binding of Lewis bases and/or hydrolytic decomposition. This can be accomplished by the use of sterically demanding substituents, such as mesityl (Mes) or 2,4,6-(CF3)3C6H2 (FMes),10,11 or by physical constraint, via incorporation in a rigid, planar structure.12 While the latter strategy prevents the formation of a four-coordinate boron species by inhibiting structural deformation, the former builds a propeller-like structure around the empty pz-orbital and obstructs the attack of nucleophiles, such as water, via the formation of a protective cage. Only small anions, such as fluoride and cyanide, can overcome the steric bulk and bind to the free pz-orbital at the boron.13–15 For this reason, triarylborane acceptors are often used as selective F− and CN− sensors.16–18 Three-coordinate boron species are also applied as emitting/electron transporting materials19–31 in organic light emitting diodes,32 and redox-active materials.33–37 Furthermore, the electron deficiency of BAr3 acceptor units make them especially useful in intramolecular charge transfer compounds, when conjugated to a π-donor (D) moiety.38–46 As excitation induced charge transfer properties increase the two-photon absorption (TPA) probability, triarylborane acceptors have great potential for use in TPA and for two-photon excited fluorescence (TPEF).47,48
TPA is the simultaneous absorption of two photons via a virtual state, which is proportional to the square of the light intensity, whereas one-photon absorption is a linear process obeying Beer's Law.49–53 For this reason, two-photon absorption enables excitation of molecules within a very small volume (∼femto liter) at the focus of a laser beam, which is useful for many applications. There is thus a strong demand for efficient two-photon absorption dyes for microfabrication,54–57 three-dimensional data-storage,58–60 optical power limiting,61–63 laser up-conversion,64,65 photodynamic therapy66,67 and biological imaging.47,68–70 Especially for the latter application, relatively few small organic molecules have been studied, as they need to be highly specific for their biological target, highly photostable, water-stable and at least somewhat water-soluble to stain cells and tissues. In addition, the two-photon brightness (σ2Φf), where σ2 is the two-photon absorption cross-section (comparable to an extinction coefficient) in GM and Φf is the fluorescence quantum yield, should be at least 50 GM to observe bright two-photon microscope images.47 Such probes have been applied as sensors of biomolecules (e.g. ions, reactive oxygen species (ROS), reactive sulphur species (RSS) and reactive nitrogen species (RNS)) and changes inside cells (pH, viscosity and polarity).71,72 However, two-photon dyes for monitoring and visualizing mitochondria and lysosomes are still limited.
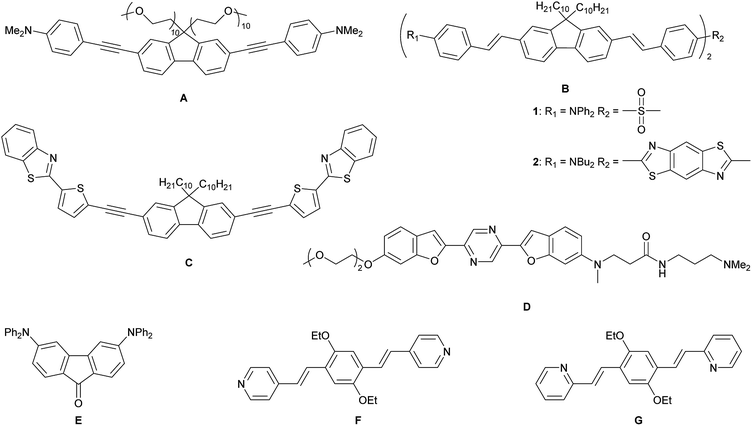
Lysosomes are acidic (pH 4.5–5.0) organelles in eukaryotic cells and are responsible for intracellular digestion degradation, secretion, plasma membrane repair, cell signalling, energy metabolism and endocrine regulation.73,74 Lysosomes are filled with more than 60 enzymes, the synthesis of which is controlled by nuclear genes. Mutations of these genes lead to lysosomal storage diseases, such as neurodegenerative disorders, e.g. Parkinson's disease and Alzheimer's disease, cardiovascular diseases and cancer.75 As they are acidic organelles, usually organic bases, such as morpholine, pyridine and dimethylamino groups, lead to accumulation in lysosomes. These terminal groups are typically attached to common dyes, such as naphthalene,76–79 1,8-naphthalimide,80–88 indole,89 coumarin,90 chromene91 and fluorenone.92 Most of them are lysosome trackers,76,77,85,91,92 while others can sense Zn2+,81 HClO,79,82 H2S,86,89 thiols,83 NO,80 β-galactosidase,88 pH78,84,87 or polarity90 within the lysosome. While all of the above-mentioned dyes are dipolar, quadrupolar chromophores for two-photon imaging of lysosomes are rare. It should be noted that efficient two-photon absorbing dyes bear the common structural motifs of dipolar, push–pull systems (D–A), quadrupolar (D–π–D, A–π–A) or octupolar systems (DA3, AD3), with the latter two being often more efficient than dipoles.49,53 While the TPA properties of many quadrupoles have been studied, only a handful of those are used for imaging, especially of the lysosome. All eight such compounds which are, to our knowledge, the only ones, are depicted in Scheme 1.
 |
| | Scheme 1 Previously reported quadrupolar chromophores for two-photon excited fluorescence (TPEF) imaging of lysosomes.93–98 | |
In 2010, Belfield and co-workers reported93 the first quadrupolar TPEF imaging agent for lysosomes (A) which exhibited a very high two-photon brightness of 431 GM in toluene. The dye localized in the lysosome, was found to be non-toxic and photostable. One year later, the same group reported three other compounds, B1, B2 and C, with σ2Φf of 1444, 1887 and 788 GM, respectively, in cyclohexane.94,95 In the more polar solvent THF, the two-photon brightness of B1 decreased to 1/3 and for compound C, σ2Φf decreased to 1/9 of its original value in a “5 wt% aqueous DMSO” solution. The brightness is usually much smaller in aqueous solution, due to the reduced quantum yield. Nevertheless, the three compounds show remarkably large values, due to their large conjugation lengths, but have relatively high molecular weights. Furthermore, these compounds are not water-soluble and must be premixed with Pluronic F-127, which is a block copolymer based on ethylene oxide and propylene oxide that is used for drug delivery. It encapsulates the chromophores upon formation of micelles facilitating dye up-take by the cells. In 2015, Cho's group reported the water-soluble dye D.96 Due to its negligible fluorescence in water, they measured its TPEF properties in a dioxane-water mixture and reported a brightness of 116 GM. The fluorenone dye E does not show bright fluorescence in water (Φf = 0.07); thus, its TPEF properties were measured in toluene (σ2Φf = 150 GM).97 The small size of the molecule might be the reason for its more modest two-photon brightness. Compounds F and G are two pH-sensitive dyes for lysosome imaging.98 The pyridine moiety acts as a ratiometric sensor for pH changes, as the protonation of the pyridine enhances intramolecular charge transfer. The two-photon cross-section increases, while the quantum yield drops at lower pH values. Therefore, the two-photon brightness maxima occur at pH 7 for F (130 GM) and at pH 3 for G (99 GM). Unfortunately, the selectivity of the dyes in the lysosome is less than excellent, as co-localization experiments showed an overlap with LysoTracker™ at lower pH, but also distribution in the cytosol at neutral pH.
As we have shown40,99–102 that quadrupolar compounds (A–π–A), with three-coordinate boron moieties as acceptors, exhibit large TPA cross-sections, we wished to apply them for imaging, but only a few water-soluble triarylboranes were known. Gabbaï and co-workers used trimethylammonio groups at the para-positions of triarylboranes to achieve water-solubility for sensing of cyanide in aqueous solution.103 They and other groups also introduced cationic phosphonio substituents onto triarylboranes for further anion sensing studies.104–106 Yang and co-workers were the first to report water-soluble three-coordinate boron compounds for imaging purposes. They substituted a triarylborane with polyethylene glycol chains for ATP sensing in the cytoplasm and cell membrane.107 Furthermore, they could sense H2S with a CuII-cyclen-substituted triarylborane.108 They reported cell-membrane permeability and a preferential distribution at mitochondria,108 while the same compound, without CuII binding, was used one year later to stain nucleoli and cytoplasm.109 However, a two-photon brightness of only 30 GM in DMSO was measured for this compound. Two other triarylboranes with piperazine in the para-position were recently reported. They are water-soluble to some degree and were found to stain nucleoli as well as the nuclear membrane, nuclear matrix, nuclear pore and the cytoplasm, while binding to RNA.110 Further three-coordinate boron containing dyes were used for imaging, although they were not water-soluble.111–115 By loading them into nanogels, they became cell membrane permeable, stained the cytoplasm and could be applied as temperature, viscosity, pH, H2O2 and biothiol sensors.111–115 Another intracellular “turn-on”-sensor for thiophenol, based on a triarylborane moiety, was very recently published by Thilagar and co-workers.116 In 2016, our group reported the only water-soluble quadrupolar three-coordinate boron compound for imaging (2TM), which exhibited a very reasonable two-photon brightness of 285 GM in MeCN (Scheme 2).117
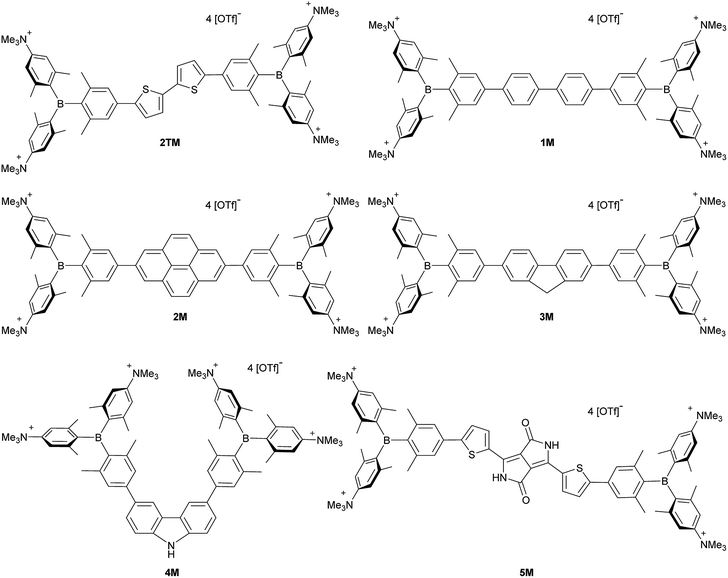 |
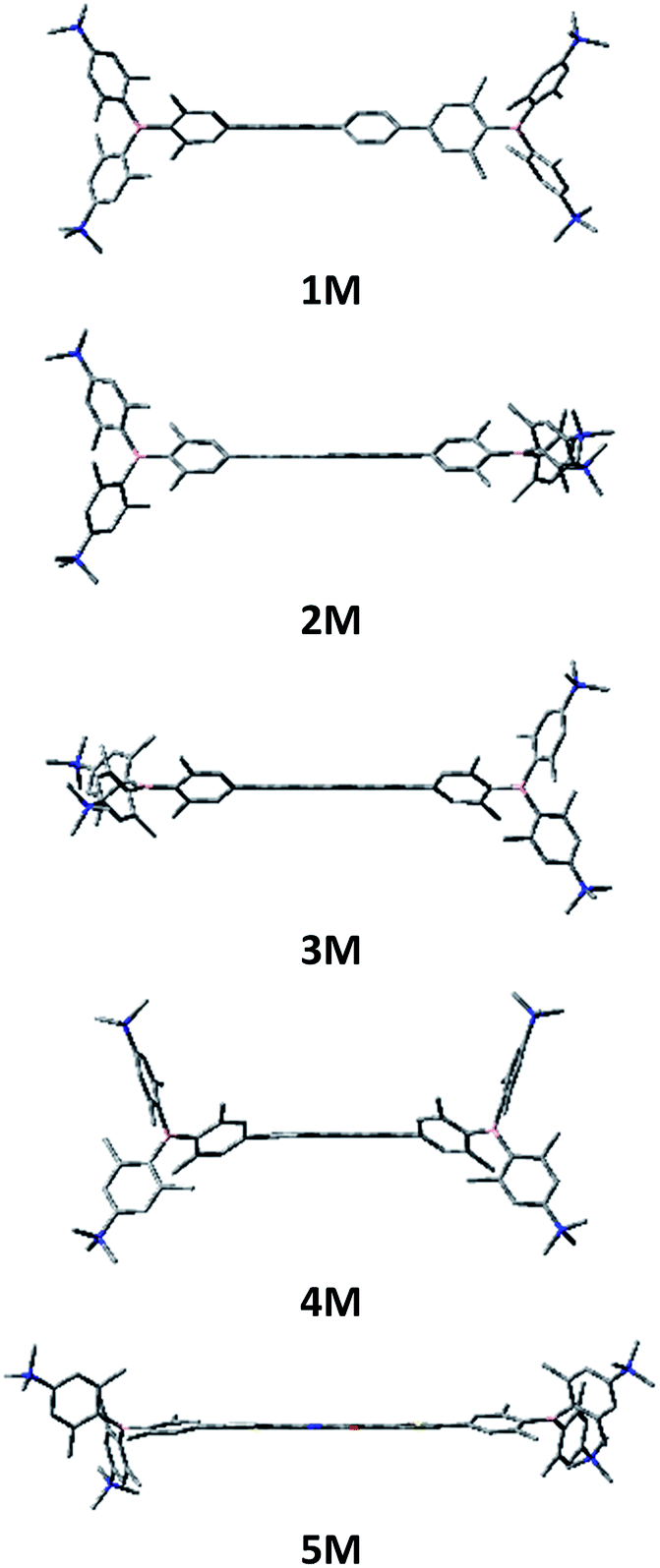
| | Scheme 2 Previously reported quadrupolar chromophore for TPEF imaging 2TM and quadrupolar target molecules 1M–5M for cell imaging. | |
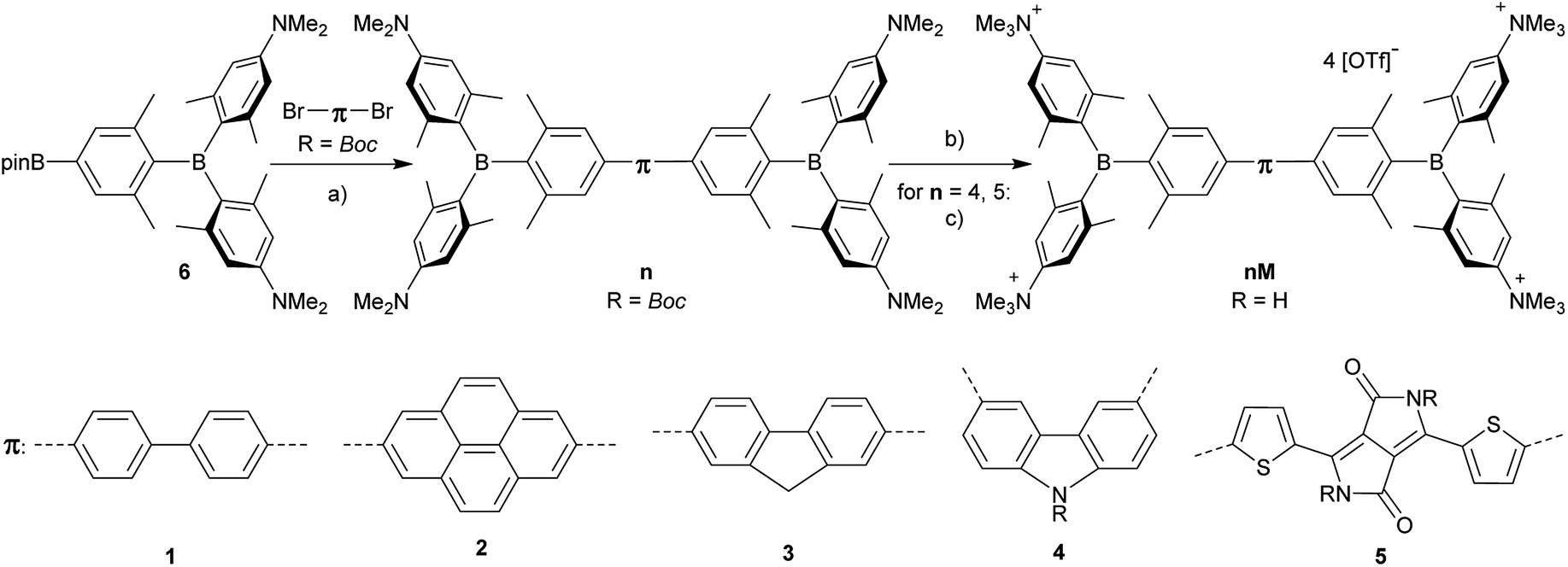
With our very promising initial results with 2TM, we thus decided to optimize our imaging dye. Tuning the emission colour, enhancing the quantum yield, TPA cross-section and photostability, maintaining low cyto-toxicity, determining co-localization as well as examining the cellular uptake pathway were our main goals. For this purpose, we designed the new dyes 1M–5M shown in Scheme 2, all of which contain trimethylammonio groups for water-solubility.103 Due to the inductive withdrawing character of the ammonium cations, these triarylborane moieties are much stronger acceptors than the normally used aryldimesitylborane. In the present study, we explored the role of the π-bridge in our A–π–A system, employing 4,4′-biphenyl, 2,7-pyrenyl, 2,7-fluorenyl, 3,6-carbazolyl and 5,5′-di(thien-2-yl)-3,6-diketopyrrolopyrrolyl bridges. We thus introduced more rigid π-bridges, to compare with the biphenyl compound 1M, to achieve more planar ground state structures, resulting in a better delocalized π-system and therefore enhanced TPA/TPEF properties. Furthermore, enhanced donor character was introduced into the π-bridge via the carbazole, while incorporation of the dithienyl-diketopyrrolopyrrole moiety leads to an A–D–A–D–A-type chromophore, allowing enhanced intramolecular charge transfers. The diketopyrrolopyrrole moiety is known for its high photostability and TPA cross-section,118,119e.g. when connected to two porphyrin groups (up to 4000 GM@910 nm in CH2Cl2).120,121
In order to synthesize the desired water-soluble tetra-ammonio chromophores 1M–5M it was necessary to prepare their neutral tetra-amino precursors 1–5. Compounds 1–5, containing four strong π-donor Me2N groups conjugated with the boron acceptor moieties, exhibit entirely different photophysical properties than those of their tetra-methylated derivatives. As the neutral compounds 1–5 are fundamentally interesting in their own right, we first describe their behaviour before that of the tetracationic chromophores of interest for imaging purposes.
Results and discussion
Synthesis
The synthesis of the neutral compounds 1–5 was achieved via Suzuki–Miyaura coupling of the different dibrominated π-bridges with our borylated triarylborane 6117 using Pd2(dba)3 as the catalyst, SPhos as the ligand and KOH as the base (Scheme 3). The neutral compounds were methylated with MeOTf in dichloromethane and the products 1M–3M precipitated in almost quantitative yields. The methylation needs to be carried out in basic glassware, such as soda-lime glass, as otherwise the reaction does not go to completion, and the product is contaminated with the compound in which only three of the amine groups are methylated. When using the two Boc-protected bridges, carbazole 4 and dithienyl-diketopyrrolopyrrole 5, the reaction time needs to be carefully controlled, as too short a reaction time leads to incomplete methylation, whereas too long a reaction time results in deprotection and subsequent methylation of the amine in the bridge. The latter two Boc-protected tetracationic compounds were subsequently deprotected with triflic acid to yield the final compounds 4M and 5M.
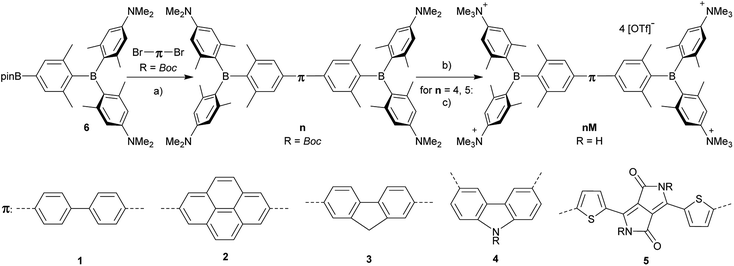 |
| | Scheme 3 Synthesis of compounds 1M–5M. (a) [Pd2(dba)3] (dba = trans,trans-dibenzylideneacetone), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos), KOH, toluene, H2O, 85 °C; (b) MeOTf, CH2Cl2; (c) TfOH, MeOH. | |
Linear optical properties and TD-DFT calculations of the neutral precursors 1–5
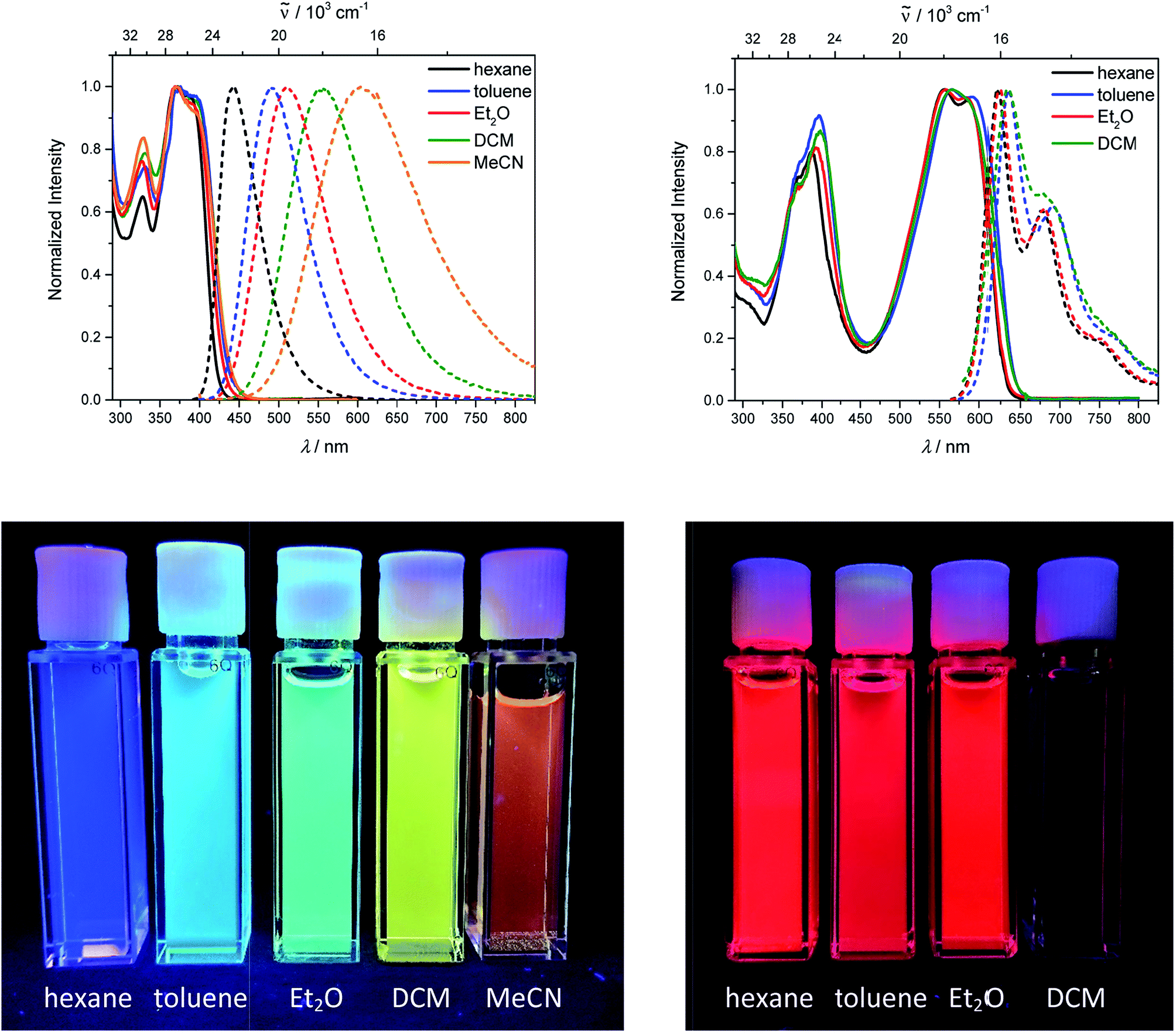
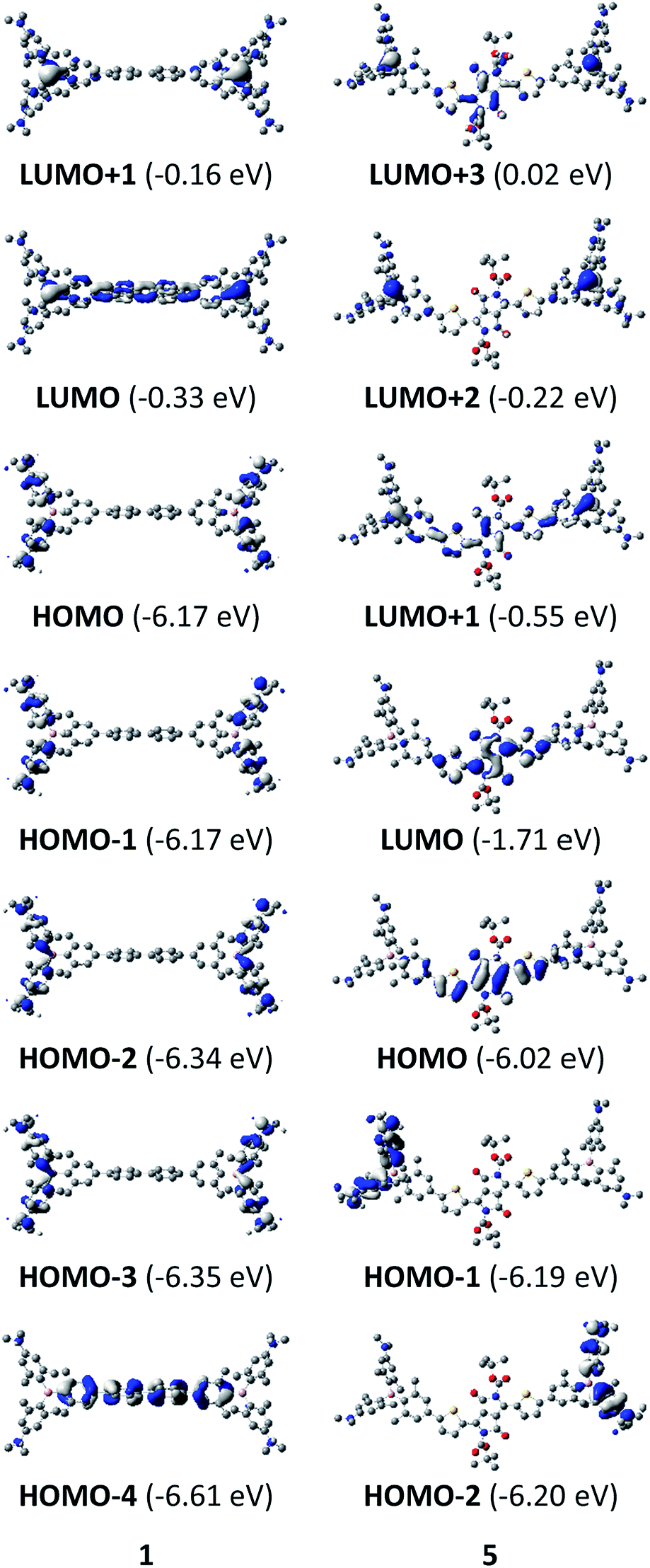
The neutral chromophores behave in a very similar manner, with the exception of compound 5. The absorption spectra of 1–4 (Fig. 1 and S1,‡Table 1) show a low energy band corresponding to charge transfer from the N,N-dimethylaminoxylyl donor to the π-bridge and boron acceptor, as confirmed by TD-DFT calculations, vide infra. As compounds 1–4 are so similar, we discuss compound 1 in detail as an example. Thus, for the biphenyl compound 1, the above described absorption band occurs at 392 nm (calculated at 354 nm) resulting from the coincidental overlap of the weak S1 ← S0 and stronger S2 ← S0 transitions (Table 2). The HOMO−1 and HOMO are nearly degenerate, and both orbitals are localized at the N,N-dimethylaminoxylyl groups, while the LUMO and LUMO+1 are located at the π-bridge and the boron atoms (Fig. 2). The higher energy absorption bands have charge-transfer character, but with increasing energy the π–π* character at the π-bridge becomes increasingly dominant. The S3 ← S0 and S5 ← S0 transitions at 372 and 328 nm, respectively, were calculated to be at 335 and 296 nm, with contributions of HOMO−4 of 11 and 55%, respectively (Table 2). HOMO−4 is delocalized over the whole π-bridge (Fig. 2). Very similar results were calculated for compounds 2–4. All compounds show different high energy absorption bands, as the π–π* contributions of the π-bridges become more and more important. For example, compound 2 shows one additional absorption band at 342 nm, which is calculated to arise from the S6 ← S0 transition from HOMO−6 to LUMO, and is a classic pyrene π–π* transition, with the typical nodal plane through the 2,7-positions (Fig. S3‡).35,36,122,123 The dithienyl-diketopyrrolopyrrole dye 5 is an exception, as the HOMO is located at the π-bridge (Fig. 2). Therefore, the S1 ← S0 transition is a locally excited (LE) HOMO to LUMO transition at the π-bridge, while higher energy transitions show the same charge transfer character as noted above for 1–4 (Table 2). For 5, the HOMO−1 and HOMO−2 are nearly degenerate and are each localized at two N,N-dimethylaminoxylyl groups. Note that the TD-DFT calculations were carried out for the geometry optimized (lowest energy) structure and not for the highest possible symmetry (Ci) of these molecules. Therefore, the corresponding S2 ← S0 and S4 ← S0 transitions are isoenergetic and exhibit charge transfer character from the nitrogens to the boron atom.
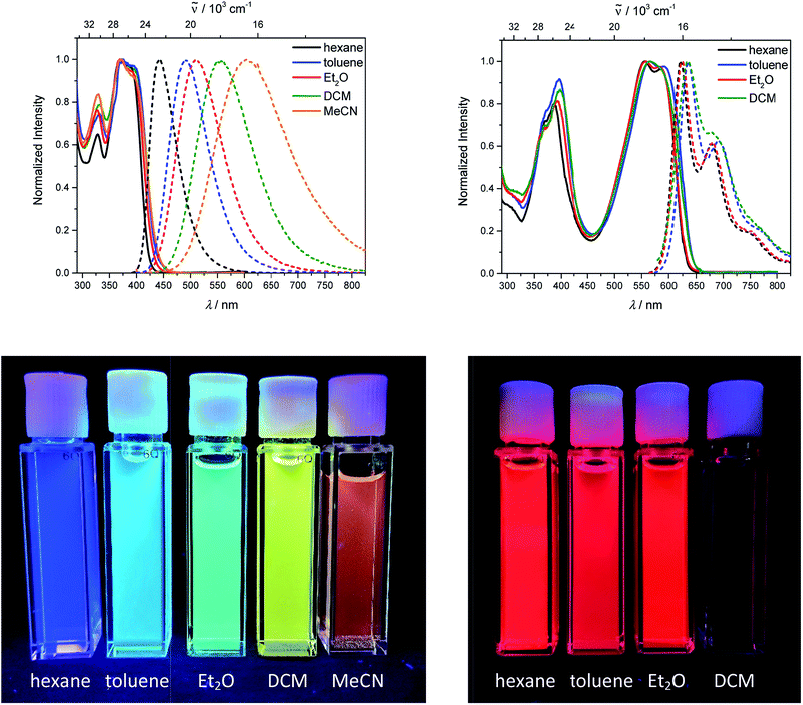 |
| | Fig. 1 Absorption and emission spectra (top) and pictures of the solutions under UV irradiation (bottom) of 1 (left) and 5 (right) in various solvents (hexane: black, toluene: blue, diethylether: red, dichloromethane: green, MeCN: orange). | |
Table 1 Photophysical data of the compounds 1 and 5 in various solvents
|
|
solvent |
λ
abs/nm |
ε/M−1 cm−1 |
λ
em/nm |
Stokes shift/cm−1 |
Φ
f
|
τ/ns |
k
r/108 s−1 |
knr/108 s−1 |
|
1
|
Hexane |
372 |
69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
442 |
4300 |
0.14 |
1.5 |
0.9 |
5.8 |
| Toluene |
376 |
|
493 |
6300 |
0.20 |
3.4 |
0.6 |
2.3 |
| Et2O |
369 |
|
511 |
7500 |
0.24 |
5.9 |
0.4 |
1.3 |
| DCM |
373 |
|
556 |
8800 |
0.30 |
8.8 |
0.3 |
0.8 |
| MeCN |
371 |
|
603 |
10400 |
0.08 |
2.5 |
0.3 |
3.7 |
|
5
|
Hexane |
557 |
59000 |
622 |
1900 |
0.57 |
2.4 |
2.4 |
1.8 |
| Toluene |
566 |
|
635 |
1900 |
0.55 |
2.3 |
2.4 |
1.9 |
| Et2O |
558 |
|
624 |
1900 |
0.40 |
2.0 |
2.0 |
3.0 |
| DCM |
568 |
|
635 |
1900 |
0.004 |
2.3 |
0.02 |
4.3 |
Table 2 TD-DFT-calculated photophysical data for 1 and 5 at the CAM-B3LYP/6-31G(d) level in hexanea
|
|
Transition (f) |
E/eVb |
λ/nmb |
Dominant componentsc |
|
Transitions with modest to high oscillator strength f are displayed in this table. Others are shown in the ESI.
Values in parentheses are experimental absorption maxima in hexane.
Components with greater than 10% contribution shown. Percentage contribution approximated by 2 × (ci)2 × 100%, where ci is the coefficient for the particular ‘orbital rotation’.
|
|
1
|
S1 ← S0 (0.054) |
3.50 (3.16) |
354 (392) |
LUMO+1 ← HOMO−1 (44%), LUMO ← HOMO (38%) |
| S2 ← S0 (0.736) |
3.50 (3.16) |
354 (392) |
LUMO ← HOMO−1 (38%), LUMO+1 ← HOMO (44%) |
| S3 ← S0 (1.496) |
3.70 (3.33) |
335 (372) |
LUMO ← HOMO−4 (11%), LUMO+1 ← HOMO−3 (29%), LUMO ← HOMO−2 (37%) |
| S5 ← S0 (0.778) |
4.19 (3.78) |
296 (328) |
LUMO ← HOMO−4 (55%), LUMO+1 ← HOMO−3 (15%) |
|
5
|
S1 ← S0 (1.739) |
2.31 (2.23) |
537 (554) |
LUMO ← HOMO (94%) |
| S2 ← S0 (0.294) |
3.35 (3.20) |
370 (388) |
LUMO ← HOMO−2 (33%), LUMO+1 ← HOMO−2 (20%), LUMO+2 ← HOMO−2 (27%), LUMO+3 ← HOMO−2 (10%) |
| S4 ← S0 (0.260) |
3.38 (3.20) |
367 (388) |
LUMO ← HOMO−1 (28%), LUMO+1 ← HOMO−1 (24%), LUMO+2 ← HOMO−1 (26%), LUMO+3 ← HOMO−1 (13%) |
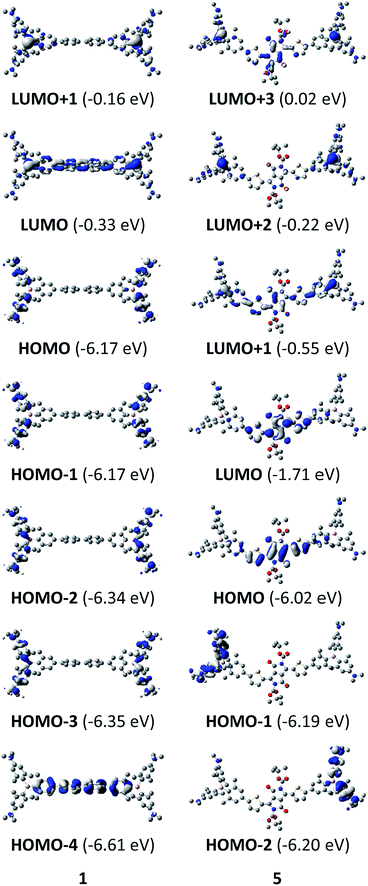 |
| | Fig. 2 DFT (CAM-B3LYP/6-31G(d))-calculated relevant orbitals for 1 and 5. Hydrogen atoms are omitted for clarity. Surface isovalue: ± 0.03 [ea0−3]1/2. Orbital energy differences are not direct approximations of excitation energies. They are provided here for comparison between the compounds. | |
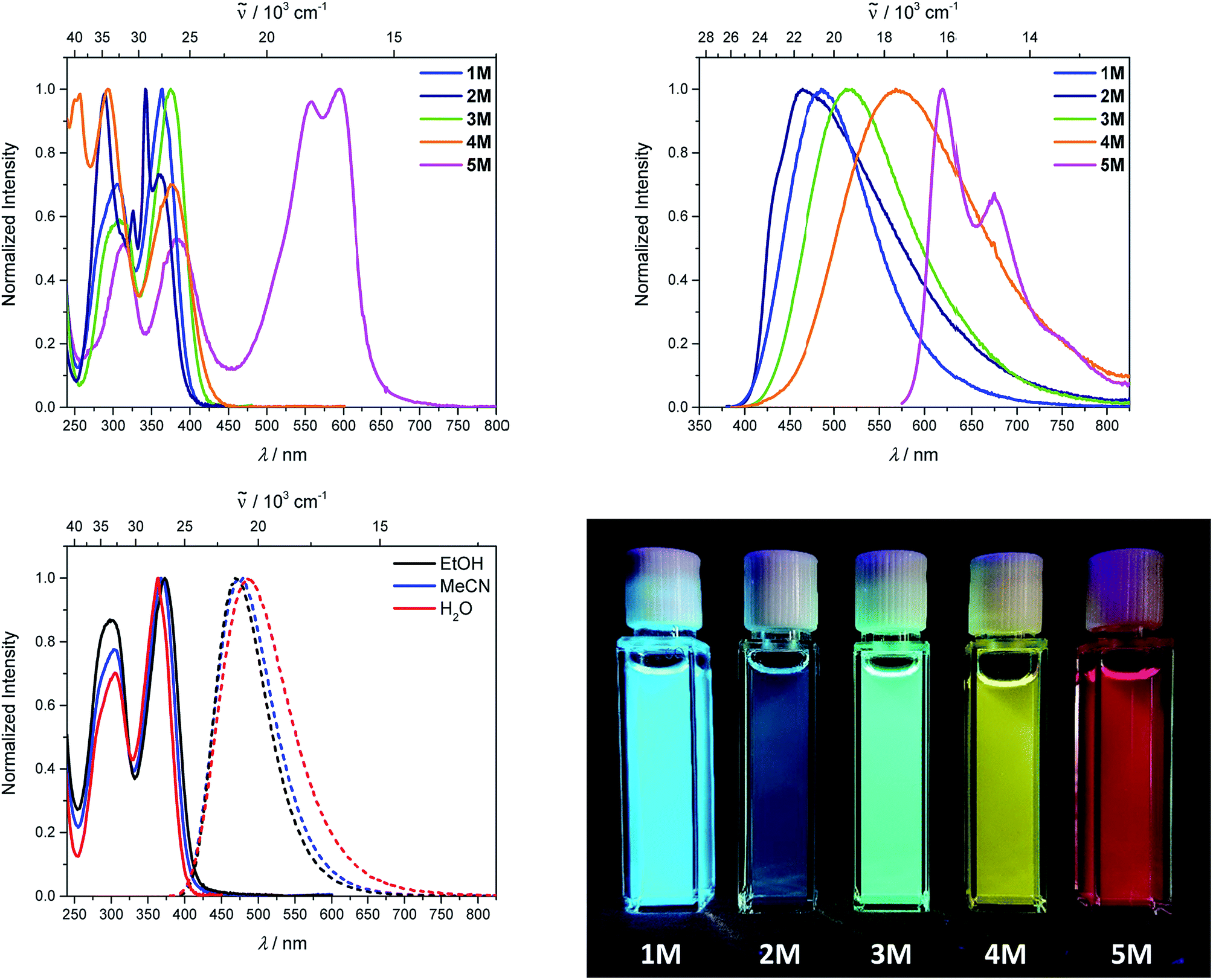
As the lowest energy absorption bands of 1–4 have charge-transfer character, their emission spectra display strong solvatochromism. Upon going from nonpolar hexane (λem,max = 442 nm) to polar MeCN (λem,max = 603 nm), the emission maximum of 1 is bathochromically shifted by 6040 cm−1, which results in an increase of the Stokes shift by 6100 cm−1, i.e., from 4300 to 10400 cm−1 (Fig. 1 and Table 1). This positive solvatochromism with increasing solvent polarity results from a large dipole moment in the excited state. As the lowest energy absorption band results from N,N-dimethylaminoxylyl-to-boron charge transfer for 1–4, which all of those compounds have in common, the emission spectra are identical regardless of the nature of the π-bridge. The emission band of 4 is slightly blue-shifted, as the LUMO is a little higher in energy (∼0.15 eV) than for 1–3. The LUMO of compound 4 is more localized at the boron than the bridge, because the carbazole bridge also acts as a donor. The fluorescence quantum yields and lifetimes are essentially the same for 1–4 (Table 1 and S1‡). Interestingly, they do not follow the expected dependence on solvent polarity. Thus, with increasing solvent polarity, the excited state is more stabilized as shown by the bathochromic shift of the emission maximum. As ΔG00 decreases, following the energy gap law,124 it is expected that the nonradiative decay constant knr should increase and therefore the quantum yield should decrease. Our compounds 1–4 show the opposite behaviour. With increasing solvent polarity, the nonradiative decay constant decreases and the quantum yield is enhanced. Also, the experimentally determined fluorescence lifetimes increase with increasing solvent polarity, while the radiative decay constants kr decrease with decreasing emission energy in qualitative accordance with the Strickler–Berg equation.125 This formula predicts a proportionality of the radiative decay constant kr with the cube of the fluorescence wavenumber ṽf3. Furthermore, in MeCN, compounds 1–4 do not follow the aforementioned trend. In this solvent, the quantum yields are smaller, and the fluorescence lifetimes shorter compared with DCM solutions. This behaviour was observed previously for nitrogen donor – boron acceptor compounds43,126–128 and has its origin in symmetry breaking in the excited state. The symmetry breaking is more enhanced in polar solvents than nonpolar solvents, leading to the unusual solvent behaviour seen above.129 The dithienyl-diketopyrrolopyrrole dye 5 is again an exception. As the low-energy absorption is an LE transition, this compound shows no solvatochromism in the solvents examined. In all solvents it emits pink light (∼630 nm), and the quantum yield is ca. 0.55, but drops significantly in DCM, as the non-radiative decay rate rises.
Linear optical properties and TD-DFT calculations of the tetracationic chromophores 1M–5M
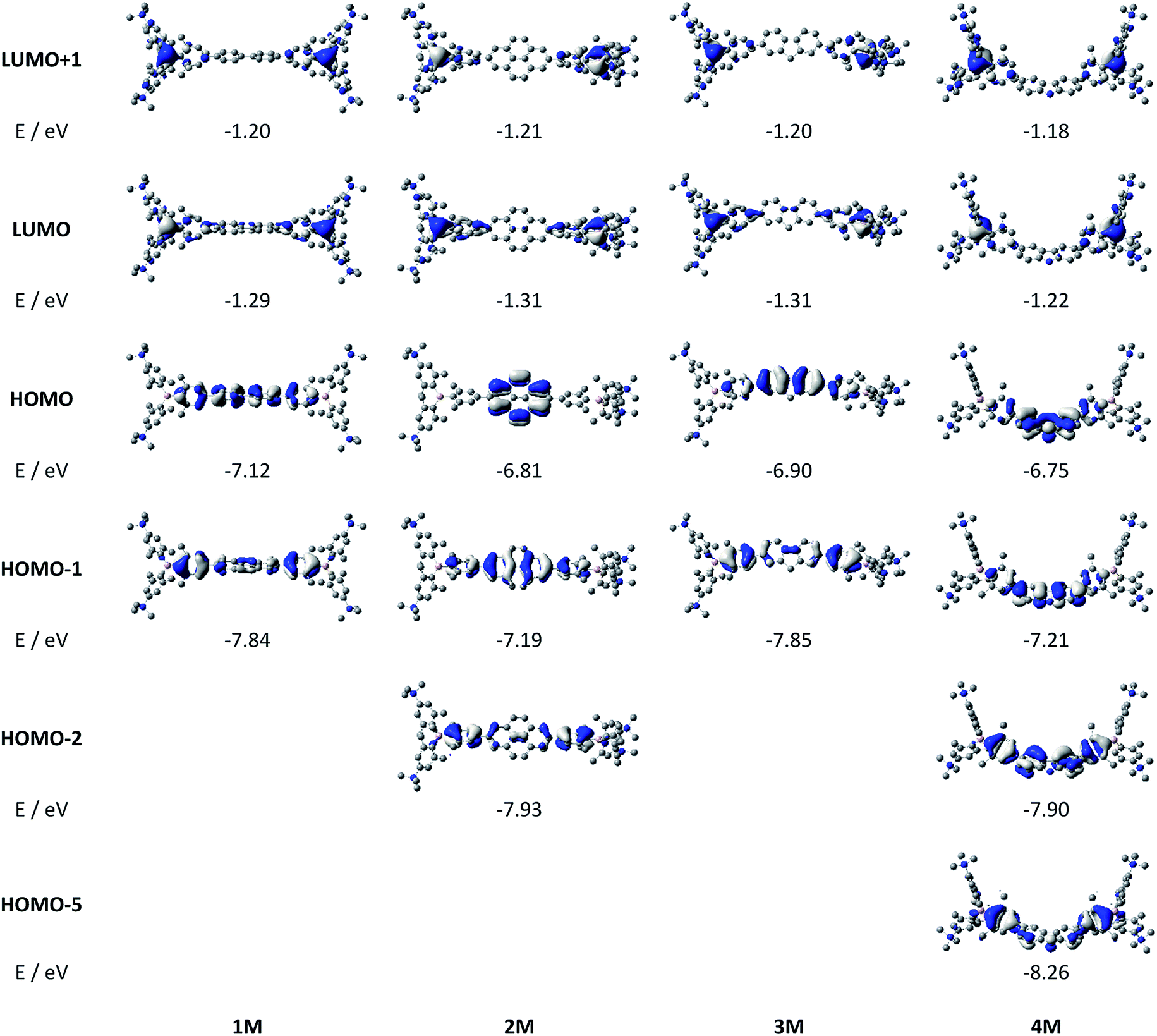
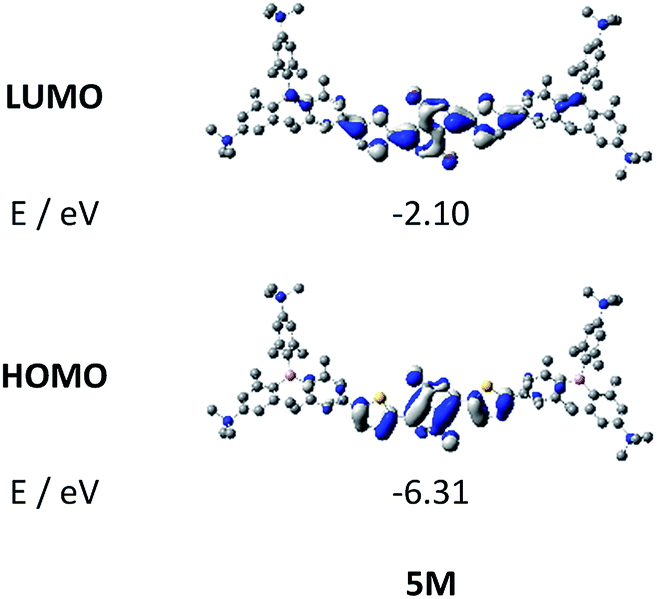
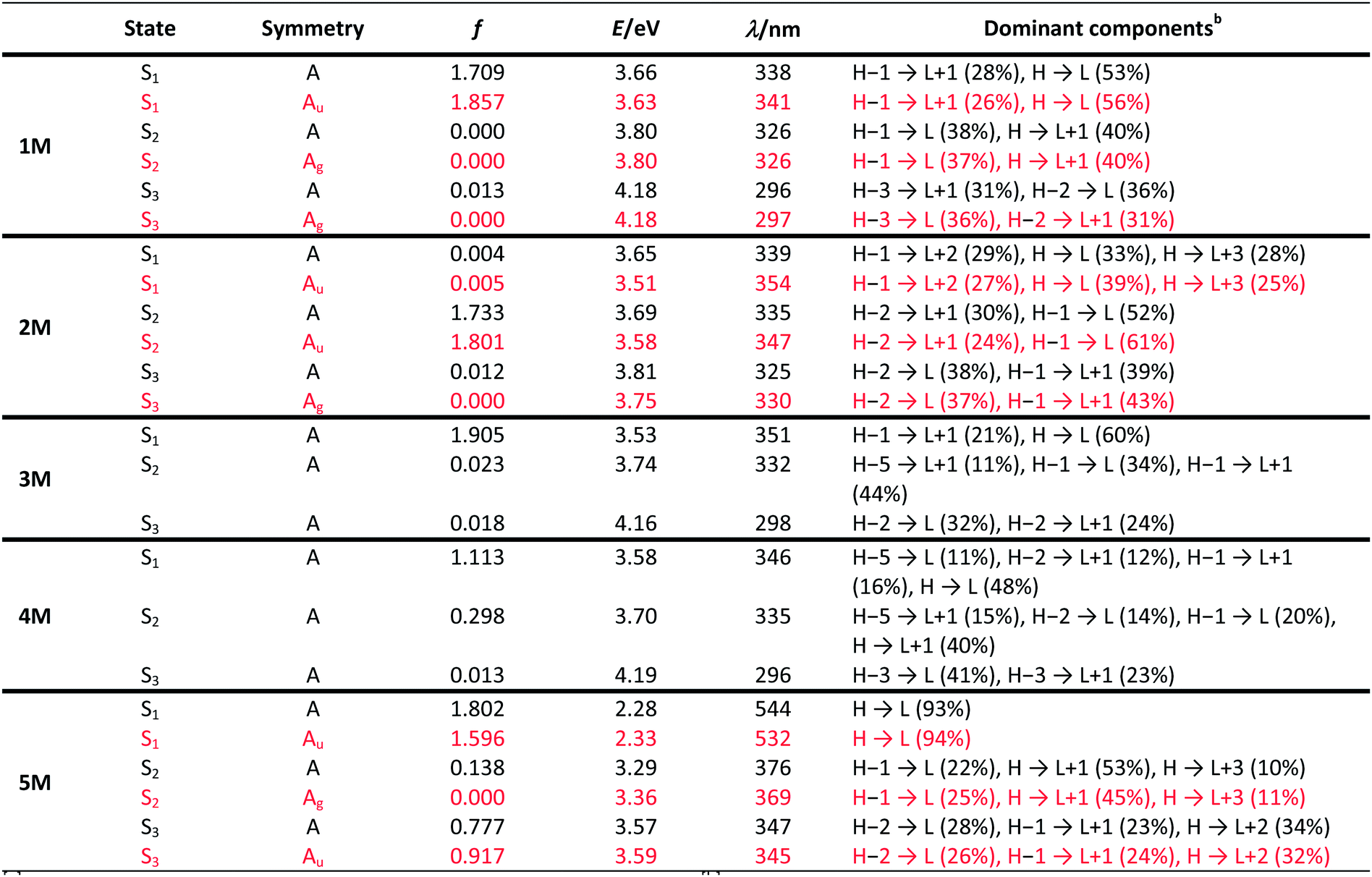
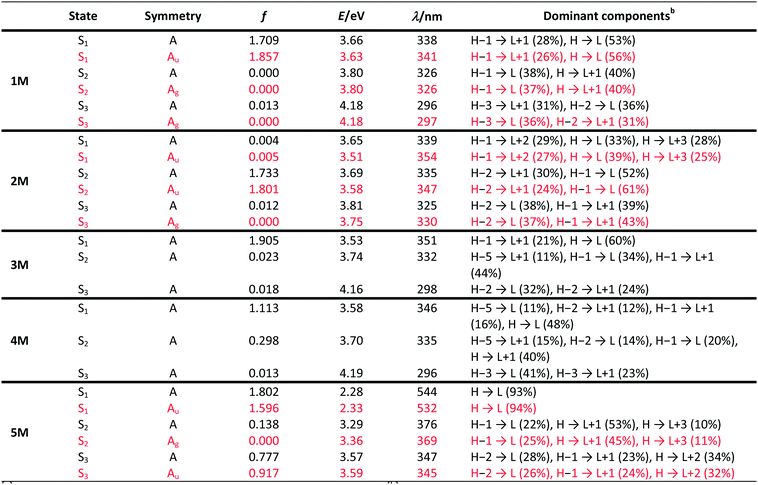
Upon methylation of the neutral precursors 1–5, and subsequent deprotection of 4 and 5, the charge transfer from the amine to the boron moiety is no longer present, so the linear optical properties of the chromophores 1M–5M are completely different from those of 1–5. Fig. 3 shows the absorption spectra of 1M–5M in water. Due to solubility issues, compound 2M was dissolved in 10% MeCN in water. The various absorption bands are attributed to the π–π* transitions of the individual π-bridges. TD-DFT calculations show that the computed low-energy absorption bands at 338, 351, 335, 346 and 544 nm, respectively, for 1M–5M (experimentally: 363, 375, 361, 376 and 594 nm, respectively) are located on the π-bridge (Table 4 and Fig. 4 and 5). For the biphenyl (1M) and fluorene (3M) compounds, this is an S1 ← S0 transition, with LUMO+1 ← HOMO−1 and LUMO ← HOMO contributions, where the HOMO and HOMO−1 are located at the π-bridge and the LUMO and LUMO+1 are mostly localized at the boron atoms (Table 4 and Fig. 4). Pyrene derivative 2M has the same behaviour, but the transition is S2 ← S0 with the main contributions being LUMO+1 ← HOMO−2 and LUMO ← HOMO−1. As the HOMO is located only at the pyrene, and has a nodal plane through the substituted 2,7-positions, it does not take part in the first allowed, low energy, transition. The LUMO+2 ← HOMO transition is the S4 ← S0 absorption, which is a higher energy absorption band of 2M at 316 nm (experimentally: 342 nm) (Table S10‡ and Fig. 4). In the case of the carbazole-bridged derivative 4M, the calculations indicate that the S1 ← S0 absorption has LUMO ← HOMO−5 (11%), LUMO+1 ← HOMO−2 (12%), LUMO+1 ← HOMO−1 (16%) and LUMO ← HOMO (48%) contributions. For 1M–4M, the HOMOs which contribute are located at the π-bridges and LUMO and LUMO+1 are mainly localized at the boron atom. The S1 ← S0 transition of the dithienyl-diketopyrrolopyrrole dye 5M is a simple LUMO ← HOMO LE transition localized at the π-bridge, with a small contribution from the borons. Due to the strong acceptor strength of the boron moiety, which lowers the energies of the virtual orbitals, the low-energy absorption maxima are red-shifted by up to 4334 cm−1 compared with those of the analogous compounds 1A–5A (Scheme 4). Compounds 1A–4A were previously reported,122,130–132 whereas compound 5A was synthesized as part of the present study.
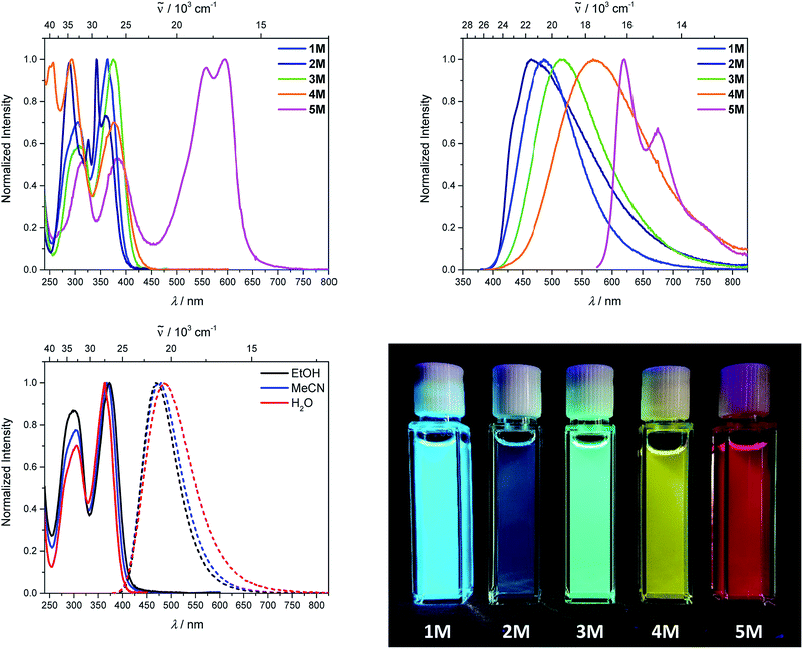 |
| | Fig. 3 Absorption (top, left) and emission spectra (top, right) of 1M–5M in water. Compound 2M was dissolved in 10% MeCN in water. Absorption and emission spectra of 1M in various solvents (EtOH: black, MeCN: blue, H2O: red) (bottom, left) and picture of the solutions of 1M–5M in MeCN under UV irradiation. | |
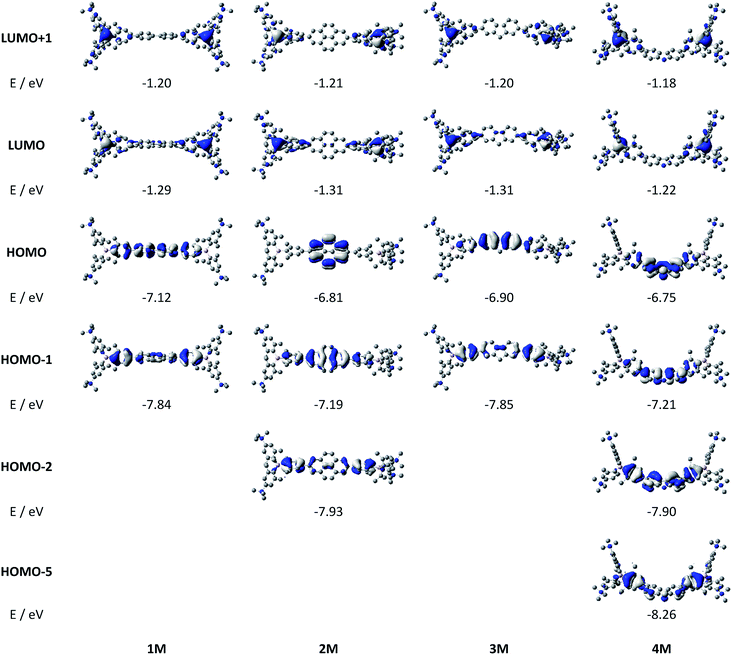 |
| | Fig. 4 DFT (CAM-B3LYP/6-31G(d))-calculated relevant orbitals for 1M–4M. Hydrogen atoms are omitted for clarity. Surface isovalue: ± 0.03 [ea0−3]1/2. | |
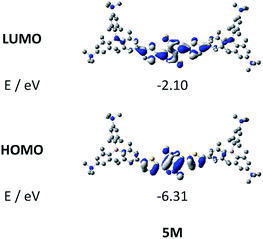 |
| | Fig. 5 DFT (CAM-B3LYP/6-31G(d))-calculated frontier orbitals for 5M. Hydrogen atoms are omitted for clarity. Surface isovalue: ± 0.03 [ea0−3]1/2. | |
 |
| | Scheme 4 Non-boron containing analogues; 1A was measured in acetone,1302A in toluene,1223A in ethyl acetate,1324A in MeCN131 and 5A in DMSO. | |
As the nature of the transitions vary somewhat for the various π-bridges, the emission colour can be tuned from blue to pink (Fig. 3). The emission maxima shift from 467 nm (2M) to 620 nm (5M) and follow the trend of the HOMO energy, as the LUMO energy stays nearly constant and is mainly boron centred (except for 5M). Furthermore, for compound 2M the HOMO−1 needs to be considered, as the HOMO is only localized on the pyrene and is not involved in the strongest low-energy S2 ← S0 excitation (S1 ← S0 has an oscillator strength near zero). The higher the HOMO energy, the more bathochromically shifted the emission band. Our variety of π-bridged water-soluble, quadrupolar three-coordinate boron chromophores thus provides a wide colour range spanning most of the visible spectrum. Furthermore, the compounds are not solvatochromic, as shown in Fig. 3 for 1M as an example. This proves that the transitions do not involve a significant change in dipole moment.§ The only observed effect is a broadening of the emission spectra in more polar solvents. Using Jortner's theory,133 the full width at half maximum of the bell-shaped curve of a transition depends on the reciprocal value of the reorganization energy of the solvent, so the broadening of the emission bands in more polar solvents is expected. The strong π-acceptor boron moieties also shift the emission maxima compared to those of the non-boron analogues 1A–5A (Scheme 4) between 509 and 8916 cm−1, depending on the contribution of the boron atoms.
The fluorescence quantum yields and lifetimes were measured in aqueous solution (Table 3). As the fluorescence of the carbazole compound 4M is very weak in aqueous solution, the lifetime could not be determined. For all other compounds 1M–3M and 5M the radiative and non-radiative decay constants were calculated and the radiative decay constants are very similar for all compounds, except for 2M. This may be due to the use of 10% MeCN to improve solubility. The variation in the quantum yields is thus due to differences in the non-radiative decay rates. Apart from 4M, all compounds show remarkably high fluorescence quantum yields in aqueous solution, especially compound 1M. In other solvents such as EtOH or MeCN, the fluorescence quantum yields increase, as the non-radiative decay rates decrease or the radiative decay rates increase for all compounds.
Table 3 One- and two-photon photophysical data of compounds 1M–5M in various solvents
|
|
Solvent |
λ
abs/nm |
ε/M−1 cm−1 |
λ
em/nm |
Stokes shift/cm−1 |
Φ
f
|
τ/ns |
k
r/108 s−1 |
knr/108 s−1 |
λ
TPA,max/nm |
σ
2/GM |
|
Measured in 10% MeCN in water.
not measurable.
|
|
1M
|
EtOH |
373 |
|
470 |
5500 |
0.71 |
4.9 |
1.4 |
0.6 |
|
|
| MeCN |
368 |
57000 |
478 |
6300 |
0.73 |
5.2 |
1.4 |
0.5 |
720 |
72 |
| H2O |
364 |
|
486 |
6900 |
0.58 |
6.6 |
0.9 |
0.6 |
|
|
|
2M
|
EtOH |
371 |
|
465 |
5400 |
0.20 |
14.3 |
0.1 |
0.6 |
|
|
| MeCN |
368 |
61000 |
462 |
5500 |
0.12 |
12.6 |
0.1 |
0.7 |
750 |
79 |
| H2Oa |
365 |
|
467 |
6000 |
0.12 |
19.5 |
0.1 |
0.4 |
|
|
|
3M
|
EtOH |
389 |
|
492 |
5400 |
0.71 |
5.0 |
1.4 |
0.6 |
|
|
| MeCN |
383 |
62000 |
501 |
6100 |
0.61 |
5.3 |
1.2 |
0.7 |
730 |
162 |
| H2O |
375 |
|
513 |
7200 |
0.33 |
3.7 |
0.9 |
1.8 |
|
|
|
4M
|
EtOH |
397 |
|
565 |
7500 |
0.45 |
7.4 |
0.6 |
0.7 |
|
|
| MeCN |
385 |
33000 |
568 |
8400 |
0.38 |
7.9 |
0.5 |
0.8 |
760 |
134 |
| H2O |
376 |
|
568 |
9000 |
0.03 |
—b |
— |
— |
|
|
|
5M
|
EtOH |
599 |
|
624 |
6700 |
0.40 |
2.2 |
1.8 |
2.7 |
|
|
| MeCN |
593 |
50000 |
617 |
6600 |
0.56 |
2.9 |
1.9 |
1.5 |
740 |
4560 |
| H2O |
594 |
|
620 |
7100 |
0.13 |
1.1 |
1.2 |
7.9 |
|
|
Two-photon absorption
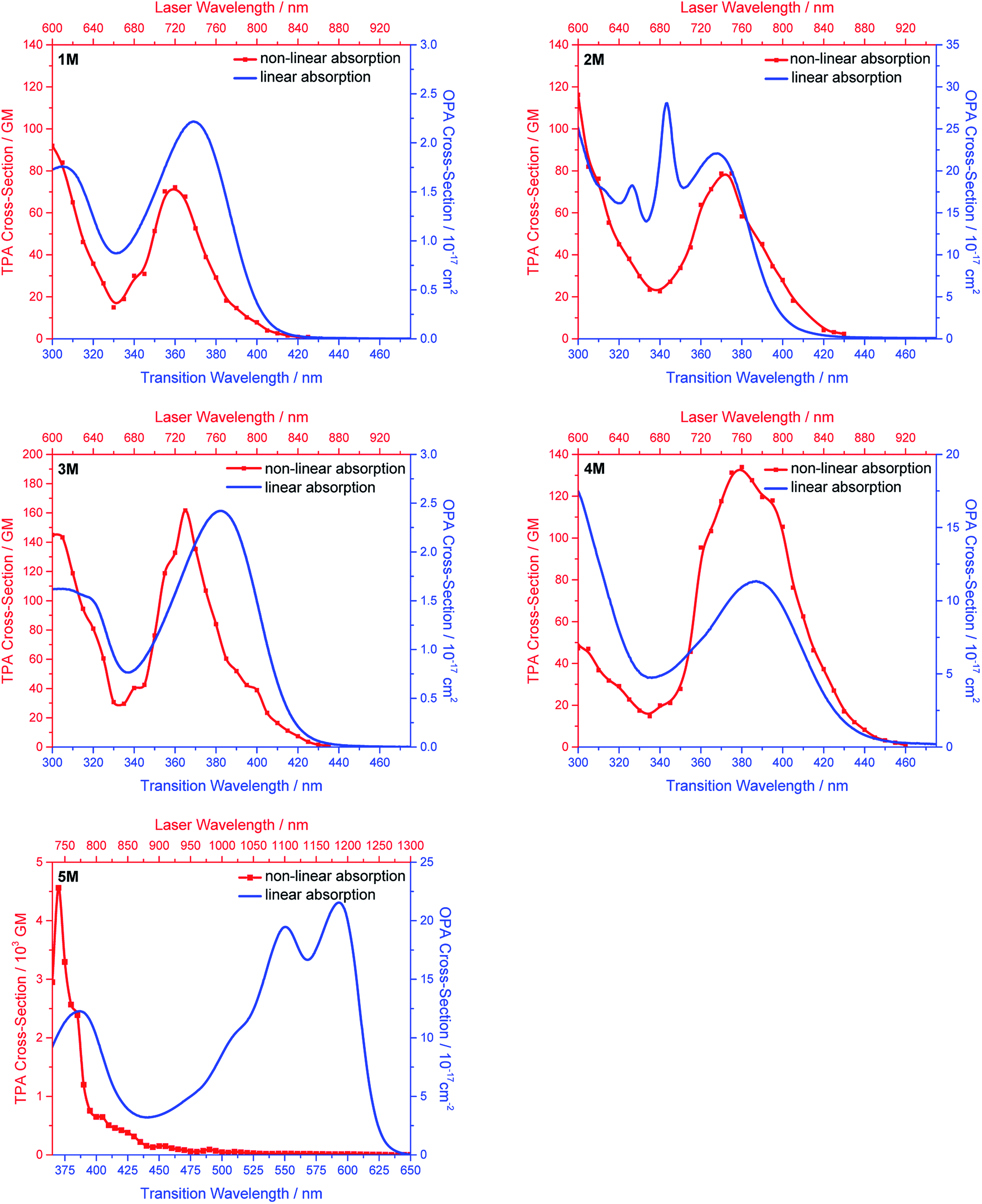
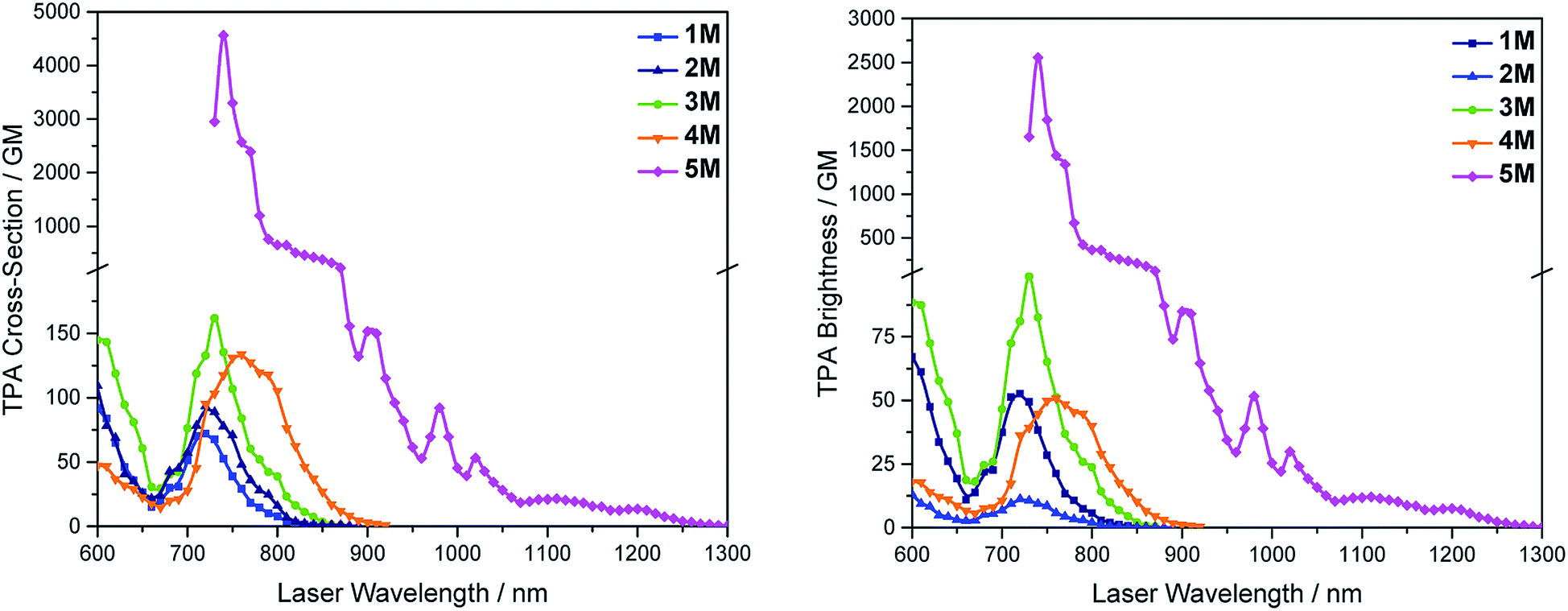
We measured the two-photon absorption spectra of 1M–5M in MeCN, as the polarity within cells is more similar to MeCN than to water,134,135 using the two-photon excited fluorescence technique. Following the electronic selection rules for centrosymmetric molecules (Ci symmetry), 1M, 2M and 5M, the TPA maximum does not occur at the one-photon absorption maximum, as the S1 ← S0 transition (Au ← Ag symmetry) is symmetry forbidden for two-photon absorption, but is located at higher energy where TPA allowed transitions of Ag ← Ag symmetry occur (S2 ← S0 transition for 1M and 5M and S3 ← S0 transition for 2M). The other two molecules 3M and 4M have C2v as their highest possible symmetry, thus lacking an inversion centre, which is why all transitions are one- and two-photon allowed. Therefore, we applied Ci symmetry for 1M, 2M and 5M and performed TD-DFT calculations (Table 4, red), to obtain more insight into which transitions are one- and or two-photon allowed. In Fig. 6, in which the TPA and rescaled one-photon absorption (OPA) are compared, for 1M we observed a TPA maximum at 720 nm, corresponding to S2 ← S0 which has Ag symmetry, and therefore is two-photon allowed but one-photon forbidden. The first excited state is, however, slightly TPA allowed as indicated by the shoulder, because of vibrational coupling of the Au state with an au vibrational mode, which makes the overall wavefunction gerade.136,137 From calculations on 2M in Ci symmetry, we can assign the two-photon allowed transition to the S3 ← S0 transition (Ag ← Ag symmetry). However, the experimental spectrum shows a TPA maximum at 750 nm, which is at the energy of the S1 ← S0 transition. This transition has a very small oscillator strength (f = 0.005) and is therefore not observable in the OPA spectrum, although the first excited state is ungerade. It might be TPA allowed because of vibrational coupling of the 1Au and/or 2Au state with an au vibrational mode, as the energy difference between S1 and S3 is only 1936 cm−1.136,137 Compound 5M shows a TPA maximum at 740 nm, which correlates with the second excited state. This state is gerade and therefore the transition should be only TPA allowed. Excitation to the ungerade first excited state is forbidden for the two photon process, and is therefore only observed in the OPA spectrum at 600 nm. Vibrational coupling in this molecule is very unlikely as the first and second excited states are separated by 8308 cm−1.136,137 The two other molecules 3M and 4M have no inversion centres; therefore, all transitions are both OPA and TPA allowed. However, the transitions have different oscillator strengths. For compound 3M the TPA maximum at 730 nm corresponds to the S2 state, which has a higher oscillator strength for TPA than OPA. The S1 state has a lower oscillator strength for TPA and is indicated by the shoulder. For compound 4M the TPA maximum at 760 nm corresponds to the S2 state, but the transition to the first excited state is also highly allowed. Therefore, the two-photon cross-sections for both transitions are not very different, while the one-photon cross-sections are. In the following discussion, we use the geometry-optimized structures to explain the magnitudes of the TPA cross-sections. As expected, the biphenyl compound 1M has the smallest TPA cross-section, being 72 GM, as this compound is the most twisted in its ground state structure (Fig. 7 and 8). The twist angles between the xylylene and phenylene rings are ca. 37° and the angle between the two phenyl rings of the flexible biphenyl bridge is also 37° (Table S13‡). Conjugation within the π-system in 1M is less efficient due to the rotational degree of freedom around the central C–C bond and, therefore, the TPA cross-section is reduced. With increasing planarity in 2M–4M, in which two phenyl rings are rigidified by incorporation into the pyrene, fluorene or carbazole moieties, the two-photon cross-sections are increased to 79, 162, and 134 GM, respectively. The ground state structures show that the angles between the xylylene group and the π-bridge are again ca. 37° for the three compounds, while the two “phenylene” rings have no twist at all, as they are constrained. To explain the rather different results for the three compounds, we must discuss the different ways, beside co-planarity, to improve the two-photon absorption cross-section. It was shown that increasing the length or efficiency of a conjugated system leads to an enhanced two-photon absorption cross-section.49–53 Therefore, compound 4M should have the smallest value, as the xylylene groups are linked via the 3,6-positions rather than the 2,7-positions of the π-bridge, which leads to a less efficient conjugation. Furthermore, increasing intramolecular charge transfer enhances the two-photon absorption cross-section.49–53 It is known that A–π–D–π–A systems are more efficient than A–π–A systems.49–53 The donor-strength of the π-bridge can be correlated with the HOMO energy, which rises from pyrene (−7.19 eV (HOMO−1)) to fluorene (−6.90 eV) to carbazole (−6.75 eV). In the case of pyrene, the HOMO−1 must be considered as the HOMO has a nodal plane at the 2,7-positions. Therefore, the two-photon cross-section should be enhanced from pyrene 2M, to fluorene 3M to carbazole 4M. The latter one 4M, however, has a shorter conjugation length than 3M, and the effect of the reduced conjugation (vide supra) lowers the TPA cross-section. The dithienyl-diketopyrrolopyrrole compound 5M has by far the highest two-photon absorption value, being 4560 GM. Its conjugated π-system is elongated, and the calculated ground state structure is almost planar.118 The angles between the xylylene and the thiophene groups are 15°, and thus much smaller than in the other compounds, and the twist between the thiophenes and the diketopyrrolopyrrole is only 2–4°. Furthermore, the two-photon brightness shows the same trend, with the exception that compound 1M has a higher value than 2M and 4M, due to its much higher fluorescence quantum yield (Fig. 7). Overall, the two-photon brightness of 5M is exceptionally high, being 2545 GM in MeCN. Furthermore, we compared the two-photon absorption of 5M with its boron-free analogue 5A. While the linear optical properties (absorption and emission) of 5M were only slightly red-shifted compared to those of 5A, the TPA data are quite different. The two-photon absorption cross-section of 5M is ca. 8.7 times higher than that of its analogue 5A in DMSO solution (Fig. S4‡). These data show that our boron-based acceptor moiety strongly enhances the TPA properties.
Table 4 TD-DFT-calculated photophysical data for 1M–5M at the CAM-B3LYP/6-31G(d) level in watera
|
Black: without symmetry constraints, red: in Ci symmetry.
Components with greater than 10% contribution shown. Percentage contribution approximated by 2 × (ci)2 × 100%, where ci is the coefficient for the particular ‘orbital rotation’.
|
|
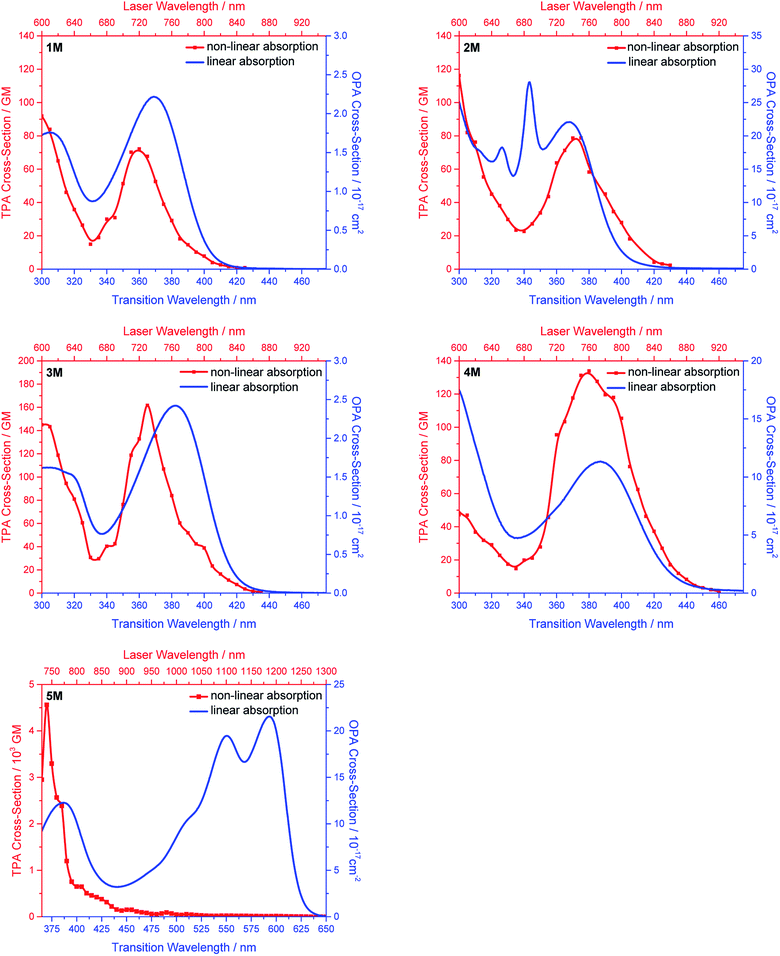 |
| | Fig. 6 One-photon absorption (blue) and two-photon absorption spectra (red) of 1M (first row, left), 2M (first row, right), 3M (second row, left), 4M (second row, right) and 5M (third row, left) in MeCN. | |
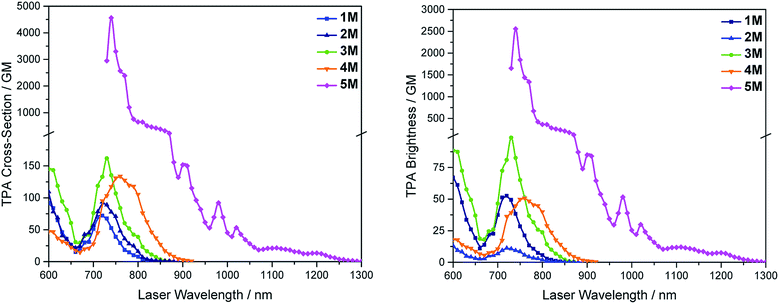 |
| | Fig. 7 Two-photon absorption spectra (left) and two-photon brightness (σ2Φf) (right) of 1M–5M in MeCN. | |
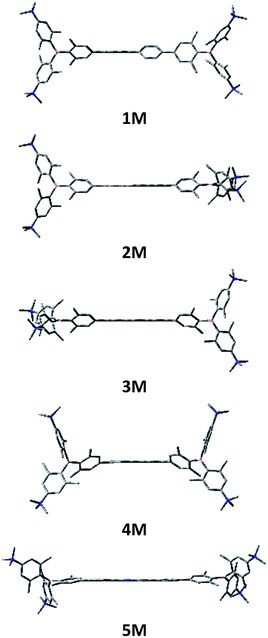 |
| | Fig. 8 Side view of the geometries of the DFT-optimized S0 states of 1M–5M at the B3LYP/6-31G(d) level of theory. Atom colour code: carbon (grey), boron (pink), sulphur (yellow), nitrogen (blue), oxygen (red). Hydrogen atoms are omitted for clarity. | |
Imaging
As none of the neutral compounds 1–5 are soluble in Dulbecco's Modified Eagle Medium (DMEM), they formed nanoparticles in that medium and were not taken up by HeLa or HepG2 cells (Fig. S6‡). In addition, pre-mixing of chromophores 1–5 with Pluronic F-127 was not successful. The compounds did dissolve in that medium, yet no cellular uptake was observable (Fig. S7‡).
Thus, the methylated species 1M–5M, designed to be water-soluble, were used for cell imaging. We treated HeLa cells with 500 nM concentrations of all 5 compounds. Visualization with a confocal laser scanning fluorescence microscope showed cellular uptake for 1M–5M (Fig. 9 and S8–S11‡). Co-staining experiments with commercially available LysoTrackers™ confirmed their localization in acidic intracellular compartments such as endosomes and lysosomes. The Pearson values (Rr), indicative of the degree of co-localization, were all higher than 0.73, while for 4M and 5M values of 0.83 and 0.81, respectively, were reached. Furthermore, cell viability experiments were performed to investigate the potential of chromophores 1M–5M for live-cell imaging. HeLa cells were treated with serial dilutions of 1M–5M and the cell metabolic activity was studied using a colourimetric (MTT) assay (Fig. S13‡). These confirmed that compounds 1M–5M did not influence the cell viability at concentrations as high as 5 μM after 24 h incubation time, and some of them were non-toxic to cells even at higher concentrations (10 μM). As the dithienyl-diketopyrrolopyrrole dye 5M has the most red-shifted absorption and emission bands and, by far, the highest TPA cross-section and brightness, we focused on this compound for further imaging experiments.
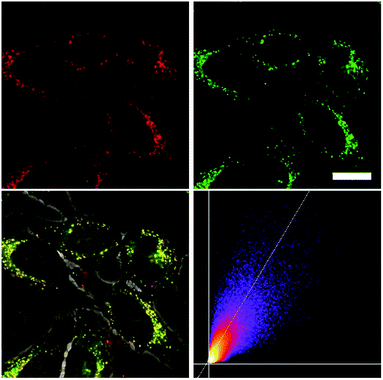 |
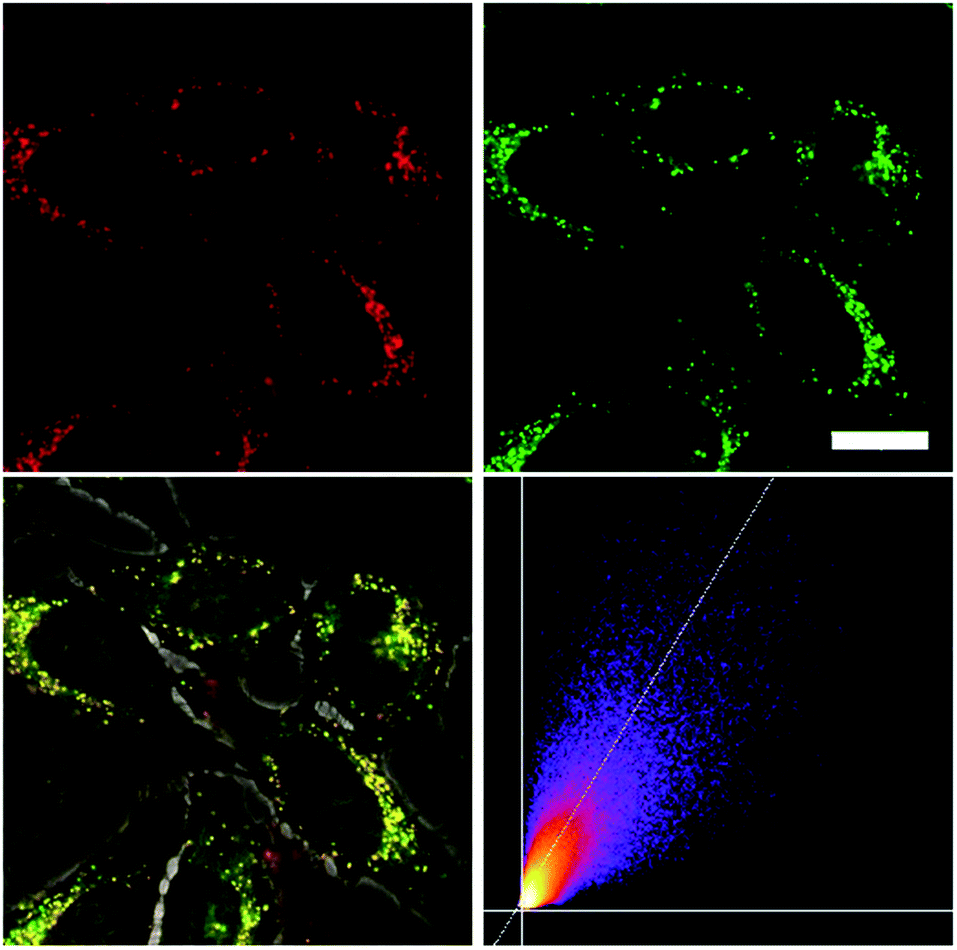
| | Fig. 9 Co-staining experiment of HeLa cells with 5M and LysoTracker™ Green. The cells were loaded with 5M (500 nM, 2 h) and LysoTracker™ Green (100 nM, 20 min) under 5% CO2 at 37 °C. Fluorescence images of 5M (top, left, λex = 559 nm; λem = 570–670 nm) and LysoTracker™ Green (top, right, λex = 473 nm; λem = 490–540 nm). The merged image of the bright field image and both fluorescence images (bottom, left), and the correlation plot of the intensities (bottom, right, Rr = 0.81), show co-localization of the dye 5M in the lysosomes. Scale bar: 20 μM. | |
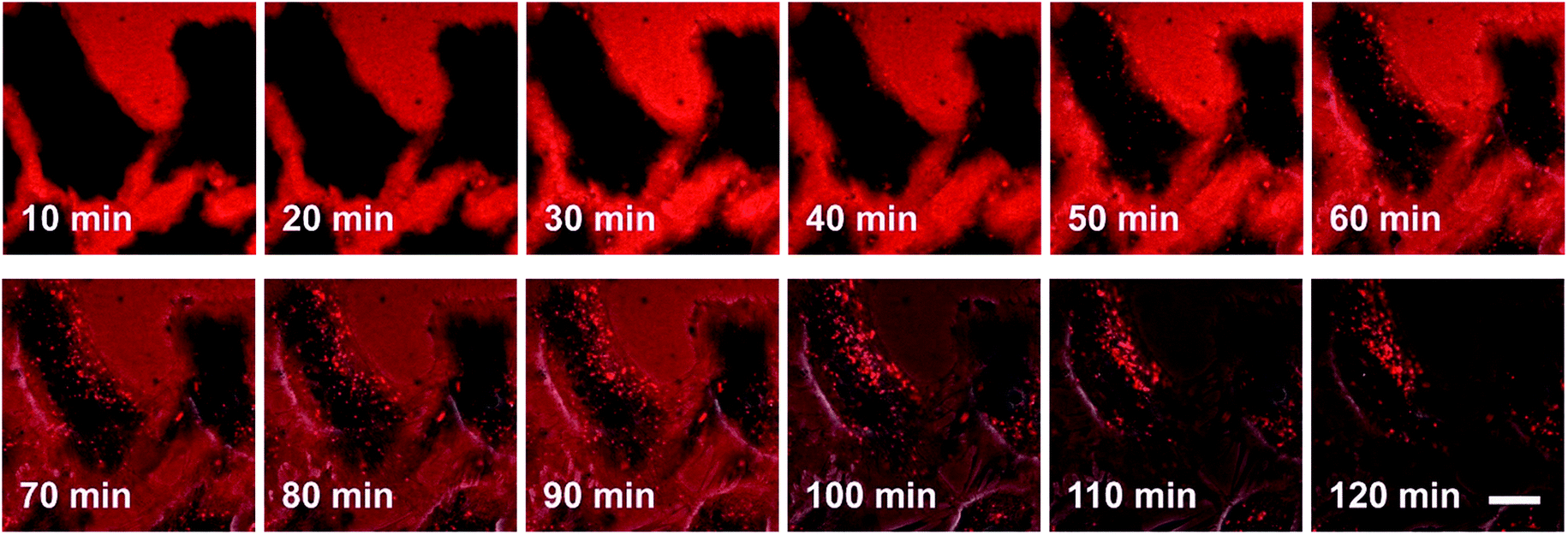
The process by which cell internalization of our dye 5M takes place was observed via time-lapse confocal microscopy for 2 h (Fig. 10). The cultured medium of HeLa cells was replaced with the dye-containing DMEM and images were recorded every 10 min without any washing process. The dye first attaches to the cell membrane and, after 50 min, small bright spots in the intracellular region are observed. With further incubation time, additional chromophore enters the cell and the signal-to-noise ratio improves as the residual dye in the medium is consumed. Furthermore, HeLa cells were stained at 4 °C or at 37 °C with the presence of 0.1% NaN3 (Fig. S12‡); both sets of conditions inhibit endocytosis. Both experiments showed much lower fluorescence intensity in comparison with the control experiment, therefore suggesting endocytosis as the mechanism by which 5M enters cells.
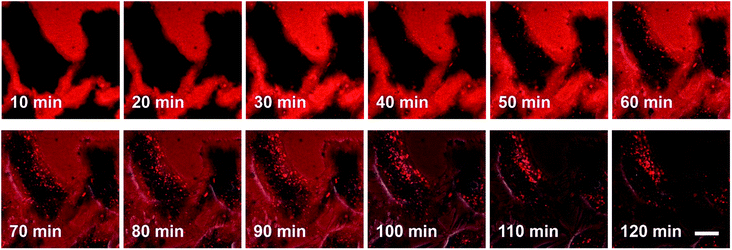 |
| | Fig. 10 Confocal microscope images of HeLa cells at 37 °C with 5M (500 nM). Merged bright field image with fluorescence image (λex = 559 nm; λem = 570–670 nm) between 10 min and 120 min after staining. Scale bar: 20 μm. | |
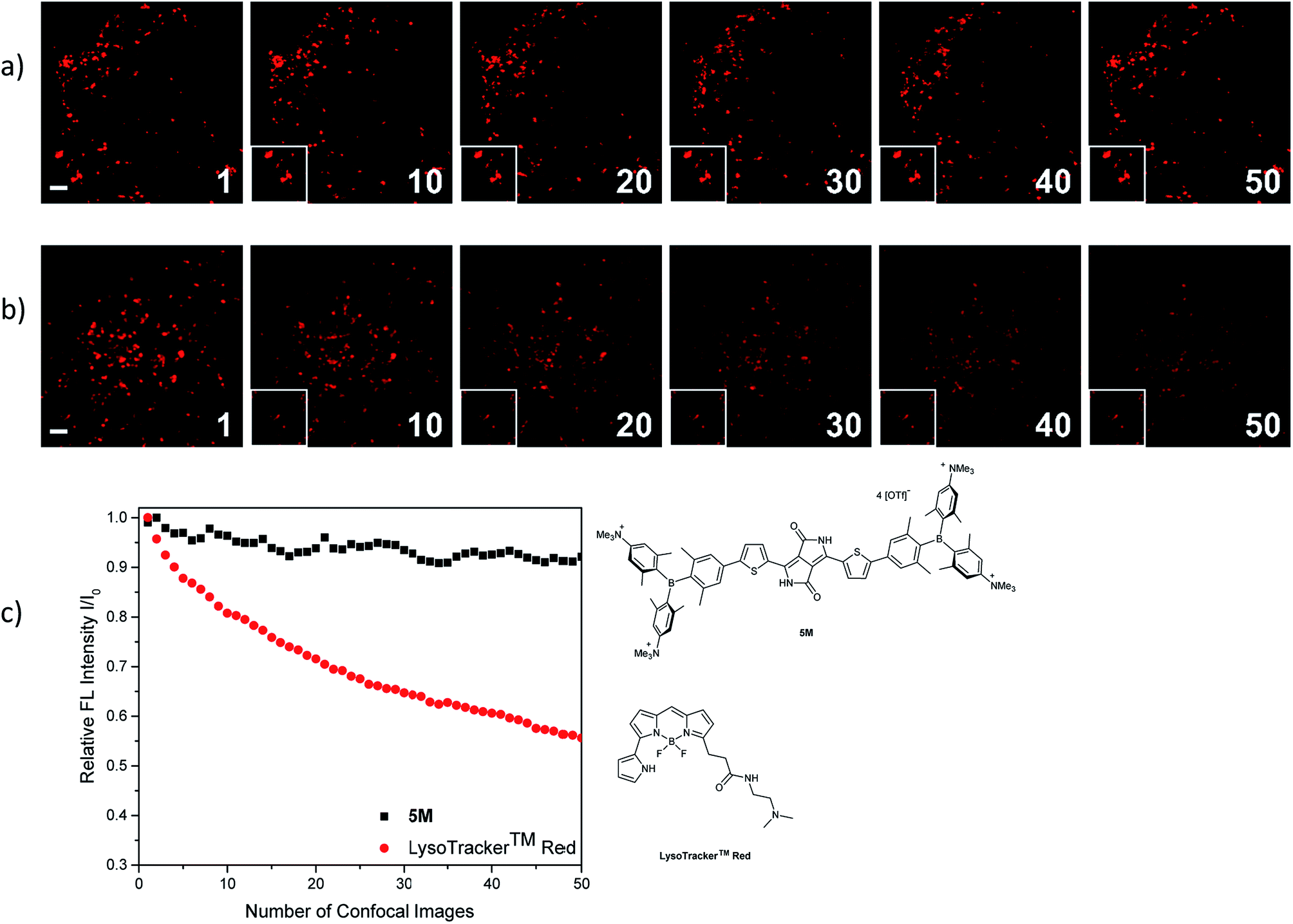
The photostability of our dye 5M was tested by repetitive imaging of HeLa cells after staining. Over 95% of the initial fluorescence intensity of 5M was retained after irradiation with an excitation laser at 561 nm for 50 images. When using the commercially available LysoTracker™ Red under the same imaging conditions, the emission intensity decreased by 45% (Fig. 11). This result revealed the outstanding photostability of 5M, which is highly desired for time-lapse imaging of live cells.
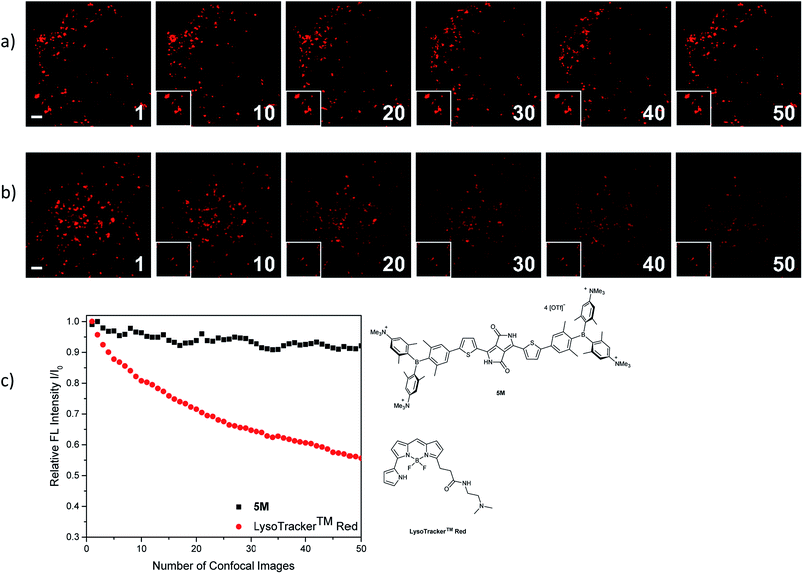 |
| | Fig. 11 Comparison of the repeated fluorescence images of HeLa cells stained with (a) 5M and (b) LysoTracker™ Red under irradiation with a confocal laser at 561 nm (WLL, output power 70%, AOTF 2%). Each number indicates the number of recorded confocal images. The rectangle (bottom, left in each picture) is the cutout of the image at n = 1. Scale bar: 2 μm. (c) Plots of integrated fluorescence intensities (I) relative to the initial value (I0) as a function of the number of recorded images. | |
As chromophore 5M has an outstanding two-photon brightness of 2545 GM (at 740 nm) and showed good imaging and cell viability properties, we also tested 5M as a two-photon excited fluorescence dye to stain HeLa cells. The two-photon imaging experiments were performed at 500 nM concentration. As clearly shown in Fig. 12, dye 5M stained the cell at the lysosomes and is a very effective two-photon imaging agent.
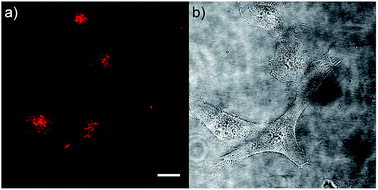 |
| | Fig. 12 (a) Two-photon excited fluorescence and (b) bright field images of HeLa cells stained with 5M (500 nM). The TPEF image was recorded under excitation at 720 nm (AOTF 38%) using an HyD detector with a bandpass filter 650/50 and an HC Fluotar L 25×0.95 W VISIR objective. Scale bar: 20 μm. | |
Conclusion
We have synthesized a series of quadrupolar A–π–A chromophores with five different π-bridges, namely biphenyl, pyrene, fluorene, carbazole and dithienyl-diketopyrrolopyrrole all containing triarylborane π-acceptor groups functionalized to enhance water solubility. While the neutral precursor molecules 1–5 are not water-soluble and are not taken up by the cells, they show interesting photophysical properties. Except for compound 5, the neutral dyes are highly solvatochromic as the excited state results from charge transfer from the nitrogens to the boron atoms. As the N,N-dimethylaminoxylyl group is present in all of the neutral chromophores, the photophysical properties of those compounds are very similar. After methylation of the four nitrogen atoms, this charge transfer is no longer possible and, therefore, the tetracationic compounds 1M–5M display completely different optical properties. The emission of the different cationic compounds covers nearly the entire visible spectrum depending on the selected π-bridge. The colour can be tuned from blue to pink over a range of 5300 cm−1, while the emission spectra of compounds themselves are not solvatochromic. The TPA cross-sections correlate with the planarity of the π-bridge, thus, the more planar the π-bridge, the higher the two-photon absorption cross-section. The two-photon absorption cross-section also correlates well with the donor ability of the π-bridge and the length of the conjugated π-system. Our dithienyl-diketopyrrolopyrrole dye 5M has the highest two-photon cross-section of 4560 GM in MeCN and due to its high fluorescence quantum yield, also exhibits a remarkable two-photon brightness of 2545 GM.
We carried out live-cell imaging with all five cationic compounds 1M–5M. All of our cationic dyes were taken up by the HeLa cells and localize at the lysosomes. Furthermore, they do not show any effect on cell viability up to concentrations of 5 μM, which is much higher than the concentration needed for imaging purposes (500 nM). Further experiments were performed with compound 5M, which shows the most red-shifted absorption and emission, the highest TPA cross-section, no cell toxicity up to 20 times the staining concentration, and a very good co-localization pattern (Rr = 0.81) with the lysosomes. We showed that this dye is taken up by the endocytosis pathway of the HeLa cells, that compound 5M is far more photostable than the commercially available LysoTracker™ Red, and that it is an excellent dye for TPEF imaging. In summary, we have designed and synthesized a series of fluorescent three-coordinate boron-containing quadrupolar dyes for one- and two-photon excited fluorescence imaging of lysosomes. Incorporation of our boron acceptor groups greatly enhances the TPA cross-sections and, via tuning of the π-bridge, we obtained two-photon absorption cross-sections up to 4560 GM and a two-photon brightness up to 2545 GM in MeCN, which are by far the highest values reported for a lysosomal imaging dye.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We are grateful for generous financial support by the Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid”, the Deutsche Forschungsgemeinschaft (DFG) (GRK 2112) and the Julius-Maximilians-Universität Würzburg. This work is partly supported by JSPS KAKENHI grant 18H03909, 18H05261, and JP16H06280 (Advanced Bioimaging Support). The authors also gratefully acknowledge the computer and data resources provided by the Leibniz Supercomputing Centre (http://www.lrz.de). We thank Prof. D. J. Tozer (Durham University) for helpful discussions regarding the TD-DFT calculations.
Notes and references
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- M. Elbing and G. C. Bazan, Angew. Chem., Int. Ed., 2008, 47, 834–838 CrossRef CAS PubMed.
- Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed.
- Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC.
- E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- X. Yin, J. Chen, R. A. Lalancette, T. B. Marder and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 9761–9765 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, M. Haehnel, M. Wehner, A. Eichhorn, L. Mailänder, M. Meier, J. Brand, F. Brede, K. Müller-Buschbaum, H. Braunschweig and T. B. Marder, Chem. Sci., 2015, 6, 5922–5927 RSC.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- T. W. Hudnall, C.-W. Chiu and F. P. Gabbaï, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958–3984 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- Y. Chen, D. Cao, S. Wang, C. Zhang and Z. Liu, J. Mol. Struct., 2010, 969, 182–186 CrossRef CAS.
- J. Ohshita, Y. Tominaga, D. Tanaka, Y. Ooyama, T. Mizumo, N. Kobayashi and H. Higashimura, Dalton Trans., 2013, 42, 3646–3652 RSC.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Eur. J. Org. Chem., 2017, 2017, 4552–4561 CrossRef CAS.
- D. Mutaguchi, K. Okumoto, Y. Ohsedo, K. Moriwaki and Y. Shirota, Org. Electron., 2003, 4, 49–59 CrossRef CAS.
- G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS.
- Z. M. Hudson, C. Sun, M. G. Helander, Y.-L. Chang, Z.-H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930–13933 CrossRef CAS PubMed.
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231–15235 CrossRef CAS PubMed.
- I. S. Park, M. Numata, C. Adachi and T. Yasuda, Bull. Chem. Soc. Jpn., 2016, 89, 375–377 CrossRef CAS.
- Y. Liu, G. Xie, K. Wu, Z. Luo, T. Zhou, X. Zeng, J. Yu, S. Gong and C. Yang, J. Mater. Chem. C, 2016, 4, 4402–4407 RSC.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- Y. H. Lee, S. Park, J. Oh, J. W. Shin, J. Jung, S. Yoo and M. H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 24035–24042 CrossRef CAS PubMed.
- J. E. Leffler, G. B. Watts, T. Tanigaki, E. Dolan and D. S. Miller, J. Am. Chem. Soc., 1970, 92, 6825–6830 CrossRef CAS.
- T. Noda and Y. Shirota, J. Am. Chem. Soc., 1998, 120, 9714–9715 CrossRef CAS.
- M. Kinoshita and Y. Shirota, Chem. Lett., 2001, 30, 614–615 CrossRef.
- A. Shuto, T. Kushida, T. Fukushima, H. Kaji and S. Yamaguchi, Org. Lett., 2013, 15, 6234–6237 CrossRef CAS PubMed.
- G. Turkoglu, M. E. Cinar and T. Ozturk, Molecules, 2017, 22, 1522 CrossRef PubMed.
- W. Kaim and A. Schulz, Angew. Chem., Int. Ed., 1984, 23, 615–616 CrossRef.
- W. Kaim, N. S. Hosmane, S. Záliš, J. A. Maguire and W. N. Lipscomb, Angew. Chem., Int. Ed., 2009, 48, 5082–5091 CrossRef CAS PubMed.
- L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS PubMed.
- J. Merz, J. Fink, A. Friedrich, I. Krummenacher, H. H. Al Mamari, S. Lorenzen, M. Haehnel, A. Eichhorn, M. Moos, M. Holzapfel, H. Braunschweig, C. Lambert, A. Steffen, L. Ji and T. B. Marder, Chem.–Eur. J., 2017, 23, 13164–13180 CrossRef CAS PubMed.
- L. Ji, I. Krummenacher, A. Friedrich, A. Lorbach, M. Haehnel, K. Edkins, H. Braunschweig and T. B. Marder, J. Org. Chem., 2018, 83, 3599–3606 CrossRef CAS PubMed.
- J. C. Doty, B. Babb, P. J. Grisdale, M. Glogowski and J. L. R. Williams, J. Organomet. Chem., 1972, 38, 229–236 CrossRef CAS.
- Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- J. C. Collings, S.-Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L.-O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS PubMed.
- L. Weber, D. Eickhoff, T. B. Marder, M. A. Fox, P. J. Low, A. D. Dwyer, D. J. Tozer, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Chem.–Eur. J., 2012, 18, 1369–1382 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed.
- Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Steffen, L. E. Longobardi, D. W. Stephan, C. Lambert and T. B. Marder, Chem. Sci., 2015, 6, 308–321 RSC.
- S.-Y. Li, Z.-B. Sun and C.-H. Zhao, Inorg. Chem., 2017, 56, 8705–8717 CrossRef CAS PubMed.
- M. Ito, E. Ito, M. Hirai and S. Yamaguchi, J. Org. Chem., 2018, 83, 8449–8456 CrossRef CAS PubMed.
- M. Meier, L. Ji, J. Nitsch, I. Krummenacher, A. Deißenberger, D. Auerhammer, M. Schäfer, T. B. Marder and H. Braunschweig, Chem.–Eur. J., 2019, 25, 4707–4712 CrossRef CAS PubMed.
- H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed.
- C. C. Jiménez, A. Enríquez-Cabrera, O. González-Antonio, J. Ordóñez-Hernández, P. G. Lacroix, P. Labra-Vázquez, N. Farfán and R. Santillan, Inorganics, 2018, 6, 131 CrossRef.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641–4678 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
-
M. Rumi, S. Barlow, J. Wang, J. W. Perry and S. R. Marder, in Photoresponsive Polymers I, ed. S. R. Marder and K.-S. Lee, Springer, Berlin, 2008, vol. 213, pp. 1–95 Search PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- H. M. Kim and B. R. Cho, Chem. Commun., 2009, 153–164 Search PubMed.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I. Y. S. Lee, D. McCord-Maughon, J. Qin, H. Röckel, M. Rumi, X.-L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51 CrossRef CAS.
- S. Kawata, H.-B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697 CrossRef CAS PubMed.
- W. Zhou, S. M. Kuebler, K. L. Braun, T. Yu, J. K. Cammack, C. K. Ober, J. W. Perry and S. R. Marder, Science, 2002, 296, 1106–1109 CrossRef CAS PubMed.
- S. Juodkazis, V. Mizeikis and H. Misawa, J. Appl. Phys., 2009, 106, 051101 CrossRef.
- D. A. Parthenopoulos and P. M. Rentzepis, Science, 1989, 245, 843–845 CrossRef CAS PubMed.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS PubMed.
- W. Dallari, M. Scotto, M. Allione, E. Samoylova, F. Pignatelli, R. Cingolani, A. Athanassiou and A. Diaspro, Microelectron. Eng., 2011, 88, 3466–3469 CrossRef CAS.
- G. S. He, J. D. Bhawalkar, C. F. Zhao and P. N. Prasad, Appl. Phys. Lett., 1995, 67, 2433–2435 CrossRef CAS.
- J. E. Ehrlich, X. L. Wu, I. Y. S. Lee, Z. Y. Hu, H. Röckel, S. R. Marder and J. W. Perry, Opt. Lett., 1997, 22, 1843–1845 CrossRef CAS PubMed.
- C. W. Spangler, J. Mater. Chem., 1999, 9, 2013–2020 RSC.
- G. S. He, R. Signorini and P. N. Prasad, Appl. Opt., 1998, 37, 5720–5726 CrossRef CAS PubMed.
- A. Abbotto, L. Beverina, R. Bozio, S. Bradamante, C. Ferrante, G. A. Pagani and R. Signorini, Adv. Mater., 2000, 12, 1963–1967 CrossRef CAS.
- W. G. Fisher, W. P. Partridge, C. Dees and E. A. Wachter, Photochem. Photobiol., 1997, 66, 141–155 CrossRef CAS PubMed.
- Y. Shen, A. J. Shuhendler, D. Ye, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2016, 45, 6725–6741 RSC.
- W. Denk, J. Strickler and W. Webb, Science, 1990, 248, 73–76 CrossRef CAS PubMed.
- P. Ning, W. Wang, M. Chen, Y. Feng and X. Meng, Chin. Chem. Lett., 2017, 28, 1943–1951 CrossRef CAS.
- Q. Zhang, X. Tian, H. Zhou, J. Wu and Y. Tian, Materials, 2017, 10, 223 CrossRef PubMed.
- S. Yao and K. D. Belfield, Eur. J. Org. Chem., 2012, 2012, 3199–3217 CrossRef CAS.
- D. Kim, H. G. Ryu and K. H. Ahn, Org. Biomol. Chem., 2014, 12, 4550–4566 RSC.
- J. A. Mindell, Annu. Rev. Physiol., 2012, 74, 69–86 CrossRef CAS PubMed.
- C. Settembre, A. Fraldi, D. L. Medina and A. Ballabio, Nat. Rev. Mol. Cell Biol., 2013, 14, 283 CrossRef CAS PubMed.
- F. M. Platt, B. Boland and A. C. van der Spoel, J. Cell Biol., 2012, 199, 723–734 CrossRef CAS PubMed.
- H. M. Kim, M. J. An, J. H. Hong, B. H. Jeong, O. Kwon, J.-Y. Hyon, S.-C. Hong, K. J. Lee and B. R. Cho, Angew. Chem., Int. Ed., 2008, 47, 2231–2234 CrossRef CAS PubMed.
- J. H. Han, S. K. Park, C. S. Lim, M. K. Park, H. J. Kim, H. M. Kim and B. R. Cho, Chem.–Eur. J., 2012, 18, 15246–15249 CrossRef CAS PubMed.
- H. J. Kim, C. H. Heo and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 17969–17977 CrossRef CAS PubMed.
- L. Yuan, L. Wang, B. K. Agrawalla, S.-J. Park, H. Zhu, B. Sivaraman, J. Peng, Q.-H. Xu and Y.-T. Chang, J. Am. Chem. Soc., 2015, 137, 5930–5938 CrossRef CAS PubMed.
- H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486–17489 CrossRef CAS PubMed.
- H.-J. Lee, C.-W. Cho, H. Seo, S. Singha, Y. W. Jun, K.-H. Lee, Y. Jung, K.-T. Kim, S. Park, S. C. Bae and K. H. Ahn, Chem. Commun., 2016, 52, 124–127 RSC.
- B. Zhu, P. Li, W. Shu, X. Wang, C. Liu, Y. Wang, Z. Wang, Y. Wang and B. Tang, Anal. Chem., 2016, 88, 12532–12538 CrossRef CAS PubMed.
- J. Fan, Z. Han, Y. Kang and X. Peng, Sci. Rep., 2016, 6, 19562 CrossRef CAS PubMed.
- L. Zhou, Y. Liu, S. Hu, H. Wang, H. Sun and X. Zhang, Tetrahedron, 2016, 72, 4637–4642 CrossRef CAS.
- H. Chen, Y. Tang, H. Shang, X. Kong, R. Guo and W. Lin, J. Mater. Chem. B, 2017, 5, 2436–2444 RSC.
- W. Feng, Z. Mao, L. Liu and Z. Liu, Talanta, 2017, 167, 134–142 CrossRef CAS PubMed.
- W. Luo, H. Jiang, X. Tang and W. Liu, J. Mater. Chem. B, 2017, 5, 4768–4773 RSC.
- J. Huang, N. Li, Q. Wang, Y. Gu and P. Wang, Sens. Actuators, B, 2017, 246, 833–839 CrossRef CAS.
- Y. Liu, F. Meng, L. He, K. Liu and W. Lin, Chem. Commun., 2016, 52, 7016–7019 RSC.
- J. Jiang, X. Tian, C. Xu, S. Wang, Y. Feng, M. Chen, H. Yu, M. Zhu and X. Meng, Chem. Commun., 2017, 53, 3645–3648 RSC.
- J. H. Son, C. S. Lim, J. H. Han, I. A. Danish, H. M. Kim and B. R. Cho, J. Org. Chem., 2011, 76, 8113–8116 CrossRef CAS PubMed.
- M. Tian, Y. Sun, L. Guo, R. Zhang, G. Zhang, R. Feng, X. Li, H. Zhang, L. He, X. Yu and X. He, Sens. Actuators, B, 2017, 243, 955–962 CrossRef CAS.
- X. Wang, D. M. Nguyen, C. O. Yanez, L. Rodriguez, H.-Y. Ahn, M. V. Bondar and K. D. Belfield, J. Am. Chem. Soc., 2010, 132, 12237–12239 CrossRef CAS PubMed.
- S. Yao, H.-Y. Ahn, X. Wang, J. Fu, E. W. Van Stryland, D. J. Hagan and K. D. Belfield, J. Org. Chem., 2010, 75, 3965–3974 CrossRef CAS PubMed.
- C. D. Andrade, C. O. Yanez, M. A. Qaddoura, X. Wang, C. L. Arnett, S. A. Coombs, J. Yu, R. Bassiouni, M. V. Bondar and K. D. Belfield, J. Fluoresc., 2011, 21, 1223–1230 CrossRef CAS PubMed.
- C. S. Lim, S. T. Hong, S. S. Ryu, D. E. Kang and B. R. Cho, Chem.–Asian J., 2015, 10, 2240–2249 CrossRef CAS PubMed.
- A. L. Capodilupo, V. Vergaro, E. Fabiano, M. De Giorgi, F. Baldassarre, A. Cardone, A. Maggiore, V. Maiorano, D. Sanvitto, G. Gigli and G. Ciccarella, J. Mater. Chem. B, 2015, 3, 3315–3323 RSC.
- S. Chen, M. Zhao, J. Su, Q. Zhang, X. Tian, S. Li, H. Zhou, J. Wu and Y. Tian, Dyes Pigm., 2017, 136, 807–816 CrossRef CAS.
- Z. Yuan, N. J. Taylor, R. Ramachandran and T. B. Marder, Appl. Organomet. Chem., 1996, 10, 305–316 CrossRef CAS.
- M. Charlot, L. Porrès, C. D. Entwistle, A. Beeby, T. B. Marder and M. Blanchard-Desce, Phys. Chem. Chem. Phys., 2005, 7, 600–606 RSC.
- C. D. Entwistle, J. C. Collings, A. Steffen, L.-O. Pålsson, A. Beeby, D. Albesa-Jové, J. M. Burke, A. S. Batsanov, J. A. K. Howard, J. A. Mosely, S.-Y. Poon, W.-Y. Wong, F. Ibersiene, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, J. Mater. Chem., 2009, 19, 7532–7544 RSC.
- L. Ji, R. M. Edkins, L. J. Sewell, A. Beeby, A. S. Batsanov, K. Fucke, M. Drafz, J. A. K. Howard, O. Moutounet, F. Ibersiene, A. Boucekkine, E. Furet, Z. Liu, J.-F. Halet, C. Katan and T. B. Marder, Chem.–Eur. J., 2014, 20, 13618–13635 CrossRef CAS PubMed.
- C.-W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60–61 CrossRef CAS PubMed.
- Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 3363–3369 CrossRef CAS PubMed.
- T. Agou, M. Sekine, J. Kobayashi and T. Kawashima, Chem.–Eur. J., 2009, 15, 5056–5062 CrossRef CAS PubMed.
- K. C. Song, K. M. Lee, N. V. Nghia, W. Y. Sung, Y. Do and M. H. Lee, Organometallics, 2013, 32, 817–823 CrossRef CAS.
- X. Li, X. Guo, L. Cao, Z. Xun, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2014, 53, 7809–7813 CrossRef CAS PubMed.
- J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed.
- J. Liu, S. Zhang, C. Zhang, J. Dong, C. Shen, J. Zhu, H. Xu, M. Fu, G. Yang and X. Zhang, Chem. Commun., 2017, 53, 11476–11479 RSC.
- J. Liu, S. Li, S. Zhang, C. Shen, J. Zhu, G. Yang and X. Zhang, Sens. Actuators, B, 2018, 261, 531–536 CrossRef CAS.
- J. Liu, X. Guo, R. Hu, J. Xu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2015, 87, 3694–3698 CrossRef CAS PubMed.
- X. Guo, X. Zhang, S. Wang, S. Li, R. Hu, Y. Li and G. Yang, Anal. Chim. Acta, 2015, 869, 81–88 CrossRef CAS PubMed.
- B. Chen, G. Feng, B. He, C. Goh, S. Xu, G. Ramos-Ortiz, L. Aparicio-Ixta, J. Zhou, L. Ng, Z. Zhao, B. Liu and B. Z. Tang, Small, 2016, 12, 782–792 CrossRef CAS PubMed.
- J. Liu, C. Zhang, J. Dong, J. Zhu, C. Shen, G. Yang and X. Zhang, RSC Adv., 2017, 7, 14511–14515 RSC.
- J. Liu, C. Zhang, J. Dong, J. Zhu, C. Shen, G. Yang and X. Zhang, New J. Chem., 2017, 41, 4733–4737 RSC.
- S. Pagidi, N. K. Kalluvettukuzhy and P. Thilagar, Langmuir, 2018, 34, 8170–8177 CrossRef CAS PubMed.
- S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC.
- A. Nowak-Król, M. Grzybowski, J. Romiszewski, M. Drobizhev, G. Wicks, M. Chotkowski, A. Rebane, E. Górecka and D. T. Gryko, Chem. Commun., 2013, 49, 8368–8370 RSC.
- J. Schmitt, V. Heitz, A. Sour, F. Bolze, H. Ftouni, J.-F. Nicoud, L. Flamigni and B. Ventura, Angew. Chem., Int. Ed., 2015, 54, 169–173 CrossRef CAS PubMed.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658–5665 CrossRef CAS PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
- R. Stahl, C. Lambert, C. Kaiser, R. Wortmann and R. Jakober, Chem.–Eur. J., 2006, 12, 2358–2370 CrossRef CAS PubMed.
- E. Sakuda, Y. Ando, A. Ito and N. Kitamura, J. Phys. Chem. A, 2010, 114, 9144–9150 CrossRef CAS PubMed.
- A. Ito, K. Kawanishi, E. Sakuda and N. Kitamura, Chem.–Eur. J., 2014, 20, 3940–3953 CrossRef CAS PubMed.
- S. Amthor, C. Lambert, S. Dümmler, I. Fischer and J. Schelter, J. Phys. Chem. A, 2006, 110, 5204–5214 CrossRef CAS PubMed.
- H.-J. Jo, C.-B. Kim, T.-Y. Ryoo, B.-K. Ahn and K.-Y. Park, Bull. Korean Chem. Soc., 2010, 31, 3749–3754 CrossRef CAS.
- N. Agarwal, P. K. Nayak, F. Ali, M. P. Patankar, K. L. Narasimhan and N. Periasamy, Synth. Met., 2011, 161, 466–473 CrossRef CAS.
- B. Kobin, S. Behren, B. Braun-Cula and S. Hecht, J. Phys. Chem. A, 2016, 120, 5474–5480 CrossRef CAS PubMed.
- J. Cortes, H. Heitele and J. Jortner, J. Phys. Chem., 1994, 98, 2527–2536 CrossRef CAS.
- M. Li, J. Fan, H. Li, J. Du, S. Long and X. Peng, Biomaterials, 2018, 164, 98–105 CrossRef CAS PubMed.
- N. Jiang, J. Fan, F. Xu, X. Peng, H. Mu, J. Wang and X. Xiong, Angew. Chem., Int. Ed., 2015, 54, 2510–2514 CrossRef CAS PubMed.
- D. Scherer, R. Dörfler, A. Feldner, T. Vogtmann, M. Schwoerer, U. Lawrentz, W. Grahn and C. Lambert, Chem. Phys., 2002, 279, 179–207 CrossRef CAS.
- B. Strehmel, S. Amthor, J. Schelter and C. Lambert, ChemPhysChem, 2005, 6, 893–896 CrossRef CAS PubMed.
- A. D. Buckingham, Q. Rev., Chem. Soc., 1959, 13, 183–214 RSC.
-
D. J. Griffiths, in Introduction to Electrodynamics, Prentice Hall, Inc., New Jersey, 3rd edn, 1999, pp. 146–159 Search PubMed.
Footnotes |
| † Dedicated to Dr Jean-François Halet on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00793h |
| § We are aware that the dipole moment μ in our charged compounds is origin dependent and not an observable.138,139 For simplification, we use the term dipole moment to describe the electron density distribution in our charged compounds. Thus, the terms dipole and quadrupole are used accordingly. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Evripidis
Michail
b,
Chenguang
Wang
c,
Hiroaki
Ogasawara
a,
Evripidis
Michail
b,
Chenguang
Wang
c,
Hiroaki
Ogasawara