
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformer-dependent self-assembled metallacycles with photo-reversible response†
Mengqi
Li‡
a,
Li-Jun
Chen‡
b,
Zhipeng
Zhang
a,
Qianfu
Luo
a,
Hai-Bo
Yang
 *b,
He
Tian
a and
Wei-Hong
Zhu
*a
*b,
He
Tian
a and
Wei-Hong
Zhu
*a
aKey Laboratory for Advanced Materials, Institute of Fine Chemicals, Shanghai Key Laboratory of Functional Materials Chemistry, Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: whzhu@ecust.edu.cn
bShanghai Key Laboratory of Green Chemistry and Chemical Processes, Chang-Kung Chuang Institute, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: hbyang@chem.ecnu.edu.cn
First published on 25th March 2019
Abstract
Discrete, well-defined metallacycles and metallacages with stimuli-responsive behaviors have been largely predominated by the organic donor/metal acceptor paradigm with spontaneous formation of coordination bonds. However, light-driven self-assembly systems usually show relatively low utilization yield of photons and low fatigue resistance. Given that almost no example illustrates the different self-assembly behaviors of antiparallel and parallel conformers in the traditional photochromic diarylethene (DAE) system, here we have for the first time constructed a unique series of photoactive conformer-dependent metallacycles, focusing on the characterization and comparison of self-assembly behavior in different ligand conformers with different di-platinum(II) acceptors. Their photoswitchable scaffold sizes and shapes are precisely controlled by photochromically separable parallel or anti-parallel conformers via coordination-driven self-assembly. The ap-conformer and closed form provide larger bending angles upon coordination with di-Pt(II) acceptors into hexagon [6 + 6] or [3 + 3] while the p-conformer only can form smaller polygon cycles. Notably, in contrast with the non-photoactive parallel conformer, the reversible interconversion of anti-parallel ring-open and ring-closed conformer metallacycles can be achieved by alternate irradiation with UV and visible light, respectively, along with a relatively high conversion ratio and good fatigue resistance. This work provides a potential way to construct smart materials for use in sensing, catalysis and drug delivery systems.
Introduction
Coordination-driven self-assembly has provided a bottom-up basis for mimicking stimuli-responsive behaviors in artificial systems.1–13 Among external stimuli such as pH, redox potential and magnetic field,14–21 light is well considered as a perfect external trigger to control structural transformations in supramolecular entities since photon input is a clean and precise spatiotemporal control in a remote and nondestructive manner.22–28 Among the most ideal candidates, photochromic diarylethene (DAE) derivatives29–39 can undergo reversible transformation between open- and closed-ring states, along with light-induced changes in their optical and electronic properties. Recent efforts have been devoted to constructing light-driven supramolecular systems such as light-controlled interconversion between a self-assembled triangle and a rhombicuboctahedral sphere and uptake or release of anionic guests using a photochromic coordination cage.40,41 Recently, we have also achieved the construction of multibisthienylethene hexagons with photoreversible supramolecule-to-supramolecule interchanges between the specific photochromic ring-open and ring-closed states.23 However, due to the low-efficiency separation of anti-parallel and parallel conformers, DAE-based self-assemblies usually show relatively low quantum yield of photons. Meanwhile, a rare example has illustrated the different self-assembly behavior of anti-parallel and parallel conformers.40 The mixed configuration of anti-parallel and parallel conformers usually brings complexity and difficulties to the research. Understanding the different self-assembly behavior would also contribute to the construction of various supramolecular systems based on diarylethenes.Incorporation of the sterically hindered bisbenzo(thiadiazole) ethene bridge by our group can completely block the conformer interchange,42–45 the two conformers can be easily separated using a column chromatograph, and the resulting anti-parallel conformer exhibits high photocyclization quantum yields (Φo-c) as well as excellent thermal stability. With this in mind, here we report a unique series of conformer-dependent metallacycles based on sterically hindered ethene bridge diarylethenes as donor building blocks, resulting from self-assembly with different di-platinum(II) ligand acceptors. As a consequence, unusual discrete, well-defined supramolecular multidithienylethene metallacycles were established with several characteristics: (i) the conformer-dependent photoswitchable scaffold sizes and shapes precisely modulated by photochromic parallel or anti-parallel conformers via coordination-driven self-assembly, (ii) the uncommon conformer-based self-sorting supramolecular behavior, and (iii) the reversible transformation between anti-parallel and ring-closed conformers with a relatively high conversion ratio and good fatigue resistance via alternate irradiation with UV and visible light. We focused on characterizing the self-assembly behavior between different photoresponsive ligand conformers with different di-Pt(II) acceptors, particularly for comparing the photo-reversible response of self-assembled metallacycles.
Results and discussion
Conformer separation of photoswitchable ligands
For the traditional photochromic DAE series, the open form can be divided into two conformers: anti-parallel (ap-) configuration with C2 symmetry and parallel (p-) configuration with mirror symmetry. According to the Woodward–Hoffmann rule, only the ap-conformer can undergo photocyclization while the p-conformer cannot. Due to the typical rapid rotation of flexible aryl groups, two conformers cannot be separated efficiently in the typical photochromic DAE system. In fact, most DAE derivatives show relatively low photocyclization quantum yields which limits their practical device application.46–49 To solve the problem, our group has recently introduced a sterically hindered bisbenzo(thiadiazole) ethene bridge and large bulky benzothiophene unit to limit the rotation of the aryl group, even successfully separating ap- and p-conformers.42 Generally, light-controlled self-assemblies usually show relatively low fatigue resistance and quite low utilization yield of photons.19,50,51 For common DAE series, the mixture of ap- and p-conformers also limits the photoresponse efficiency.40 Hence, separation of two conformers of the DAE series by introducing a sterically hindered ethene bridge can be expected to make a breakthrough to the limitation.For developing photo-induced metallacycles via self-assembly behavior, we borrowed the sterically hindered ethene bridge DAE concept to develop a photoactive organic ligand donor (PY, Scheme 1). That is, we incorporated a bulky aryl group (pyridine-substituted benzothiophene) into the hindered ethene bridge of bisbenzo(thiadiazole). The target photoswitchable molecule PY was synthesized via Suzuki coupling between the corresponding dibromo substituted intermediate and pyridine borate. Taken together with the large bulky effect, the rotation between the ethene bridge and the side chain group is completely blocked due to the high rotation barrier, thus leading to the successful separation of ap- and p-conformers (ap-PY and p-PY, Scheme 1) by common silica gel chromatography. The closed form c-PY was prepared by UV irradiation of ap-PY.44
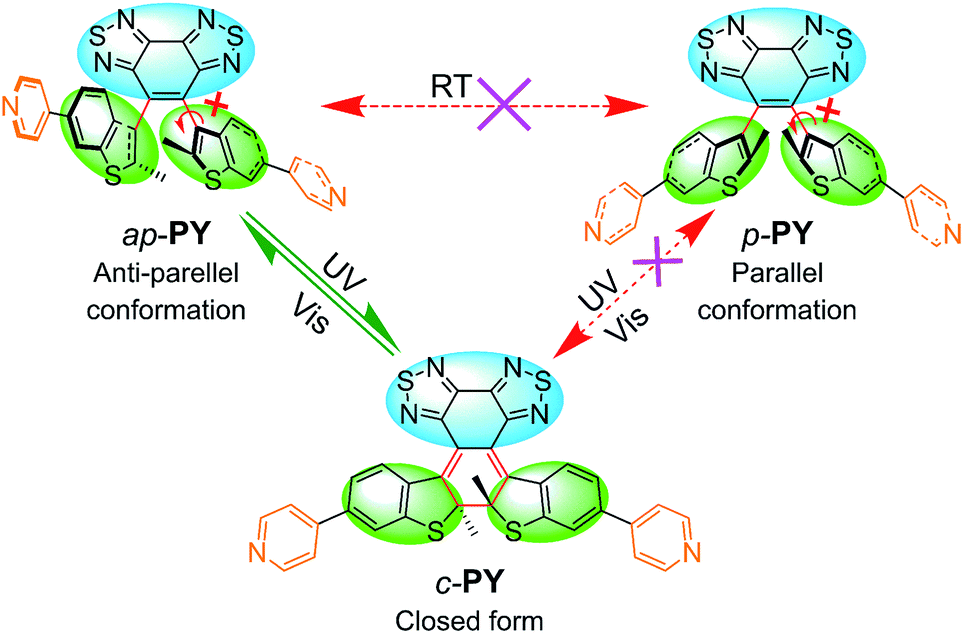 | ||
| Scheme 1 Chemical structures and isomerism of PY. The conversion relationship of ap-PY, p-PY and c-PY with a sterically hindered bisbenzo(thiadiazole) ethene bridge (blue unit) and bulky aryl group (green unit). Note: “ap” and “p” represent the anti-parallel and parallel conformation, respectively, while “c” represents the closed form. Two open forms are thermally isolated (red dashed arrows) due to blocked isomerization of high rotation barrier. The parallel ring open isomer is photochemically inert (red dashed arrows), and only the anti-parallel conformer is able to reversibly transform to the closed form (olive arrows), according to the Woodward–Hoffmann rule on electrochemical cyclization of a 4n + 2 π-electron system. | ||
Self-assembly of parallel conformers into triangle and rhombus metallacycles
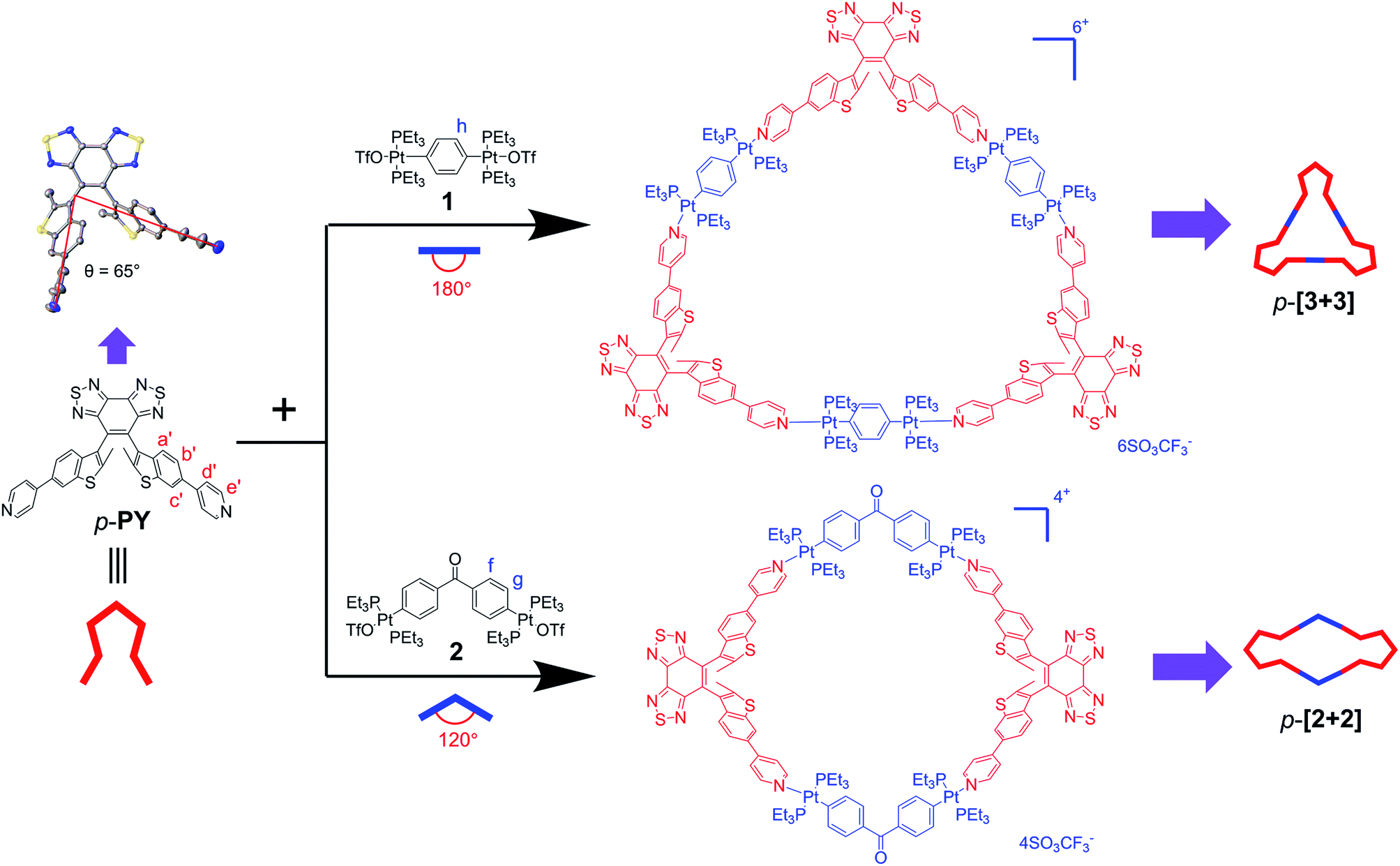
Due to the different conformation of these isomers, it is attractive enough to study the difference of self-assembly behavior with the three separated conformers ap-PY, p-PY and c-PY. According to the “directional bonding” model and the “symmetry interaction” model,1,52 the final geometry of the resulting individual two-dimensional polygons is dependent upon the value of turning angle with their angular components. That is, the shape and size of metallacycles have a critical effect upon different bending angles of photoresponsive conformer ligands and di-platinum acceptors. As a result of the successful separation of ap-PY, p-PY and c-PY, we have an opportunity to compare the self-assembly difference between ap- and p-conformers.Our previous study23 showed that for traditional diarylethenes, compared to parallel conformers, anti-parallel is the preferable conformation to self-assemble with di-platinum acceptors into a two-dimensional polygon. Here, given that we established successful separation of ap-PY and p-PY, we have an opportunity to study the self-assembly behavior of parallel conformers.
First, we focused on the self-assembly of p-PY with the diplatinum ligand di-Pt(II) 1 (Scheme 2). The triangle p-[3 + 3] can be easily obtained by stirring mixtures of p-PY with 180° di-Pt(II) acceptor 1 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in CD2Cl2 (Scheme S1†). The quantitative self-assembly formation of a discrete triangle of p-[3 + 3] was confirmed through multinuclear NMR (1H and 31P), showing a single discrete assembly with high symmetry (Fig. 1). In the simple 1H NMR pattern of p-[3 + 3], the pyridine protons showed a significant downfield shift (ca. 0.15 and 0.49 ppm for He′ and Hd′ respectively) with respect to those of p-PY due to the loss of electron density upon complexation of the pyridine N atom with the Pt(II) metal center. At the same time, the protons in the benzothiophene unit also exhibited a downfield shift for Ha′, Hb′ and Hc′, respectively (Fig. 1a). Moreover, the 31P NMR spectrum clearly displayed a single peak indicating a high symmetry structure of p-[3 + 3]. Meanwhile, an upfield shift was found from the corresponding starting platinum acceptor to metallacycles by ca. 8.05 ppm for p-[3 + 3], which can be ascribed to the increase in the electron density of the P atom upon coordination. This change as well as the decrease in the coupling of flanking 195Pt satellites (ΔJ = 152.5 Hz) is consistent with the electron back-donation from Pt (Fig. 1b). Here the well-defined signals of both 1H and 31P NMR fully support the formation of a perfect structure as a sole assembly product.
:1 molar ratio in CD2Cl2 (Scheme S1†). The quantitative self-assembly formation of a discrete triangle of p-[3 + 3] was confirmed through multinuclear NMR (1H and 31P), showing a single discrete assembly with high symmetry (Fig. 1). In the simple 1H NMR pattern of p-[3 + 3], the pyridine protons showed a significant downfield shift (ca. 0.15 and 0.49 ppm for He′ and Hd′ respectively) with respect to those of p-PY due to the loss of electron density upon complexation of the pyridine N atom with the Pt(II) metal center. At the same time, the protons in the benzothiophene unit also exhibited a downfield shift for Ha′, Hb′ and Hc′, respectively (Fig. 1a). Moreover, the 31P NMR spectrum clearly displayed a single peak indicating a high symmetry structure of p-[3 + 3]. Meanwhile, an upfield shift was found from the corresponding starting platinum acceptor to metallacycles by ca. 8.05 ppm for p-[3 + 3], which can be ascribed to the increase in the electron density of the P atom upon coordination. This change as well as the decrease in the coupling of flanking 195Pt satellites (ΔJ = 152.5 Hz) is consistent with the electron back-donation from Pt (Fig. 1b). Here the well-defined signals of both 1H and 31P NMR fully support the formation of a perfect structure as a sole assembly product.
 | ||
| Scheme 2 Graphical representation of self-assembly of p-PY with 180° and 120° acceptors 1 and 2 with ORTEP representation of X-ray single crystal structures of p-PY drawn with 50% probability. The red line shows the bending angle of two pyridines. | ||
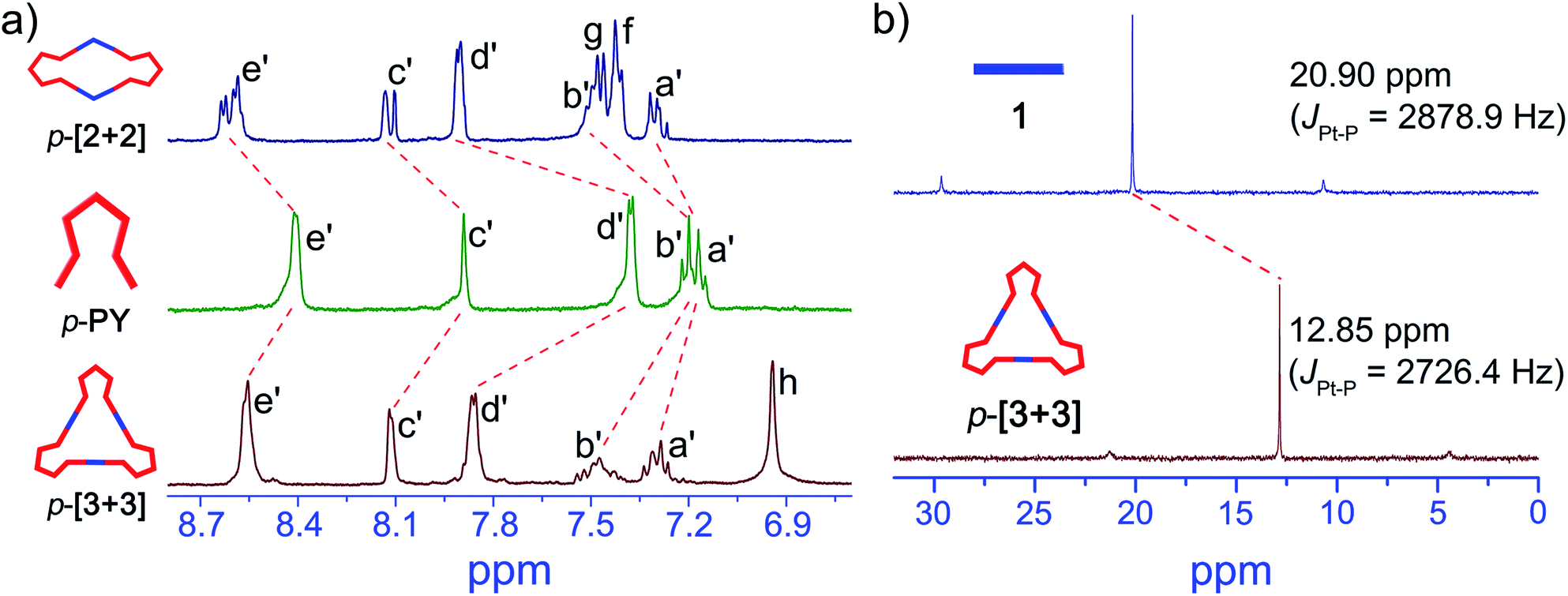 | ||
| Fig. 1 Characterization of p-[3 + 3] and p-[2 + 2]. (a) Partial 1H NMR spectra (400 MHz, 293 K) of p-[2 + 2], p-PY and p-[3 + 3] in CD2Cl2. (b) 31P NMR spectra (161.9 MHz, CD2Cl2, 293 K) of 180° di-Pt(II) acceptor 1 and p-[3 + 3]. | ||
The stoichiometry in formation of discrete triangles can be investigated by electrospray ionization time of flight mass spectrometry (ESI-TOF-MS), providing further support to the existence of p-[3 + 3]. The ESI-TOF-MS spectrum of p-[3 + 3] (Fig. 2a) shows peaks at m/z = 1728.11, 1258.36, and 977.50 (Fig. 2a; S1 and S2 in the ESI†), exactly corresponding to [M-3OTf]3+, [M-4OTf]4+ and [M-5OTf]5+. The peaks were isotopically resolved and agreed very well with their calculated theoretical distribution of the assigned [3 + 3] assembly.
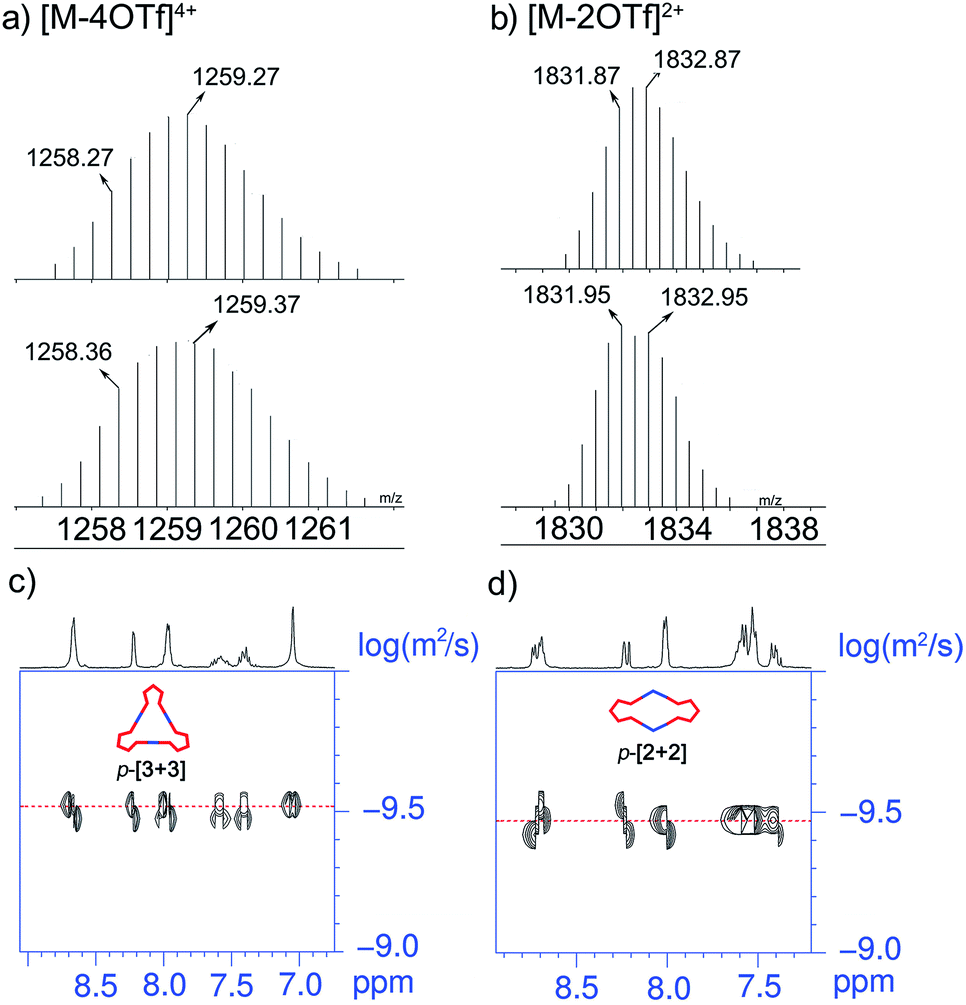 | ||
| Fig. 2 Theoretical (top) and experimental (bottom) ESI-TOF-MS results of p-[3 + 3] of (a) [M-4OTf]4+ and p-[2 + 2] of (b) [M-2OTf]2+. Partial 2D DOSY spectra of (c) p-[3 + 3] and (d) p-[2 + 2] (400 MHz, CD2Cl2, 293 K). | ||
Then we further studied the self-assembly of p-PY with smaller angle (120°) di-Pt(II) acceptor 2, which could produce smaller metallacycles. A rhombus p-[2 + 2] with a smaller size was obtained with the quantitative self-assembly of p-PY with 120° di-Pt(II) acceptor 2 (Scheme S1†). The quantitative self-assembly formation of rhombus p-[2 + 2] was confirmed by multinuclear NMR (1H and 31P) spectroscopy (Fig. 1a and S3†). The 1H NMR spectra indicated a similar downfield shift in both pyridine and benzothiophene. The 31P NMR spectra also showed an upfield shift from initial di-platinum acceptor 2, along with a decrease in the coupling of flanking 195Pt satellites (ΔJ = 132.5 Hz, Fig. S3†). However, there was an unexpected phenomenon in the 1H and 31P NMR spectra of p-[2 + 2] compared with those of p-[3 + 3]. In the 1H NMR spectrum of p-[2 + 2], the He′ of pyridines showed two doublets near 8.61 ppm while Hc′ of benzothiophene showed two singlets at about 8.12 ppm. Moreover, the signal of the phosphine group in the 31P NMR spectrum also displayed two very close singlets. Considering the hindered rotation of the Pt–N (pyridine) bond, this would also be attributed to the hindered rotation between the bisbenzo(thiadiazole) ethene bridge and benzothiophene units, thus resulting in large ring tension induced by small cycles for protons of pyridine and benzothiophene units.53–55 Different from p-[2 + 2], p-[3 + 3] possesses a larger size of metallacycles which reduces the steric hindrance between the ethene bridge and the benzothiophene unit.
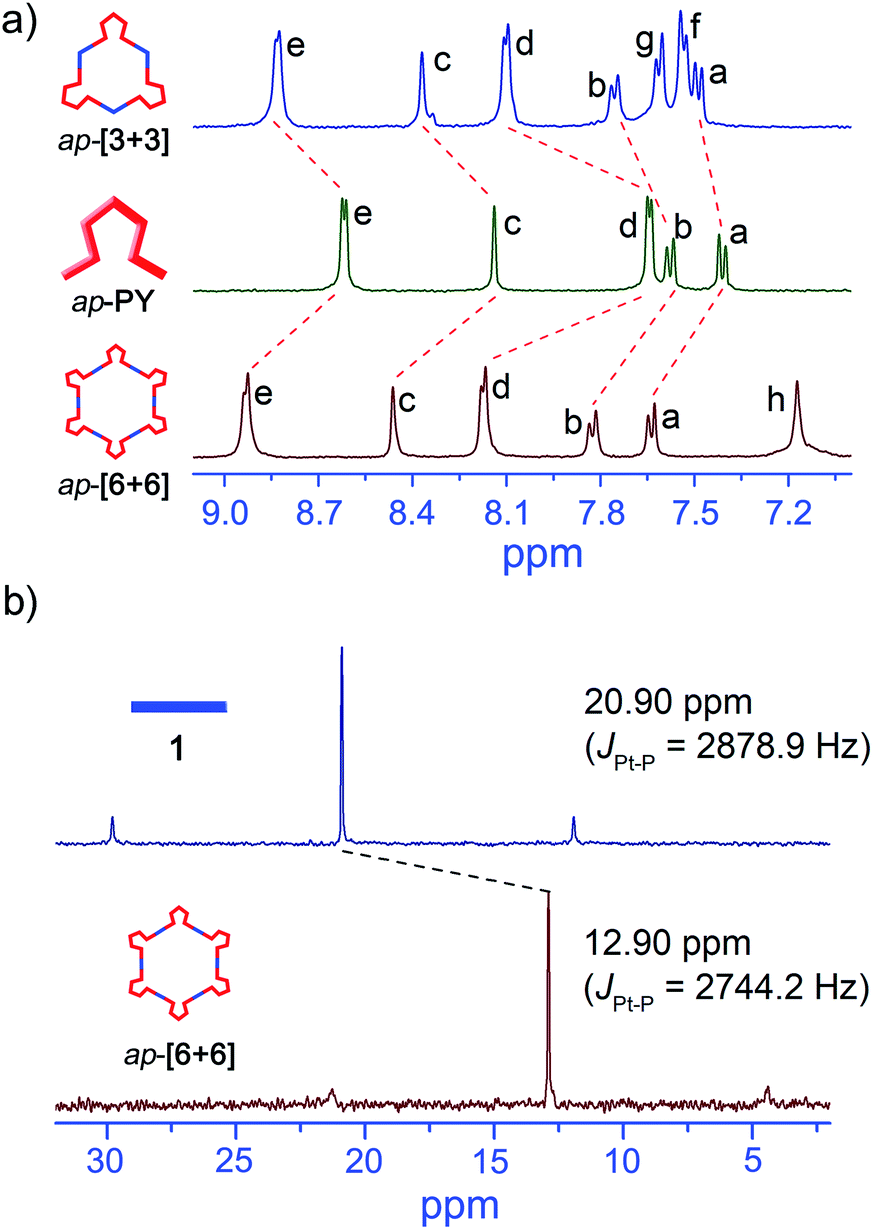
Interestingly, upon changing the di-Pt(II) acceptor from 1 (180°) to 2 (120°), we have successfully obtained discrete metallacycles with different sizes and shapes, which can be attributed to the different bending angles of di-Pt(II) acceptors. Two dimensional (2D) diffusion-ordered 1H NMR spectroscopy (DOSY) was applied as an advanced technique to measure the size of the supramolecular system in solution. The observation of a single band at logD = −9.45 and −9.50 also confirmed the formation of a single self-assembled product (Fig. 2c and d). Meanwhile, the diffusion coefficient of p-[3 + 3] (3.16 × 10−10 m2 s−1) is obviously smaller than that of p-[2 + 2] (3.55 × 10−10 m2 s−1), respectively. The size of the two metallacycles can also be calculated according to the Stocks–Einstein equation (eqn (1)):
| D = KbT/6πηrs | (1) |
Self-assembly of anti-parallel conformers and the closed form into hexagonal metallacycles
Next, we focused on self-assembly of ap-PY with the diplatinum ligand to study the difference between the two conformers in self-assembly based on the successful conformer separation (Scheme 3). Still based on the established method, the large assembled hexagon ap-[6 + 6] can be easily obtained (Scheme S2†). The quantitative formation of hexagon ap-[6 + 6] was confirmed by multinuclear NMR (1H and 31P) and ESI-TOF-MS (Fig. 3 and S5†). The simple 1H NMR pattern of ap-[6 + 6] showed a similar down-field shift to the parallel conformer-based metallacycles in both pyridine and benzothiophene units due to the loss of electron density upon coordination to the Pt(II) metal center. Meanwhile, the 31P NMR spectrum showed a sharp single peak shifted upfield by 8.00 ppm from initial di-platinum acceptor 1, along with a decrease in the coupling of flanking 195Pt satellites (ΔJ = 134.7 Hz, Fig. 3b), indicative of the discrete and high symmetry structure. The corresponding isotope patterns of ESI-TOF-MS observed were in good agreement with the calculated m/z ratios, which further confirmed the formation of larger metallacycles ap-[6 + 6] by changing the conformer of the unassembled ligand.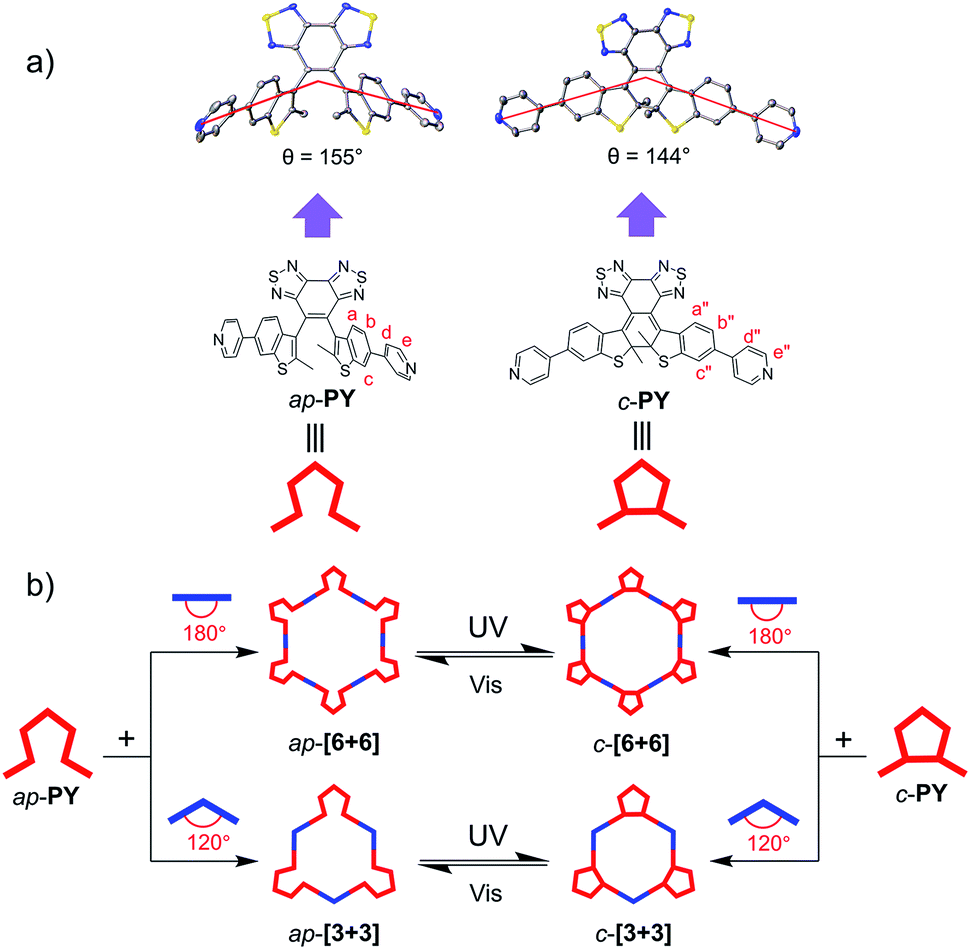 | ||
| Scheme 3 Graphical representation of self-assembly of ap-PY and c-PY with 180° and 120° di-Pt(II) acceptors 1 and 2, and structural transformation of hexagons with ORTEP representation of X-ray single crystal structures of ap-PY and c-PY drawn with 50% probability. The red line shows the bending angle of two pyridines. | ||
 | ||
| Fig. 3 Characterization of hexagon ap-[3 + 3] and ap-[6 + 6]. (a) Partial 1H NMR spectra (400 MHz, 293 K) of ap-[3 + 3], ap-PY and ap-[6 + 6] in a mixture of CD2Cl2 and acetone-d6. (b) 31P NMR spectra (161.9 MHz, CD2Cl2, 293 K) of 180° di-Pt(II) acceptor 1 and ap-[3 + 3]. | ||
Then we further studied the self-assembly of ap-PY with smaller angle (120°) di-Pt(II) acceptor 2. The discrete smaller cyclotrimer ap-[3 + 3] can also be obtained straightforward. Multinuclear NMR (1H and 31P) and ESI-MS-TOF evidenced the stoichiometry of formation of ap-[3 + 3] as well (Fig. 3a, S6 and S7†). Upon self-assembly to form highly symmetrical hexagon ap-[3 + 3], 31P NMR also exhibited a single peak shifted upfield from initial di-platinum acceptor 2 (Fig. S6†). Also in ESI-TOF-MS, the peaks at m/z = 1039.51 and 1336.62, exactly corresponding to [M-5OTf]5+ and [M-4OTf]4+, respectively, are in good agreement with the theoretical distribution of the discrete nature of hexagon ap-[3 + 3] (Fig. S6 and S7†).
Regarding the ap-metallacycles, we also performed 2D 1H DOSY to visualize the size of metallacycles. The observation of a single band of both ap-[6 + 6] and ap-[3 + 3] indicated the discrete structure of these two metallacycles. The diffusion coefficients (D) were about 9.12 × 10−11 and 1.29 × 10−10 m2 s−1, respectively, in CD2Cl2, corresponding to objects with diameters of about 10.9 and 7.7 nm for ap-[6 + 6] and ap-[3 + 3], showing the larger size of ap-[6 + 6] (Fig. 4). Moreover, compared with the p-conformer upon coordination to the same di-Pt(II) acceptor, ap-conformer-based metallacycles possess a larger size and different shapes, suggestive of the different self-assembly behavior between ap- and p-conformers.
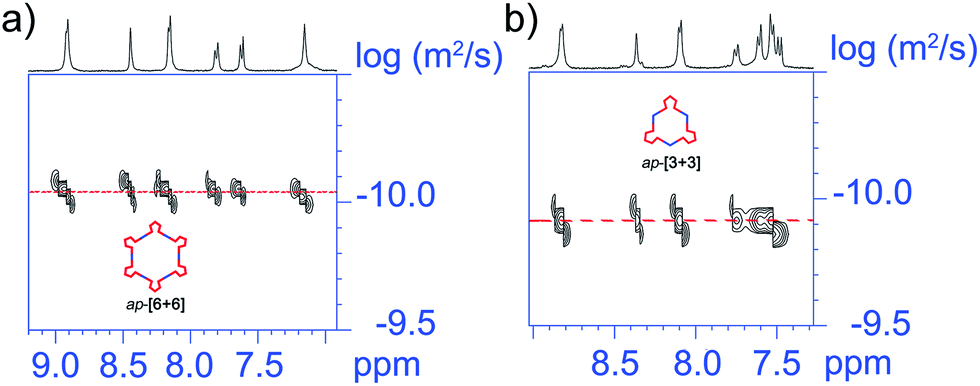 | ||
| Fig. 4 Partial 2D DOSY spectrum of (a) ap-[6 + 6] and (b) ap-[3 + 3], indicating that the calculated molecular diameters are about 10.9 and 7.7 nm for ap-[6 + 6] and ap-[3 + 3], respectively. | ||
Upon changing the photoresponsive ligands and di-Pt(II) acceptors with different bending angles, we obtained a series of metallacycles with different sizes and different shapes. As for p-PY, given that the sterically hindered ethene bridge and benzothiophene unit can block the rotation of side units, the two pyridine units in the parallel conformer p-PY can only extend to the same sides (65°, Scheme 2). This factor limits the bending angles of p-PY. In contrast, the pyridine units in the anti-parallel conformer ap-PY can extend to different sides with larger angle (155°, Scheme 3), thereby forming larger polygon cycles. Meanwhile, it is notable that compared to the parallel conformer, ap-conformer-based metallacycles possess much simpler 1H NMR patterns because of the larger polygon cycles, leading to lower ring tension and reduce the hindered rotation of the Pt–N (pyridine) bond as well as bisbenzo(thiadiazole) ethene bridge between benzothiophene units.41,55
Furthermore, hexagon c-[6 + 6] can also be constructed by the closed form donor c-PY through a similar method (Scheme S3†). 1H and 31P NMR and ESI-TOF-MS confirmed the quantitative formation of the discrete structure of hexagon c-[6 + 6] (Fig. S8–S11†). 2D 1H DOSY measurements were also performed for c-[6 + 6], along with a diameter of about 11.7 nm fitting to the size of ap-[6 + 6] (Fig. S12 and S13†).
Conformer-based self-sorting behavior
Obviously, the self-assembly of ap- and p-conformers with two different Pt(II) ligands exhibits a series of discrete supramolecular metallacycles. To further study the conformer-dependent self-assembly, we further investigated the behavior of the equimolar ratio mixture of ap-PY and p-PY with a stoichiometric amount of di-Pt(II) acceptor 1 in CD2Cl2 and DMSO-d6 solution. Interestingly, the resulting mixtures revealed the completely selective self-assembly of ap-[6 + 6] and p-[3 + 3]. In 1H NMR and 2D DOSY spectra (Fig. 5), only the peaks corresponding to hexagon ap-[6 + 6] and triangle p-[3 + 3] were observed, with no peaks for other mixed species. 31P NMR can also confirm the self-sorting behavior (Fig. S14†). Clearly, it is the mismatched geometrical conformation between ap-PY and p-PY that leads to completely self-sorting behavior.56–58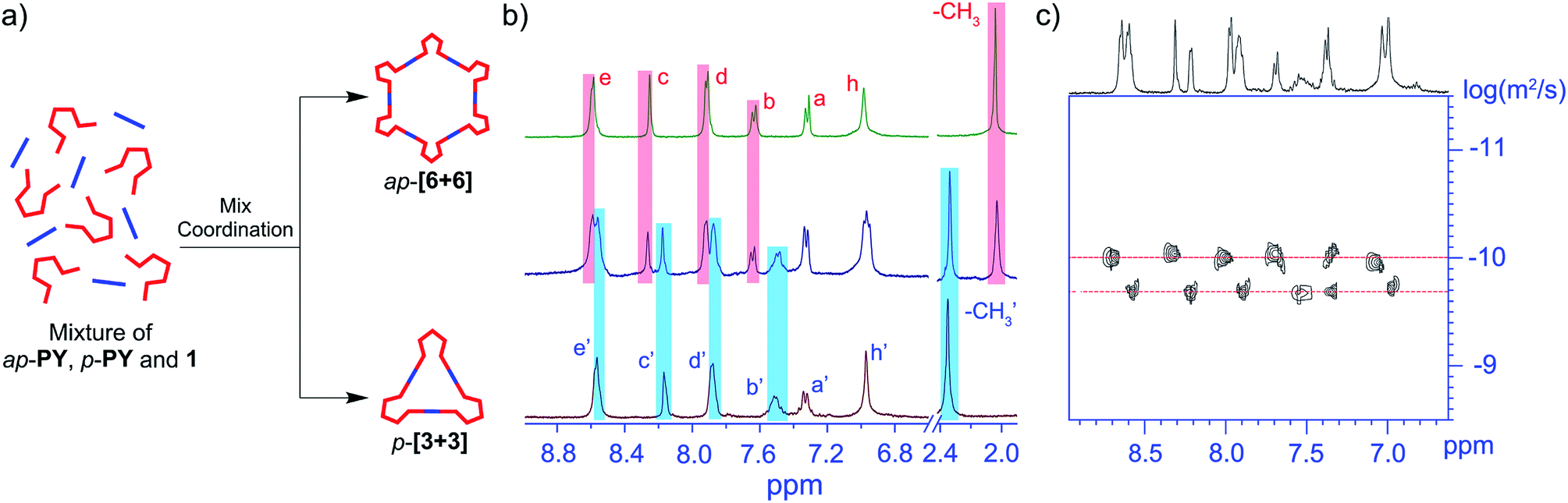 | ||
| Fig. 5 Mixed self-assembly of ap-PY and p-PY with di-Pt(II) 1 in CD2Cl2 and DMSO-d6. (a) Graphical representation of mixed self-assembly of ap-PY and p-PY with di-Pt(II) 1. (b) Partial 1H NMR spectral comparison (400 MHz, 293 K) of ap-[6 + 6], p-[3 + 3], and a mixed assembly system. (c) Partial 2D DOSY spectrum of mixed assembly system. | ||
However, compared to ap- and p-conformers, the differences of the bending angles between ap- and c-conformers are smaller. Hence, would it still show the self-sorting behavior of two distinct complexes or the two conformers be self-assembled into a statistical mixture? To solve the issue, the self-sorting behavior between ap- and c-conformers was studied. A mixture of 2.0 eq open form of ap-conformer and 1.0 eq closed form with 3.0 eq Pt salt 1 in CD2Cl2 and DMSO was prepared. Interestingly, we observed quite clear and similar spectra as shown in Fig. 6 with all peaks corresponding to ap-[6 + 6] and c-[6 + 6]. Moreover, the assembled NMR spectrum showed also the same characteristic as that of the simple mixture of ap-[6 + 6] and c-[6 + 6] (ratio = 2:1, Fig. 6 and S15†). It displayed an obvious self-sorting behavior between the open form and the closed form.
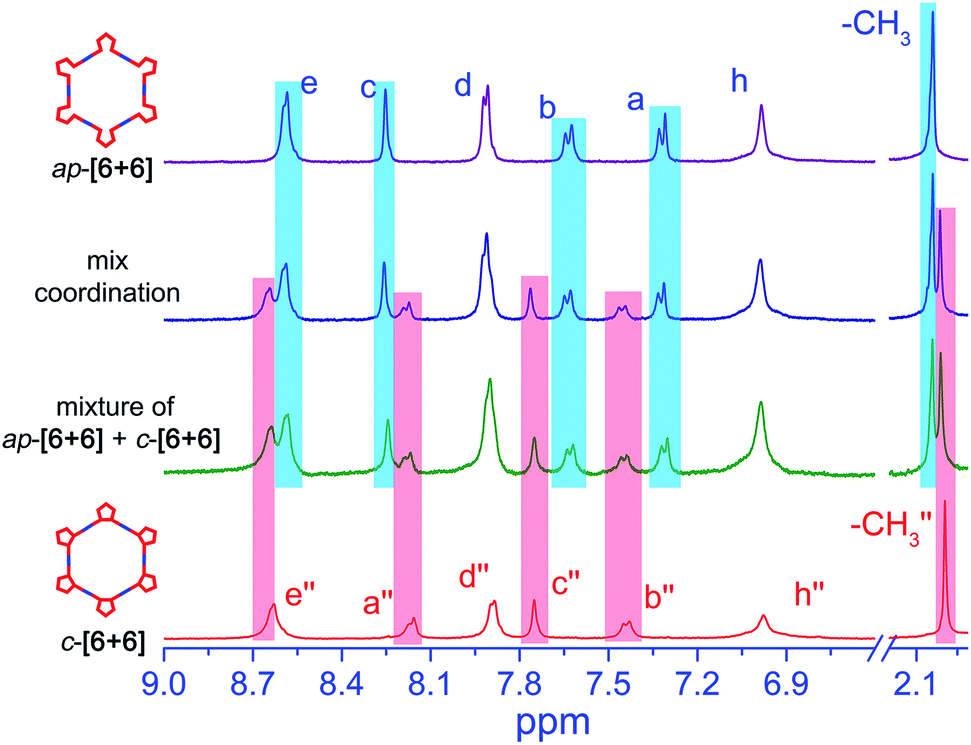 | ||
| Fig. 6 Characterization of metallacycles assembled by ap-PY and c-PY with di-Pt(II) 1. Comparison of partial 1H NMR (400 MHz, CD2Cl2 and DMSO-d6, 293 K) spectra of ap-[6 + 6], mixed coordination of 2.0 eq ap-PY and 1.0 eq c-PY with 3.0 eq di-Pt(II) 1 acceptor, mixture of ap-[6 + 6] + c-[6 + 6] (about 2:1), and c-[6 + 6]. | ||
Actually, the open form exhibits 155°, and the closed form shows 144° (Scheme 3). Though the difference is not large, the open form still assembles with an open form to result in an open-form metallacycle, and the closed form only assembles with a closed form to produce a ring-closed metallacycle. Clearly, the assembly is critically dependent upon the specific bending angles of the open-form and closed form.59 Moreover, the similar self-sorting behavior can also be found when ap-PY and p-PY or ap-PY and c-PY coordinate with Pt(II) salt 2 (Fig. S16 and S17†). The above self-sorting behavior further demonstrates the conformer-dependent self-assembly behavior.
Photo-reversible response of self-assembled metallacycles
With the supramolecular system in hand, we turn our attention to the photoresponsive performance of ligands and metallacycles. According to the Woodward–Hoffmann rule, for photoresponsive DAE derivatives, only the ap-conformer can undergo photocyclization while p-conformer cannot. That is, the photocyclization quantum yield (Φo-c) in conventional DAEs is usually limited to 50%. As expected for the p-conformer, the corresponding p-PY, p-[2 + 2] and p-[3 + 3] didn't show any change both in absorption and emission spectra under UV light irradiation in CH2Cl2 solution (Fig. S21–S23†). As for ap-configuration, both the ligand and metallacycles show excellent photoresponsive performance (Scheme 1 and Fig. 7a), especially for the photocyclization quantum yield (Φo-c) of ap-[6 + 6] and ap-[3 + 3] as high as 61.2% and 66.8% (Table 1), respectively.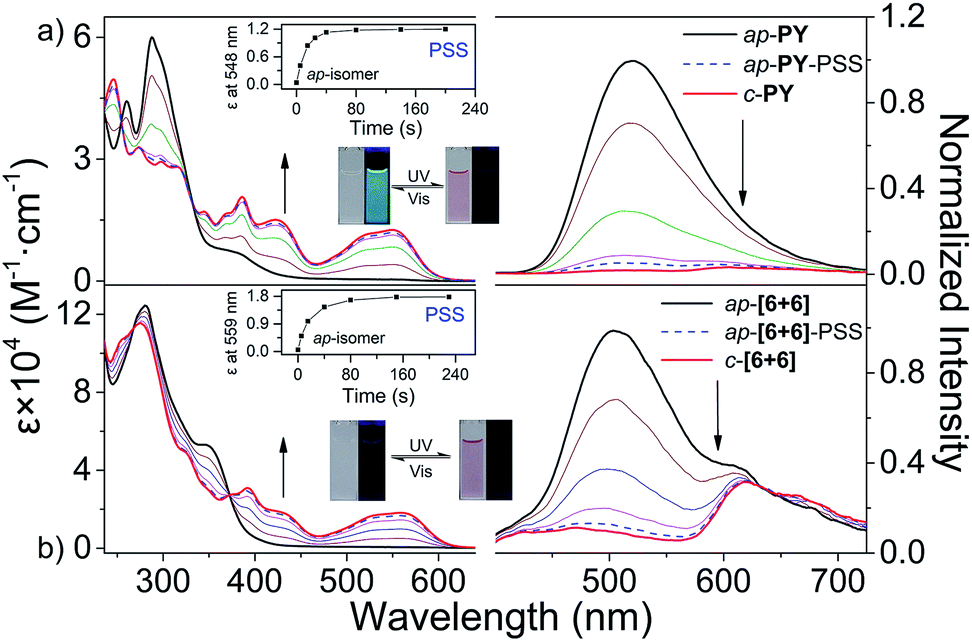 | ||
| Fig. 7 Light induced spectral change of photoresponsive metallacycles. Absorption and fluorescence spectra of (a) ap-PY and (b) ap-[6 + 6], respectively, under irradiation with UV light (λ = 302 ± 20 nm) in CH2Cl2. Excitation for fluorescence is set at each isosbestic point. The inset images show the color and emission changes triggered by UV (λ = 302 ± 20 nm) and Vis (λ > 480 nm) light and the spectral changes of extinction coefficient. | ||
| Compounds | λ abs, max (nm) [εpb/εmb (103 M−1 cm−1)] | CRo-cc [%] | CRc-oc [%] | Φ o-c [%] | Φ c-o [%] | Φ F (%) | λ em1 [nm] | τ 1 [ns] | λ em2 [nm] | τ 2 [ns] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Typical absorption maxima of the ring-open isomer in the UV region and the ring-closed isomer in the visible region, respectively. b ε p represents the molar extinction coefficient of metallacycles calculated by the concentration of the photoresponse unit and εm represents the molar extinction coefficient of metallacycles calculated by the concentration of metallacycles. c Conversion ratio from open to closed isomers (irradiation at λ = 302 ± 20 nm) and ring-open isomers (under visible light irradiation, λ > 480 nm), calculated from absorption spectra. d Quantum yields of photocyclization (Φo-c) at 313 nm and cycloreversion (Φc-o) at 517 nm. e Fluorescence quantum yield (ΦF). | ||||||||||
| ap-PY | 285 [59.9] | 94 | — | 73.4 | — | 0.46 | 518 | 0.39 | — | — |
| c-PY | 549 [12.6] | — | >99 | — | 4.6 | — | — | — | — | — |
| p-PY | 285 [66.4] | — | — | — | — | 0.33 | 494 | 0.26 | — | — |
| ap-[6 + 6] | 280 [124.5/747.0] | 90 | — | 61.2 | — | 0.10 | 503 | 1.51 | 615 | 1.74 |
| c-[6 + 6] | 559 [18.2/109.2] | — | >99 | — | 6.5 | 0.04 | — | — | 616 | 1.81 |
| p-[3 + 3] | 281 [96.2/288.6] | — | — | — | — | 0.70 | 464 | 1.40 | 570 | 1.93 |
| ap-[3 + 3] | 283 [96.3/289.6] | 93 | — | 66.8 | — | 0.14 | 481 | 0.41 | 618 | 1.50 |
| c-[3 + 3] | 562 [17.2/51.6] | — | >99 | — | 6.3 | — | — | — | — | — |
| p-[2 + 2] | 283 [68.9/137.8] | — | — | — | — | 0.8 | 467 | 1.02 | 578 | 2.05 |
Here, we mainly focus on the photoresponsive properties of self-assembly systems. Different from traditional self-assembly systems, these metallacycles (ap-[6 + 6] and ap-[3 + 3]) are a series of photo-active self-assembly systems which can transform reversibly between open- and closed-forms triggered by light (Scheme 1b). Similar to free ligand ap-PY, ap-[6 + 6] showed an intense absorption band at 280 nm corresponding to the bisbenzo(thiadiazole) ethene bridge, along with a medium shoulder peak at about 340–430 nm. Upon UV light (302 ± 20 nm) irradiation of ap-[6 + 6] in CH2Cl2 solution, the color turned red, similar to ap-PY, with the appearance of three new peaks at 559, 426 and 392 nm, showing a clear isosbestic point without a redshift during the photo-transformation process. Here the phenomenon arose from the typical photocyclization in formation of the corresponding closed metallacycles (c-[6 + 6], Fig. 7b). Compared with c-PY, the three absorption peaks of the photoresponsive c-[6 + 6] exhibited a red-shift by 11, 5 and 8 nm, indicative of an extended π conjugation upon coordination of ap-PY with acceptor 1. Moreover, the assembled metallacycles in ap-, p- and c-forms exhibited higher absorption coefficients based on each photochromic unit (Table 1 and Fig. S28†). The similar coordination metallacycle ap-[3 + 3] also exhibited excellent photoswitchable performance (Fig. S24†). Notably, the conversion yield for both different ring size hexagons in ap-[6 + 6] and ap-[3 + 3] is more than 90%, which is comparable to that for the photochromic ligand ap-PY (Table 1). It is indicative that, upon coordination-induced self-assembly with the Pt(II) acceptor, the supramolecular hexagons can still keep good photo-switchable characteristics.
Meanwhile, as powerful tools, 1H and 31P NMR spectroscopy techniques can be used to monitor the UV light-induced transformation process from ap-[6 + 6] to c-[6 + 6]. Upon irradiation with 302 ± 20 nm light, the 1H NMR spectra (Fig. S30†) during the light-induced transformation from ap-[6 + 6] to c-[6 + 6] showed distinct changes, especially in Ha shifting from 7.51 to 8.20 ppm, attributed to the formation of intramolecular hydrogen bonds between Ha and the N atom of thiadiazole. We observed clear and simple 1H NMR spectra illustrating the maintenance of the high symmetry structures during the photoisomerization process. All peaks can be ascribed to ap-[6 + 6] and c-[6 + 6], whereas no other peaks belonging to the intermediate states could be found.
Additionally, the fluorescence of ap-metallacycles can also be photo-modulated. For instance, similar to photoresponsive ligand ap-PY (Fig. 7a), metallacycle ap-[6 + 6] showed a weak green fluorescence emission band at 400–725 nm upon excitation at 375 nm along with an unexpected shoulder peak at about 615 nm (Fig. 7b). Subsequently, upon UV light irradiation at 302 ± 20 nm, the emission of open metallacycle ap-[6 + 6] converted into closed c-[6 + 6] became seriously quenched in fluorescence by 96.5% at the photostationary state (PSS). The unexpected fluorescence shoulder peak has been reported previously in a [3 + 3] system.44 Here we further study the origination of the shoulder peak in ap-[6 + 6]. The fluorescence lifetime monitored by the additional peak at around 600 nm was in the range of nanoseconds, indicative of the typical fluorescence characteristic, not phosphorescence (Table 1 and Fig. S31–S34†). The reference mixture with the di-platinum acceptor in 1:1 ratio can further confirm that the additional fluorescence still might be attributed to the self-assembled formation of metallacycles (Fig. S35†).
In this way, we can achieve the supramolecular photo-switched reversibility with the implanted photoresponsive unit, which is in stark contrast with the traditional “inactive” coordination-induced metallacycles. For both ap-[3 + 3] and ap-[6 + 6], irradiation with visible light (λ > 480 nm) can bleach the red solution and regenerate the open ring isomers. As a matter of fact, the alternating irradiation with UV (302 ± 20 nm) and visible light (λ > 480 nm) toggled the photoswitch repeatedly between the open and closed hexagon metallacycles with remarkable fatigue resistance (Fig. S36†). For both different ring size hexagons, the quantum yields of photocyclization and cycloreversion in ap-[6 + 6] and ap-[3 + 3] were almost the same (Table 1). Compared to the photochromic ligand ap-PY, the quantum yields decreased slightly upon coordination, which can be attributed to the metallacycle ring tension to some restriction in the rotation of benzothiophene units.42,60,61
Conclusions
In summary, we have for the first time constructed a unique series of conformer-dependent photoactive metallacycles, especially focusing on the characterization and comparison of self-assembly behavior in the separated photoresponsive ligand conformers with different di-platinum(II) acceptors. The scaffold sizes and shapes of photoswitchable metallacycles can be precisely controlled by photochromic parallel or anti-parallel conformers via coordination-driven self-assembly. Along with the specific conformer-based self-sorting behavior, the ap-conformer and closed form provide larger bending angles upon coordination with di-platinum(II) acceptors into hexagon [6 + 6] or [3 + 3] while p-conformer can only form smaller polygon cycles with triangle or rhombus cycles. Furthermore, taking advantage of the separated ap-conformer ligand, it possesses excellent photoresponsive performance with a relatively high conversion ratio and well-controlled reversible structure transformation, especially for realizing the photocyclization quantum yield (Φo-c) of ap-[6 + 6] and ap-[3 + 3] as high as 61.2% and 66.8%, respectively, making a breakthrough to the general limitation to 50% in conventional diarylethene systems. We can achieve the supramolecular photo-switchable reversibility with the implanted photoresponsive unit, which is in stark contrast with the traditional “inactive” coordination-induced metallacycles. This work would be a good model to develop soft smart materials for use in probing, catalysis and guest encapsulation systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W.-H. Z acknowledges financial support from the NSFC for Science Center Program (21788102), Creative Research Groups (21421004) and Key Project (21636002), NSFC/China, the National key Research and Development Program (2016YFA0200300), the Shanghai Municipal Science and Technology Major Project (2018SHZDZX03), and the Program of Introducing Talents of Discipline to Universities (B16017). H.-B. Y acknowledges the financial support of NSFC/China (No. 21625202), the 973 Program (No. 2015CB856600), STCSM (No. 16XD1401000), and the Program for Changjiang Scholars and Innovative Research Team in University.Notes and references
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151 RSC.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS PubMed.
- G. Charalambidis, E. Georgilis, M. K. Panda, C. E. Anson, A. K. Powell, S. Doyle, D. Moss, T. Jochum, P. N. Horton, S. J. Coles, M. Linares, D. Beljonne, J. V. Naubron, J. Conradt, H. Kalt, A. Mitraki, A. G. Coutsolelos and T. S. Balaban, Nat. Commun., 2016, 7, 12657 CrossRef CAS PubMed.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342 CrossRef CAS PubMed.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090 CrossRef CAS PubMed.
- K. Wu, K. Li, Y. J. Hou, M. Pan, L. Y. Zhang, L. Chen and C. Y. Su, Nat. Commun., 2016, 7, 10487 CrossRef CAS PubMed.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447 CrossRef CAS PubMed.
- A. I. d'Aquino, Z. S. Kean and C. A. Mirkin, Chem. Mater., 2017, 29, 10284 CrossRef.
- L. J. Chen, S. Chen, Y. Qin, L. Xu, G. Q. Yin, J. L. Zhu, F. F. Zhu, W. Zheng, X. Li and H. B. Yang, J. Am. Chem. Soc., 2018, 140, 5049 CrossRef CAS PubMed.
- W. Zheng, W. Wang, S. T. Jiang, G. Yang, Z. Li, X. Q. Wang, G. Q. Yin, Y. Zhang, H. Tan, X. Li, H. Ding, G. Chen and H. B. Yang, J. Am. Chem. Soc., 2018, 141, 583 CrossRef PubMed.
- W. Wang, Y. X. Wang and H. B. Yang, Chem. Soc. Rev., 2016, 45, 2656 RSC.
- S. Das, P. Ranjan, P. S. Maiti, G. Singh, G. Leitus and R. Klajn, Adv. Mater., 2013, 25, 422 CrossRef CAS PubMed.
- H. F. Cheng, A. I. d'Aquino, J. Barroso-Flores and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 14590 CrossRef CAS PubMed.
- J. Zhu, Y. Kim, H. Lin, S. Wang and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 5061 CrossRef CAS PubMed.
- J. Zhou, M. Chen and G. Diao, ACS Appl. Mater. Interfaces, 2014, 6, 18538 CrossRef CAS PubMed.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H.-F. Hsu and M. Yoshizawa, J. Am. Chem. Soc., 2013, 135, 12976 CrossRef CAS PubMed.
- W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285 CrossRef CAS PubMed.
- S. Oldknow, D. R. Martir, V. E. Pritchard, M. A. Blitz, C. W. G. Fishwick, E. Zysman-Colman and M. J. Hardie, Chem. Sci., 2018, 9, 8150 RSC.
- S. C. Wei, M. Pan, Y. Z. Fan, H. Liu, J. Zhang and C. Y. Su, Chem.–Eur. J., 2015, 21, 7418 CrossRef CAS PubMed.
- S. Chen, L. J. Chen, H. B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596 CrossRef CAS PubMed.
- H. Yotsuji, K. Higashiguchi, R. Sato, Y. Shigeta and K. Matsuda, Chem.–Eur. J., 2017, 23, 15059 CrossRef CAS PubMed.
- P. K. Kundu, S. Das, J. Ahrens and R. Klajn, Nanoscale, 2016, 8, 19280 RSC.
- Y. Han, C. Gao and X. He, Sci. China: Chem., 2011, 55, 604 CrossRef.
- K. K. Kartha, N. K. Allampally, A. T. Politi, D. D. Prabhu, H. Ouchi, R. Q. Albuquerque, S. Yagai and G. Fernández, Chem. Sci., 2019, 10, 752 RSC.
- D. Zhang, Y. Nie, M. L. Saha, Z. He, L. Jiang, Z. Zhou and P. J. Stang, Inorg. Chem., 2015, 54, 11807 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174 CrossRef CAS PubMed.
- J. Su, T. Fukaminato, J. P. Placial, T. Onodera, R. Suzuki, H. Oikawa, A. Brosseau, F. Brisset, R. Pansu, K. Nakatani and R. Métivier, Angew. Chem., Int. Ed., 2016, 55, 3662 CrossRef CAS PubMed.
- C.-W. Hsu, C. Sauvée, H. Sundén and J. Andréasson, Chem. Sci., 2018, 9, 8019 RSC.
- M. Fukagawa, I. Kawamura, T. Ubukata and Y. Yokoyama, Chem.–Eur. J., 2013, 19, 9434 CrossRef CAS PubMed.
- Y. Cai, Y. Gao, Q. Luo, M. Li, J. Zhang, H. Tian and W.-H. Zhu, Adv. Opt. Mater., 2016, 4, 1410 CrossRef CAS.
- N. M.-W. Wu, M. Ng and V. W.-W. Yam, Angew. Chem., Int. Ed., 2019, 58, 3027 CrossRef CAS PubMed.
- E. Siemes, O. Nevskyi, D. Sysoiev, S. K. Turnhoff, A. Oppermann, T. Huhn, W. Richtering and D. Wöll, Angew. Chem., Int. Ed., 2018, 57, 12280 CrossRef CAS PubMed.
- Y. Zheng, H. Sato, P. Wu, H. J. Jeon, R. Matsuda and S. Kitagawa, Nat. Commun., 2017, 8, 100 CrossRef PubMed.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catalysis, 2018, 1, 516 CrossRef.
- X. Hu, M. E. McFadden, R. W. Barber and M. J. Robb, J. Am. Chem. Soc., 2018, 140, 14073 CrossRef CAS PubMed.
- M. Han, Y. Luo, B. Damaschke, L. Gómez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Angew. Chem., Int. Ed., 2016, 55, 445 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y. S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319 CrossRef CAS PubMed.
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Angew. Chem., Int. Ed., 2014, 53, 4603 CrossRef CAS PubMed.
- W. Li, X. Li, Y. Xie, Y. Wu, M. Li, X. Y. Wu, W. H. Zhu and H. Tian, Sci. Rep., 2015, 5, 9186 CrossRef CAS PubMed.
- M. Li, L.-J. Chen, Y. Cai, Q. Luo, W. Li, H.-B. Yang, H. Tian and W.-H. Zhu, Chem, 2019, 5, 634 CAS.
- J. Yoon and A. P. de Silva, Angew. Chem., Int. Ed., 2015, 54, 9754 CrossRef CAS PubMed.
- K. Morinaka, T. Ubukata and Y. Yokoyama, Org. Lett., 2009, 11, 3890 CrossRef CAS PubMed.
- T. Nakashima, R. Fujii and T. Kawai, Chem.–Eur. J., 2011, 17, 10951 CrossRef CAS PubMed.
- M. Kamrul Hossain, M. Takeshita and T. Yamato, Eur. J. Org. Chem., 2005, 2005, 2771 CrossRef.
- T. Yamada, K. Muto, S. Kobatake and M. Irie, J. Org. Chem., 2001, 66, 6164 CrossRef CAS PubMed.
- C. S. Pecinovsky, E. S. Hatakeyama and D. L. Gin, Adv. Mater., 2008, 20, 174 CrossRef CAS.
- Y.-F. Han, G.-X. Jin and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 9263 CrossRef CAS PubMed.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS PubMed.
- X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276 CrossRef CAS PubMed.
- G. Tárkányi, H. Jude, G. Pálinkás and P. J. Stang, Org. Lett., 2005, 7, 4971 CrossRef PubMed.
- H.-B. Yang, N. Das, F. Huang, A. M. Hawkridge, D. D. Díaz, A. M. Arif, M. G. Finn, D. C. Muddiman and P. J. Stang, J. Org. Chem., 2006, 71, 6644 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784 CrossRef CAS PubMed.
- C. Gütz, R. Hovorka, C. Klein, Q. Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693 CrossRef PubMed.
- F. J. Rizzuto, M. Kieffer and J. R. Nitschke, Chem. Sci., 2018, 9, 1925 RSC.
- M. Wang, C. Wang, X. Q. Hao, X. Li, T. J. Vaughn, Y. Y. Zhang, Y. Yu, Z. Y. Li, M. P. Song, H. B. Yang and X. Li, J. Am. Chem. Soc., 2014, 136, 10499 CrossRef CAS PubMed.
- S. Aloïse, M. Sliwa, Z. Pawlowska, J. Réhault, J. Dubois, O. Poizat, G. Buntinx, A. Perrier, F. Maurel, S. Yamaguchi and M. Takeshita, J. Am. Chem. Soc., 2010, 132, 7379 CrossRef PubMed.
- M. Walko and B. L. Feringa, Chem. Commun., 2007, 43, 1745 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1848524. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00757a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |