
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinitiated carbonyl-metathesis: deoxygenative reductive olefination of aromatic aldehydes via photoredox catalysis†
Shun
Wang
,
Nanjundappa
Lokesh
,
Johnny
Hioe
,
Ruth M.
Gschwind
 * and
Burkhard
König
*
* and
Burkhard
König
*
Faculty of Chemistry and Pharmacy, University of Regensburg, D-93040 Regensburg, Germany. E-mail: ruth.gschwind@ur.de; burkhard.koenig@ur.de
First published on 18th March 2019
Abstract
Carbonyl–carbonyl olefination, known as McMurry reaction, represents a powerful strategy for the construction of olefins. However, catalytic variants that directly couple two carbonyl groups in a single reaction are less explored. Here, we report a photoredox-catalysis that uses B2pin2 as terminal reductant and oxygen trap allowing for deoxygenative olefination of aromatic aldehydes under mild conditions. This strategy provides access to a diverse range of symmetrical and unsymmetrical alkenes with moderate to high yield (up to 83%) and functional-group tolerance. To follow the reaction pathway, a series of experiments were conducted including radical inhibition, deuterium labelling, fluorescence quenching and cyclic voltammetry. Furthermore, NMR studies and DFT calculations were combined to detect and analyze three active intermediates: a cyclic three-membered anionic species, an α-oxyboryl carbanion and a 1,1-benzyldiboronate ester. Based on these results, we propose a mechanism for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond generation involving a sequential radical borylation, “bora-Brook” rearrangement, B2pin2-mediated deoxygenation and a boron-Wittig process.
C bond generation involving a sequential radical borylation, “bora-Brook” rearrangement, B2pin2-mediated deoxygenation and a boron-Wittig process.
Introduction
Alkenes are omnipresent in natural compounds and essential functional groups for many chemical transformations. Among many methods that have been developed for the generation of alkenes, olefination and metathesis reactions converting two functional groups into one alkene are particularly useful for the synthesis of complex molecules. The classic Wittig olefination uses phosphonium ylide reagents (Scheme 1A, eqn (1))1 and many related carbonyl olefination processes, such as the Horner–Wadsworth–Emmons,1b Peterson,2 Julia3 and Tebbe4 reactions utilizing different ylide or carbene precursors have been developed. Olefin cross-metathesis reactions catalyzed by metal alkylidenes allow the exchange between two olefins to form a pair of distinct alkenes (Scheme 1A, eqn (2)).5 The related catalytic olefin–carbonyl metathesis strategy for the synthesis of alkenes was discovered just recently (Scheme 1A, eqn (3)).6 | ||
| Scheme 1 Divergent functionalization of carbonyl. | ||
Contrary to these olefination and metathesis processes, catalytic bicarbonyl olefination reactions remain less developed. The McMurry reaction provides an attractive route to alkenes from two carbonyl groups (Scheme 1B).7 The net result of such a carbonyl–carbonyl olefination could be viewed as a “metathesis” process although the oxygen atom is generally not released as oxygen gas, but bounded to reagents. Classic McMurry protocols use titanium salts in combination with a reducing reagent under heating. The whole process is driven thermodynamically by the formation of strong Ti–O bonds. Despite being very effective and widely used, this system generally requires relatively harsh reaction conditions, such as the use of stoichiometric amounts of the titanium reagent and strong reductants at high temperature, lowering the overall functional group tolerance.7,8 Therefore, several milder and catalytic methods were developed. A variant using only catalytic amounts of titanium with an excess of chlorosilane was developed by Fürstner and co-workers.9 Wagner used hexachlorodisilane at high temperature (160 °C) for converting diarylmethanones into tetraarylethylenes.10 More recently, Ott et al. reported the stereoselective preparation of E-alkenes from two aldehydes by using phosphanylphosphonate.11 After that, Li described a stepwise Ru-catalyzed carbonyl–carbonyl olefination method wherein hydrazine was employed as the mediator to transform one carbonyl into its carbanion equivalent.12 Despite these advances, the direct catalytic carbonyl–carbonyl olefination in one-step and under mild reaction conditions remains a challenge.
In recent years, photoredox catalysis has evolved into an attractive alternative to traditional strategies for generating radical intermediates.13 For instance, ketyl radicals are easily accessed from carbonyl compounds by using a photoredox-mediated single-electron reduction strategy.14 The obtained ketyl radicals have been used in C–C bond formation by addition to π systems or radical–radical coupling (Scheme 1C). However, a photoredox catalyzed reductive coupling of carbonyls followed by deoxygenation yielding a carbon–carbon double bond has not been achieved so far. Herein, we report the first deoxygenative olefination coupling of aromatic aldehydes enabled by the cooperative action of bis(pinacolato)diboron and a photoredox catalytic system (Scheme 1D).
The anticipated deoxygenative olefination process requires a four-electron reduction and an efficient oxygen acceptor. Moreover, a careful design of the catalytic system is required to suppress the competing pinacol coupling.15 With these mechanistic challenges in mind, we envisioned the possibility of combining the photocatalytic carbonyl reduction system with a diboron reagent allowing an efficient McMurry-type process based on the following considerations: (1) the Lewis acidity of the boron atom of the diboronate compounds could potentially activate the carbonyl groups thus facilitating the single electron reduction process.16 (2) The formation of a strong B–O bond would provide a significant thermodynamic driving force for the deoxygenation process.16b,17
Results and discussion
Optimization of reaction condition
To examine the feasibility of our hypothesis, we chose para-tolualdehyde 1a as the model substrate in combination with bis(pinacolato)diboron (B2pin2) as the oxygen acceptor. A promising result was obtained when irradiating a mixture containing 1a, B2pin2, DIPEA, Cs2CO3, [Ir(dFCF3ppy)2dtbbpy]PF6 (1 mol%) and DMF with a blue LED lamp giving trace amounts of alkene 2a (see ESI Table S1, entry 1†). Interestingly, removing the electron donor DIPEA from the system also yielded the product in 6% yield with full conversion of 1a (Table S1, entry 2†). Moreover, the alkene 2a was not detected in the absence of either Cs2CO3 or B2pin2 (Table S1, entry 3–4†). As the formation of the product does not require an additional electron donor, we reasoned that B2pin2 may serve as the terminal reductant and the oxygen acceptor in this reaction. Next, we explored the effect of another co-catalyst, which could potentially shuttle electrons from the boron species to the photocatalytic system.18 To our delight, a significant increase in yield (39%, Table S1, entry 6†) was observed upon adding benzyl thiol (10 mol%) as co-catalyst, whereas quinuclidine proved ineffective (Table S1, entry 5†). Testing the reaction with other bases resulted in lower yields (Table S1, entry 7–15†). Further evaluation of other thiols revealed that only benzyl thiol (20 mol%) and 4-Me-benzyl thiol (20 mol%) gave comparably good yields (Table S2, entry 6 and 14†). A variety of photocatalysts were tested for this transformation; using a slightly modified catalyst [Ir(FCF3ppy)2dtbbpy]PF6 increased the yield to 74% (Z/E = 2.2/1). Screening of the solvents revealed that DMF was the optimal solvent for this reaction (Table S3†). Better yield and Z/E selectivity were achieved by employing a combination of [Ir(FCF3ppy)2dtbbpy]PF6 with 4-Me-benzyl thiol (Table S4, entry 3†). Subsequently, the effect of concentration was examined, subtle varying the concentration to 0.33 M increased the yield to 87% with a better Z/E selectivity of the product (Z/E = 2.6/1) (Table S5, entry 3†). Interestingly, small amounts of product 2a (17%, Table 1, entry 10) were also detected in the absence of the photocatalyst. Presumably, a small amount of benzaldehyde is excited by visible light in the presence of B2pin2 to form ketyl radicals that lead to the formation of product. However, further control experiments confirmed that both the photocatalyst and light were crucial for an efficient transformation (Table 1, entry 9 and 10).|
|
|||
|---|---|---|---|
| Entry | Change from standard conditions | Yield of 2a (Z and E) [%] | Z/Eb |
a Unless otherwise noted, all reactions were carried out with 1a (0.2 mmol), [Ir(FCF3ppy)2dtbbpy]PF6 (0.002 mmol), B2pin2 (0.24 mmol), Cs2CO3 (0.24 mmol), 4-Me-BnSH (0.04 mmol) in anhydrous DMF (0.6 mL) under nitrogen, irradiation with 3 W blue LED for 24 h at 25 °C. Yields were determined by crude NMR using 1,3,5-trimethoxybenzene as an internal standard.
b
Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E ratio determined by 1H NMR analysis. :E ratio determined by 1H NMR analysis.
|
|||
| 1 | None | 87 | 2.6/1 |
| 2 | Without thiol | Trace | — |
| 3 | Without B2pin2 | 0 | — |
| 4 | Without Cs2CO3 | Trace | — |
| 5 | Na2CO3 instead of Cs2CO3 | Trace | — |
| 6 | CsF instead of Cs2CO3 | Trace | — |
| 7 | DMF (1 mL) | 77 | 2.5/1 |
| 8 | DMF (0.4 mL) | 68 | 2.4/1 |
| 9 | Without light | 0 | — |
| 10 | Without photocatalyst | 17 | 3.5/1 |

Synthetic scope
With the optimized reaction conditions in hand, we then investigated the scope of this reaction with substituted aromatic aldehydes as substrates (Fig. 1). A broad range of aromatic aldehydes bearing para-(1a–1g), meta-(1h–1p) or ortho-(1q–1s) substituents reacted smoothly to afford the corresponding alkenes. Many synthetically useful functional groups including alkyl (1a, 1c and 1q), alkoxyl (1d–e, 1h, 1r and 1t), acetal (1u), silyl (1m), boronic ester (1n) are tolerated in this transformation.Importantly, the presence of acidic protons in amides (1f–g, and 1i) and amines (1j) did not interfere with the reaction, giving yields of the isolated alkenes ranging from 47% to 63%. Aromatic substituents, such as phenyl (1k, 1s) and thiophenyl (1l) furnished alkene products, albeit in lower yields. Furthermore, the reaction was compatible with halogen substituents on the benzaldehydes and gave the chloro- and fluoro-substituted alkenes in 40% and 32% yield, respectively (1o–p). However, benzaldehydes possessing strong electron-withdrawing groups, such as nitro or nitrile, were not tolerated. ortho-Substituted 2-methyl benzaldehyde gave an excellent Z/E selectivity (Z/E: 16/1) and good yield (60%). More sterically hindered groups such as methoxy (1r) and phenyl (1s) at the ortho-position showed a similarly high Z/E selectivity up to (39:1), albeit a decreased yield. This significant increase in Z/E selectivity may be attributable to the increase in triplet energy of the Z isomers caused by a larger twisting angle in the presence of an ortho-substitution, while a small increase in triplet energy in the less-congested E isomer is expected.19 Additionally, heteroarenes including benzothiophene (1v), benzofuran (1w) as well as indole (1x) performed well in this transformation. However, aliphatic aldehydes and aromatic ketones gave only trace amounts of products with low conversions, presumably due to their higher reduction potentials and steric hindrance, respectively.
 | ||
| Fig. 1 Scope of aldehydes for the homo-coupling deoxygenative olefinationa. aReaction conditions: 1 (0.2 mmol), [Ir(FCF3ppy)2dtbbpy]PF6 (0.002 mmol), B2pin2 (0.24 mmol), Cs2CO3 (0.24 mmol), 4-MeBnSH (0.04 mmol) in anhydrous DMF (0.6 mL), irradiation with 3 W blue LED for 24 h at 25 °C, isolated yields. Yields are the combined yield of Z and E isomers. bEthyl 2-mercaptopropionate (0.04 mmol) was used in place of 4-MeBnSH. cIsolated yield, average of two parallel reactions. | ||
After having established the scope of aldehydes in homo-coupling reactions, we turned our attention to more challenging cross-coupling reactions between two different aldehydes. As shown in Fig. 2, using slightly modified reaction conditions, the coupling between two different aldehydes proceeds well to give a range of unsymmetrical alkenes. Aldehydes bearing amides and amine groups at the aromatic ring react smoothly with para-tolualdehyde to give the corresponding alkenes in moderate to good yields (5a–5c). Aldehydes carrying alkoxy groups at the phenyl ring were tolerated under our reaction conditions, affording the alkenes in modest to good yields with modest Z/E selectivity (5d–5f). However, this cross-coupling reaction is sensitive to steric hindrance and ortho-substituents led to a decreased reactivity (5e). The reaction of heteroaromatic aldehydes furnished a heterocycle-containing stilbene (5g). Coupling between benzaldehyde and 3,5-dimethoxybenzaldehyde afforded the alkene in 57% yield with good selectivity (Z/E = 1/3.5).20
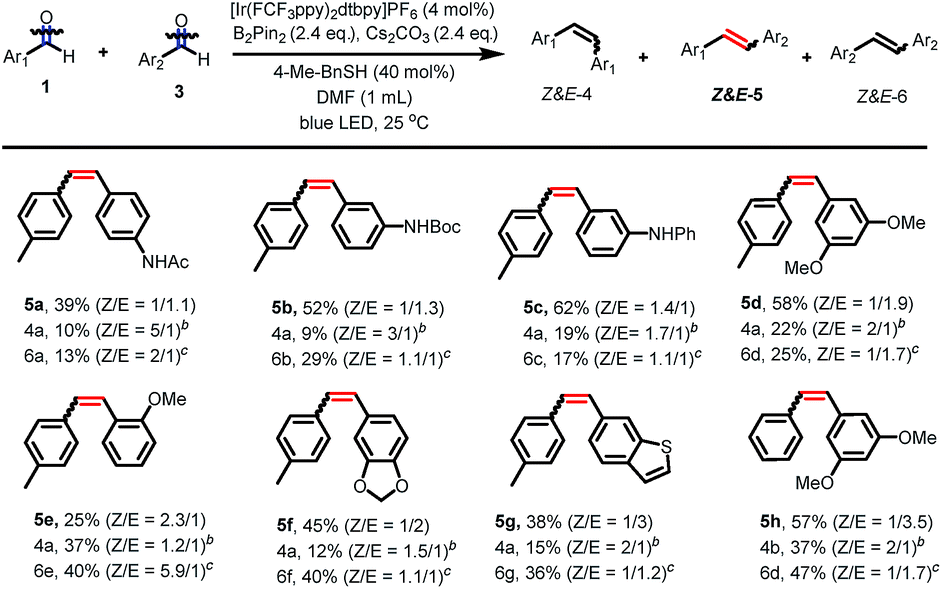 | ||
| Fig. 2 Scope of aldehydes for the cross-coupling deoxygenative olefinationa. aReaction conditions: 1 (0.1 mmol), 3 (0.15 mmol), [Ir(FCF3ppy)2dtbbpy]PF6 (0.004 mmol), B2pin2 (0.24 mmol), Cs2CO3 (0.24 mmol), 4-Me-BnSH (0.04 mmol) in anhydrous DMF (1 mL), irradiation with 3 W blue LED for 24 h at room temperature, isolated average yield of two parallel reactions. Yields refer to the combined yield of Z and E isomers. bYields and Z/E ratio values were determined with 1H NMR using 1,3,5-methoxybenzene as internal standard; based on the amount of aldehyde 1. cYields and Z/E ratio values were determined with 1H NMR using 1,3,5-methoxybenzene as internal standard; based on the amount of aldehyde 3. | ||
Mechanistic investigation
To gain insights into the reaction mechanism, a series of chemical experiments, spectroscopic investigations and in situ illumination NMR experiments21 were conducted.The initial Stern–Volmer luminescence quenching experiments revealed that phenylmethanethiolate quenches the excited state of the photocatalyst much more efficiently than the corresponding thiol, while the aldehyde and B2pin2 do not quench at all (see ESI, Fig. S6†). This indicates a potential electron transfer from the sulfur anion to the excited state of the photocatalyst. This reduced [Ir(FCF3ppy)2dtbbpy]PF6 (II) (Ir01/2III/II1/2 = −1.38 V vs. SCE in DMF, Fig. S1†) catalyst causes the single electron reduction of 1a. Even though the Ered of 1a is higher (Ered = −2.07 V vs. SCE in DMF, Fig. S3†), the decrease of reduction potential of aldehyde 1a on addition of Lewis acidic B2pin2 (Fig. S4 and S5†) facilitates this single-electron reduction.
Next, the presence of radical species in the catalytic cycle was tested by addition of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO, 1.0 eq.) to the mixture, which shows a dramatic drop in product yield (down to 8% see ESI†). This was also evidenced in the 13C NMR spectrum, which shows a line broadened signal for the carbonyl of the benzaldehyde (see ESI Fig. S9†), possibly due to exchange of the radical species (ketyl radical) with benzaldehyde.
Subsequently, a series of control experiments were performed to identify key intermediates. 1,2-Diol, benzyl alcohol and 1,2-diketone were excluded as intermediates, since using them in place of benzaldehyde did not lead to alkene formation (ESI, Scheme S1†). Furthermore, benzylboronic esters S4 and benzyloxyborate ester S5 were identified as by-products in the reaction (Fig. S7†). Most interestingly, control experiments in the presence of D2O (10.0 eq.) quenched the reaction significantly, affording the alkene in trace amounts along with the formation of deuterated (at the benzylic position) boronic esters S4-D and borate ester S5-D (Scheme 2). In contrast, with only d7-DMF as solvent, deuterium was not incorporated in the products or the boronic ester S4 and borate ester S5. These findings suggest intermediate boron-related species, such as an α-oxyboryl carbanion22 and an α-boryl carbanion23 in the reaction mechanism.
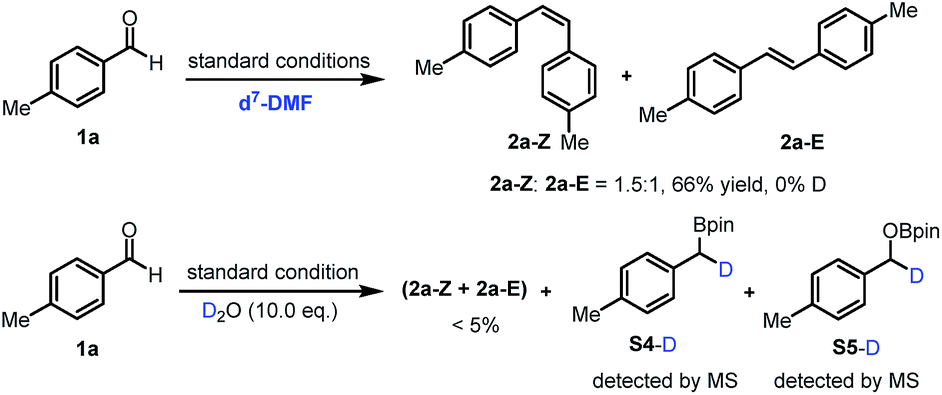 | ||
| Scheme 2 Deuteration experiments. | ||
Next, a systematic in situ illumination NMR study was carried out to directly detect these reaction intermediates. To monitor this unusual transformation from carbonyl groups to double bonds and to boost sensitivity of otherwise insensitive 13C signals, benzaldehyde specifically 13C labelled at the carbonyl position was used (see Fig. 3A). This enabled us not only to predominately track the chemical modulations at the carbonyl position, but also to identify the number of protons bound to the carbon in several intermediate species by 1H coupled 1D 13C experiments (see below); therefore, in the following only the 13C signals of the labelled carbon are discussed.
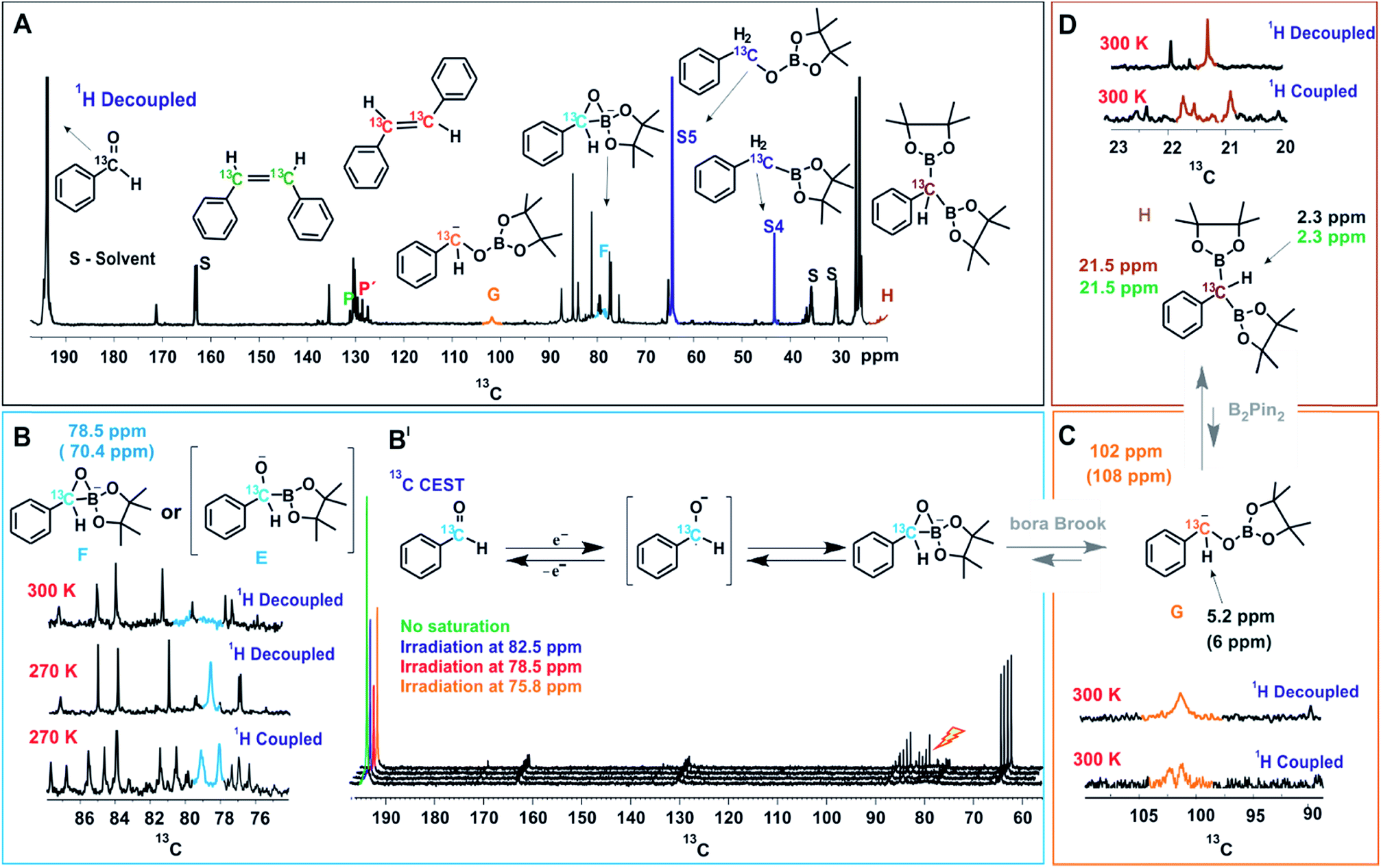 | ||
| Fig. 3 NMR Studies of the Reaction Intermediates. (A) In situ13C spectrum of the reaction mixture after 18 hours of illumination, the observed intermediate peaks are marked with respective colors and stable intermediates are directly compared to independently synthesized compounds. (B) Stabilization and characterization of transient intermediate F is achieved at low temperature (270 K) and characterized from 1H decoupled and coupled 13C spectra. (B′) 13C CEST spectra establishing initial chemical transformation between benzaldehyde and primary key intermediate F. (C) Identification of α-oxyboryl carbanion G from 13C and 1H chemical shifts (for HSQC see ESI†) and the multiplicity pattern of 13C at the benzylic position. (D) Assignment of intermediate H from the multiplicity pattern of 13C at the benzylic position and 13C and 1H chemical shifts (for HSQC see ESI†). The 1H and 13C chemical shifts of independently synthesized H are given in green. The values inside brackets are calculated chemical shift values. Unless otherwise mentioned, all spectra were measured at 300 K, in a 600 MHz NMR spectrometer. | ||
On irradiation (455 nm, blue LED) of the reaction mixture, besides starting material and product resonances, several new 13C signals are detected, indicating the generation of possible reaction intermediates. Fig. 3A shows the in situ1H decoupled 13C spectrum of the reaction mixture after 18 hours of irradiation. Besides benzaldehyde, the two products (P and P′ in Fig. 3A), and the by-products S4 and S5, several additional peaks appeared. Among them three intermediates were assigned, which are marked with G, F and H and highlighted with different colors in Fig. 3A. To identify and characterize the intermediates, a combined approach of chemical exchange information from 13C CEST (chemical exchange saturation transfer), chemical shift information and multiplicity pattern accessed from 1H coupled 1D 13C spectra was applied. Furthermore, theoretical calculations and spectra of individually synthesized intermediates were used to corroborate the assignment.
To identify the next intermediate formed from benzaldehyde, the spectra are scanned by 13C CEST NMR (detailed information about CEST is given in ESI†).24 In case there is any chemical exchange on the ms time scale with benzaldehyde, saturation can be transferred from the exchanging intermediate onto benzaldehyde and hence the intermediate becomes detectable. The most pronounced intensity drop observed for the benzaldehyde 13CO signal at 193 ppm is on saturation at 78.5 ppm (Fig. 3B′). However, at 300 K, the peak at 78.5 ppm is very broad (blue, Fig. 3B) suggesting a transient nature of the intermediate. To characterize this intermediate further, the temperature was lowered to 270 K resulting in a considerable narrowing of this 13C signal. In addition, the 1H coupled 13C spectrum reveals a 13C–H moiety by appearance of a doublet. From the literature, it was considered that the base might react with the diboron reagent to generate sp3–sp2 diboron species that participates in ketyl radical borylation25 to give α-boryl alkoxide. Furthermore, theoretical calculations predict a 13C chemical shift of 70.4 ppm for a cyclic three-membered anionic species F (Fig. 3B). The open form of the α-boryl alkoxide E was found to be energetically less favorable by theoretical calculations. However we cannot distinguish experimentally between E and F. Based on this evidence, the 13C peak at 78.5 ppm is assigned to the cyclic three-membered species F or to the α-boryl carbonyl peak of benzaldehyde indicate a fast chemical alkoxide E; thus, the above discussed line broadening of exchange with the ketyl radical. In addition, CEST exchange saturation transfer reveals a slow exchange of benzaldehyde with the cyclic three-membered anionic species F or E. Combining the experimental observations we conclude that a sequential exchange process takes place from benzaldehyde through a very short-lived ketyl radical to E or F.
Next, the assignment of the α-oxyboryl carbanion intermediate G is discussed (see Fig. 3C). In a “bora-Brook” rearrangement process this α-oxyboryl carbanion G was found to be generated from the isomerization of α-boryl alkoxide E.22 Furthermore, Nozaki et al. proposed a three-membered-ring species similar to F as the transition state of the C to O boryl migration in the “bora-Brook” rearrangement.22c Given these reports and the oxophilicity of boron (B–O bond BDE = 193 kcal mol−1),26 such a carbon to oxygen boryl migration in the cyclic three-membered anionic species F to generate α-oxyboryl carbanion G is highly probable. The aforementioned control experiments, wherein deuterium-trapped benzyloxyborate ester S5-D was observed, further corroborates the existence of G. Indeed, we could detect a relatively broad peak of very small intensity at 102 ppm in the reaction mixture corresponding to the benzylic carbon of α-oxyboryl carbanion G (Fig. 3A and C). The 1H coupled 13C spectrum shows a doublet (Fig. 3C) indicating a 13CH group and the calculated chemical shifts (13C 108 ppm and 1H 6 ppm) are in good agreement with the assignment to the benzylic carbon of α-oxyboryl carbanion G. Moreover, the essential role of DMF and Cs2CO3 in our reaction is in line with the observation by Nozaki that the presence of a polar solvent and larger alkali metal cation could enhance the nucleophilicity of the anionic oxygen atom in such processes.22c Overall, we conclude that the α-oxyboryl carbanion G is generated via a “bora-Brook” rearrangement from the cyclic three-membered anionic species F.
Previous theoretical calculations showed that the transformation of an α-oxyboryl carbanion G to a 1,1-benzyldiboronate ester H (for structure see Fig. 3D) is thermodynamically and kinetically favorable.27 Therefore, the intermediacy of H in our reaction process was examined with NMR. To identify the chemical shifts of H, a 13C–1H HSQC spectrum (see, ESI†) of the pure independently synthesized intermediate H was measured and revealed a benzylic carbon at 21.5 ppm and the corresponding proton at 2.50 ppm. Indeed, careful observation of the 13C spectra of the reaction mixture revealed a very small peak of H at 21.5 ppm (Fig. 3A and D). The assignment to H was confirmed by a doublet (–13CH) in the 1H coupled 13C spectrum (Fig. 3D) and HSQC spectra of the reaction mixture (ESI†). At present, we can only suggest that H is formed via a nucleophilic attack of the carbanion G to B2pin2 followed by a deoxygenation step. A related mechanism was proposed by Liu and Lan et al. for a borylation of an α-oxyboronic species, in which similar gem-diboron compounds are generated via an oxoanion instead of a carbanion attacking B2pin2.27
gem-Diborylalkanes, such as intermediate H, treated with a suitable base, are known to be deprotonated or mono-deborylated to boryl carbanions.23a–d,g The resulting α-boryl anions react with carbonyl groups through a boron-Wittig pathway to give olefins.23a–d,g Indeed, the olefinic product could be obtained in an independent experiment treating benzaldehyde 1d with 1,1-benzyldiboronate ester H in the presence of Cs2CO3 (Scheme 3A). Moreover, we could trap the α-boryl anion as boronic esters S4-D in the presence of D2O (10.0 eq.) (Scheme 2). This is a strong hint that an α-boryl carbanion is involved in our reaction pathway, although we could not directly identify it via in situ NMR.
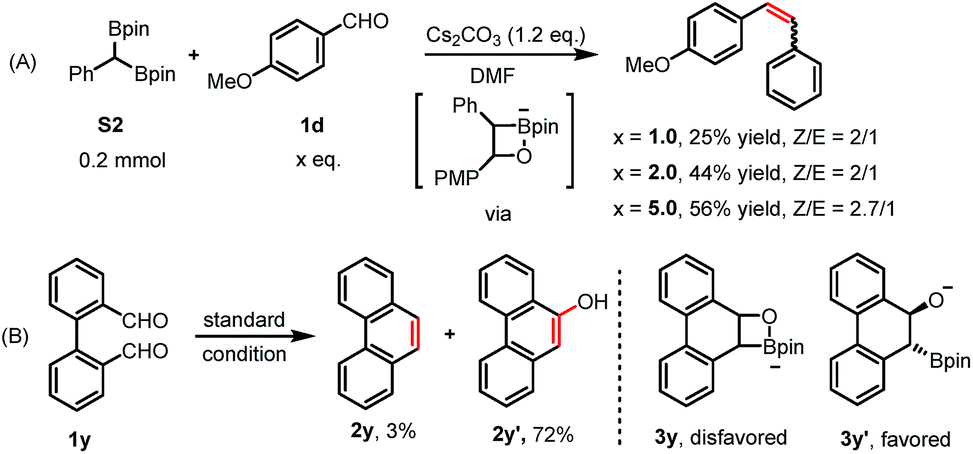 | ||
| Scheme 3 Further mechanistic studies. | ||
2,2′-Diphenyldicarboxaldehyde 1y was used to investigate a potential intramolecular olefination reaction, but 9-phenanthrenol 2y′ was isolated as the main product (72%) with only 3% of phenanthrene 2y (Scheme 3B; for more details, see ESI†).
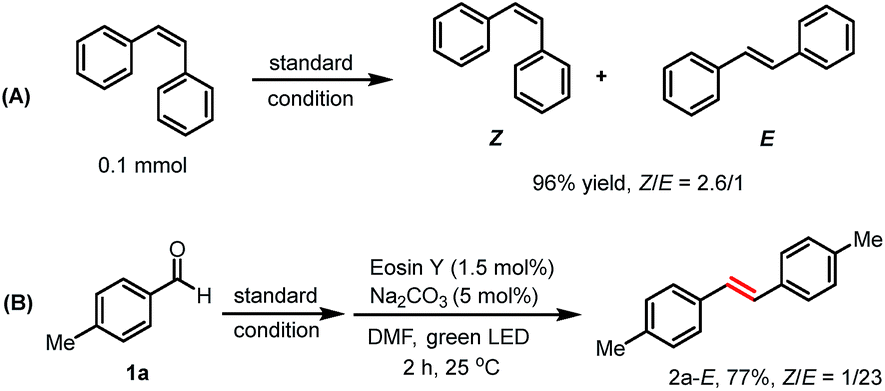
Next, we turned our attention to explore the origin of the Z/E selectivity in the reaction. By NMR, we observed that the more thermodynamically stable E isomer was formed dominantly at the early stage of the reaction. Later, a E to Z isomerization occurs, with the Z isomer being the major product (Table S8†). We rationalize the formation of E-alkenes as the major configuration at the early reaction stage as a consequence of a syn boron–oxygen elimination.23a,28 Additional evidence for the photo-induced alkene isomerization process was obtained when E-alkene was subjected to the standard reaction condition. The corresponding product was obtained in a similar Z/E ratio (Z/E = 2.6/1, 96% yield) as in our reaction system (Scheme 4A).29 We also found that alkene E-2a could be obtained in one pot using the reported photo-sensitized Z to E isomerization strategy, giving alkene E-2a in 77% (Scheme 4B).30
 | ||
| Scheme 4 Isomerization studies. | ||
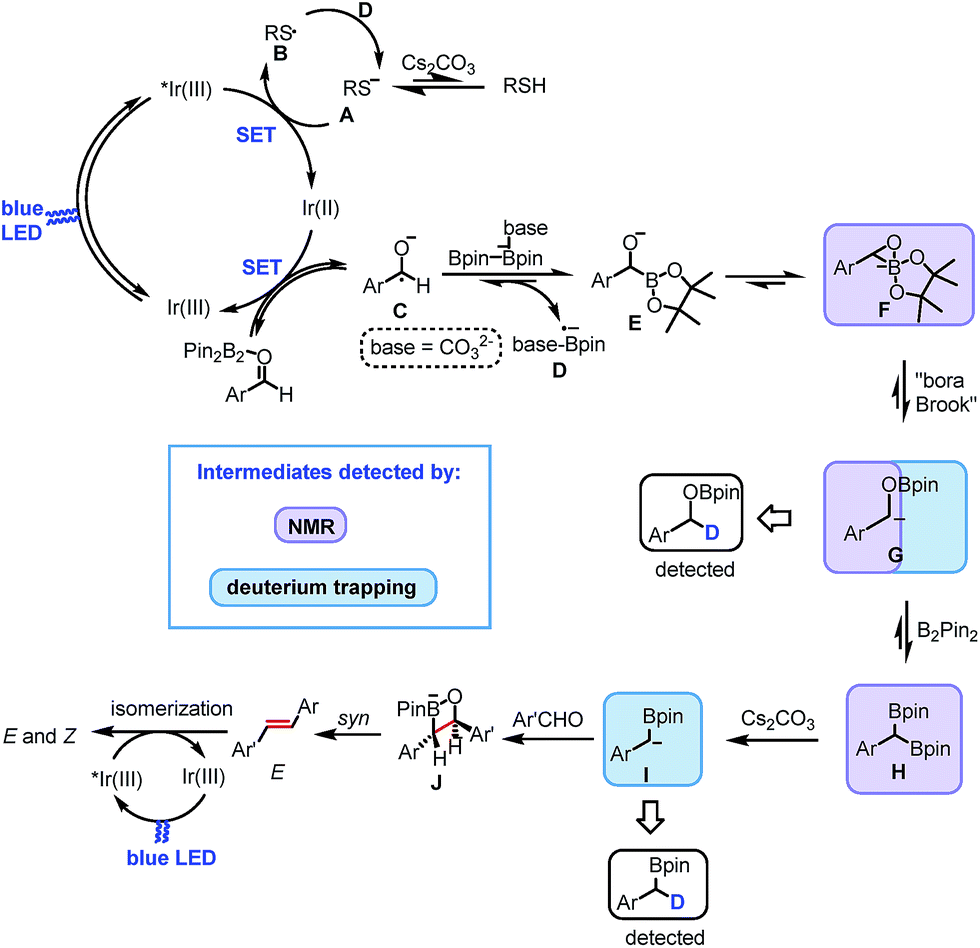
Based on the above experimental evidence and mechanistic pathways previously reported in literature, we propose the mechanism depicted in Scheme 5 for the reported photocatalytic carbonyl–carbonyl olefination of aromatic aldehydes. Initially, the photoexcited state of [IrIII(FCF3ppy)2dtbbpy]+ is reductively quenched by the sulfur anion A, formed by the deprotonation of thiol by base, affording sulfur radical B and [IrII(FCF3ppy)2dtbbpy] (Ir01/2III/II1/2 = −1.38 V vs. SCE in DMF). Single electron transfer (SET) from the Ir(II) species to benzaldehyde in the presence of B2pin2 gives the ground-state Ir(III) and ketyl radical C. Subsequent radical borylation31 of C produces radical anion D and anion E or F. The resulting radical anion D reduces the sulfur radical B back to the anion A. The three-membered cyclic anion F undergoes a “bora-Brook” rearrangement to form α-oxyboryl carbanion G. The resulting carbanion G subsequently reacts with another molecule of B2pin2 leading to the formation of 1,1-benzyldiboronate ester H. The base-promoted mono-deborylation of H gives rise to the formation of α-boryl carbanion I. After nucleophilic attack to the carbonyl group of a second aldehyde, a four-membered cyclic intermediate J is most probably formed, which affords E-alkene via a B–O syn elimination. Finally, energy transfer from the excited state of the photocatalyst to the E-alkene produces a mixture of Z and E isomers as the final product.
 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed a photoredox-catalyzed reaction for reductive carbonyl–carbonyl olefination of aromatic aldehydes using B2pin2 as both the oxygen atom trap and the terminal reductant. The reaction system provides a mild and efficient method to prepare both symmetrical and unsymmetrical diarylalkenes through intermolecular bicarbonyl olefination and tolerates a broad range of functional groups. Combining our in situ illumination NMR technique with a series of mechanistic studies, α-oxyboryl carbanion, α-boryl carbanion and 1,1-benzyldiboronate esters were detected as key intermediate species in the reaction. Furthermore, theoretical calculations corroborate the NMR observation of the cyclic three-membered anionic species involved in the “bora-Brook” rearrangement. Mechanistic studies support the hypothesis that the formation of the double bond is facilitated by a boron-Wittig process. This combination of photoredox catalysis with boron chemistry constitutes a unique example of an Umpolung strategy to convert an aldehyde into a boryl-functionalized carbanion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Science Foundation (DFG) (GRK 1626, Chemical Photocatalysis; KO 1537/18-1). This project has received funding from the European Research Council (ERC) under the European Unions Horizon 2020 research and innovation programme (grant agreement No. 741623). S. W. thanks the China Scholarship Council (CSC) for a predoctoral fellowship (CSC student number 201606280052). N. L. acknowledges ERC (ERC CoG-614182 – IonPairsAtCatalysis) for the funding. We are grateful to Dr Gregory S. Huff (University of Regensburg) for helpful discussions and writing suggestions. We further thank Dr Rudolf Vasold (University of Regensburg) for his assistance in GC-MS measurements and Ms Regina Hoheisel (University of Regensburg) for her assistance in cyclic voltammetry measurements.Notes and references
- (a) T. Takeda, Modern carbonyl olefination: Methods and applications, John Wiley & Sons, 2006 Search PubMed; (b) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- D. J. Peterson, J. Org. Chem., 1968, 33, 780–784 CrossRef CAS.
- J. B. Baudin, G. Hareau, S. A. Julia and O. Ruel, Tetrahedron Lett., 1991, 32, 1175–1178 CrossRef CAS.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS.
- (a) A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243 CrossRef CAS PubMed; (b) O. Eivgi and N. G. Lemcoff, Synthesis, 2018, 50, 49–63 CrossRef CAS; (c) Y. Vidavsky and N. G. Lemcoff, Beilstein J. Org. Chem., 2010, 6, 1106–1119 CrossRef CAS PubMed.
- (a) J. R. Ludwig and C. S. Schindler, Synlett, 2017, 28, 1501–1509 CrossRef CAS PubMed; (b) L. Ravindar, R. Lekkala, K. P. Rakesh, A. M. Asiri, H. M. Marwani and H.-L. Qin, Org. Chem. Front., 2018, 5, 1381–1391 RSC; (c) M. R. Becker, R. B. Watson and C. S. Schindler, Chem. Soc. Rev., 2018, 47, 7867–7881 RSC.
- J. E. McMurry, Chem. Rev., 1989, 89, 1513–1524 CrossRef CAS.
- A. Fürstner and B. Bogdanović, Angew. Chem., Int. Ed. Engl., 1996, 35, 2442–2469 CrossRef.
- A. Fürstner and A. Hupperts, J. Am. Chem. Soc., 1995, 117, 4468–4475 CrossRef.
- M. Moxter, J. Tillmann, M. Füser, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2016, 22, 16028–16031 CrossRef CAS PubMed.
- K. Esfandiarfard, J. Mai and S. Ott, J. Am. Chem. Soc., 2017, 139, 2940–2943 CrossRef CAS PubMed.
- W. Wei, X.-J. Dai, H. Wang, C. Li, X. Yang and C.-J. Li, Chem. Sci., 2017, 8, 8193–8197 RSC.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (e) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (f) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (g) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (h) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- (a) H. G. Yayla and R. R. Knowles, Synlett, 2014, 25, 2819–2826 CrossRef CAS; (b) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (c) N. Hoffmann, Eur. J. Org. Chem., 2017, 2017, 1982–1992 CrossRef CAS; (d) K. N. Lee and M.-Y. Ngai, Chem. Commun., 2017, 53, 13093–13112 RSC.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- (a) L. Deloux and M. Srebnik, Chem. Rev., 1993, 93, 763–784 CrossRef CAS; (b) A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed; (c) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945–962 CrossRef; (d) K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 1999, 527–538 CrossRef.
- (a) D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed; (b) S. Bae and M. K. Lakshman, J. Org. Chem., 2008, 73, 1311–1319 CrossRef CAS PubMed; (c) V. Gurram, H. K. Akula, R. Garlapati, N. Pottabathini and M. K. Lakshman, Adv. Synth. Catal., 2015, 357, 451–462 CrossRef CAS PubMed; (d) J. Kim and C. R. Bertozzi, Angew. Chem., Int. Ed., 2015, 54, 15777–15781 CrossRef CAS PubMed; (e) H. Lu, Z. Geng, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 2774–2776 CrossRef CAS PubMed; (f) K. Yang, F. Zhou, Z. Kuang, G. Gao, T. G. Driver and Q. Song, Org. Lett., 2016, 18, 4088–4091 CrossRef CAS PubMed; (g) M. Rauser, C. Ascheberg and M. Niggemann, Angew. Chem., Int. Ed., 2017, 56, 11570–11574 CrossRef CAS PubMed; (h) W. Fu and Q. Song, Org. Lett., 2018, 20, 393–396 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. S. Hamilton, Synlett, 2014, 25, 1191–1196 CrossRef PubMed; (b) N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed; (c) M. Weiser, S. Hermann, A. Penner and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2015, 11, 568–575 CrossRef CAS.
- (a) Y.-P. Zhao, L.-Y. Yang and R. S. H. Liu, Green Chem., 2009, 11, 837–842 RSC; (b) W. Cai, H. Fan, D. Ding, Y. Zhang and W. Wang, Chem. Commun., 2017, 53, 12918–12921 RSC; (c) X.-J. Wei, W. Boon, V. Hessel and T. Noël, ACS Catal., 2017, 7, 7136–7140 CrossRef CAS PubMed; (d) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275–5278 CrossRef CAS PubMed.
- However, in all cases, the homo-coupled products of two aldehydes were observed (the yields were given in Table 2).
- (a) C. Feldmeier, H. Bartling, E. Riedle and R. M. Gschwind, J. Magn. Reson., 2013, 232, 39–44 CrossRef CAS PubMed; (b) C. Feldmeier, H. Bartling, K. Magerl and R. M. Gschwind, Angew. Chem., Int. Ed., 2015, 54, 1347–1351 CrossRef CAS PubMed; (c) H. Bartling, A. Eisenhofer, B. König and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860–11871 CrossRef CAS PubMed.
- (a) D. S. Matteson, Aust. J. Chem., 2011, 64, 1425–1429 CrossRef CAS; (b) D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed; (c) H. Kisu, H. Sakaino, F. Ito, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2016, 138, 3548–3552 CrossRef CAS PubMed.
- (a) I. Marek and J.-F. Normant, Chem. Rev., 1996, 96, 3241–3268 CrossRef CAS; (b) T. Klis, S. Lulinski and J. Serwatowski, Curr. Org. Chem., 2010, 14, 2549–2566 CrossRef CAS; (c) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC; (d) C. Wu and J. Wang, Tetrahedron Lett., 2018, 59, 2128–2140 CrossRef CAS; (e) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581–10584 CrossRef CAS PubMed; (f) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed; (g) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306–1327 CrossRef CAS.
- N. Lokesh, A. Seegerer, J. Hioe and R. M. Gschwind, J. Am. Chem. Soc., 2018, 140, 1855–1862 CrossRef CAS PubMed.
- (a) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (b) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS PubMed; (c) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed; (d) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS PubMed; (e) J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed; (f) F. Sandfort, F. Strieth-Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem.–Eur. J., 2018, 24, 17210–17214 CrossRef CAS PubMed; (g) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed; (h) J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161–166 RSC; (i) G. Yan, D. Huang and X. Wu, Adv. Synth. Catal., 2018, 360, 1040–1053 CrossRef CAS.
- Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw-Hill, NewYork, NY, 15th edn, 1998 Search PubMed.
- L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257 CrossRef CAS PubMed.
- A. Pelter, D. Buss, E. Colclough and B. Singaram, Tetrahedron, 1993, 49, 7077–7103 CrossRef CAS.
- J. Metternich and R. Gilmour, Synlett, 2016, 27, 2541–2552 CrossRef CAS.
- J.-J. Zhong, Q. Liu, C.-J. Wu, Q.-Y. Meng, X.-W. Gao, Z.-J. Li, B. Chen, C.-H. Tung and L.-Z. Wu, Chem. Commun., 2016, 52, 1800–1803 RSC.
- (a) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (b) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS PubMed; (c) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed; (d) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS PubMed; (e) J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed; (f) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed; (g) J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161–166 RSC; (h) G. Yan, D. Huang and X. Wu, Adv. Synth. Catal., 2018, 360, 1040–1053 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00711c |
| This journal is © The Royal Society of Chemistry 2019 |