
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoredox Ni-catalyzed peptide C(sp2)–O cross-coupling: from intermolecular reactions to side chain-to-tail macrocyclization†
Hyelee
Lee
 *a,
Nicolas C.
Boyer
a,
Qiaolin
Deng
b,
Hai-Young
Kim‡
c,
Tomi K.
Sawyer
d and
Nunzio
Sciammetta
*a
*a,
Nicolas C.
Boyer
a,
Qiaolin
Deng
b,
Hai-Young
Kim‡
c,
Tomi K.
Sawyer
d and
Nunzio
Sciammetta
*a
aDepartment of Discovery Chemistry, MRL, Merck & Co., Inc., Boston, Massachusetts 02115, USA. E-mail: hyelee.lee@merck.com; nunzio.sciammetta@merck.com
bComputational and Structural Chemistry, MRL, Merck & Co., Inc., Kenilworth, New Jersey 07033, USA
cProcess and Analytical Research and Development, MRL, Merck & Co., Inc., Boston, Massachusetts 02115, USA
dChemistry Capabilities for Accelerating Therapeutics, MRL, Merck & Co., Inc., Boston, Massachusetts 02115, USA
First published on 12th April 2019
Abstract
Ni/photoredox (4DPAIPN) dual catalysis enabled challenging peptide C(sp2)–O coupling reactions. Successful cross-coupling reactions were demonstrated with highly functionalized alcohols including side chains of amino acids (i.e., serine, threonine, tyrosine), trans-4-hydroxy-L-proline, alkyl alcohols, alkynylated alcohols, and carbohydrates. Coupling reactions between bromobenzoyl-capped peptides containing various side chains and either a protected serine building block or a serine-containing dipeptide also proceeded efficiently. Chemoselective C–O coupling (over C–N) was achieved in intermolecular reactions in the presence of a C-terminal primary amide. Furthermore, by judicious structural design in combination with computational modeling, we demonstrated side chain-to-tail macrocyclization of peptides containing a β-turn motif via C–O coupling. The methodology developed in this work brings new opportunities for late-stage diversification of complex linear and macrocyclic peptides.
Introduction
Peptides are increasingly gaining attention as compelling therapeutic agents in drug discovery because of various attractive properties such as high binding affinities for flat or extended protein surfaces, exquisite target selectivities, good efficacy and safety profile. Recent successes in the development of peptide therapeutics and their increasing testing in clinical trials provide clear evidence that they are alternative modalities for challenging intracellular targets. They expand the druggable target space (e.g., protein–protein interactions, allosteric binding sites) that historically has been inaccessible by traditional small molecules or biologics.1In structure–activity relationship (SAR) efforts surrounding peptide-based inhibitors, medicinal chemists routinely seek alteration of amino acid sequences, incorporation of D-amino acids, unnatural amino acids, N-methylation, peptidomimetics, peptoids, etc. However, these approaches may be limited by their heavy reliance on the availability of costly building blocks. Therefore, elaboration via post-synthetic modification is of particular importance.2 Macrocyclization is often utilized to improve metabolic and chemical stability, biological activity, selectivity and pharmacokinetic properties of peptide leads.3 The most common synthetic methods4 include macrolactamization, ring-closing metathesis, azide–alkyne cycloaddition, and disulfide or thioether5 bond formations. More recently, N-arylation,6S-arylation,7 and decarboxylative conjugate addition8 have been reported (Fig. 1A). These methods allow access to new amide, C(sp2)–C(sp2), C(sp2)–N, S–S, C(sp3)–S, C(sp2)–S, and C(sp3)–C(sp3) bonds. However, some of these scaffolds suffer from poor proteolytic and chemical stability (e.g., thioether-containing peptides) and/or low cell permeability (e.g., N–H-containing cross-linkage). To address these issues, we hypothesized that the underutilized ether linkage (C(sp2)–O)9 could provide proteolytic and chemical stability and nullify hydrogen-bond donor (HBD) ability, which can considerably affect cell permeating properties of peptides, in particular passive transport. Interestingly, macroetherification has been scarcely studied in the context of cyclic peptide synthesis. This is presumably due to synthetic challenges associated with the highly functionalized nature of peptides and the inherently low nucleophilicity of oxygen compared to other heteroatoms. The few reported examples of macroetherification of peptidic substrates are Ullmann-type reactions, SNAr, SN2-type displacement, or Tsuji–Trost reactions (Fig. 1B).10 These methodologies typically require excess amounts of strong bases, high temperature, highly activated aryl halides, and/or preparation of templates. To the best of our knowledge, side chain functionalization of serine, threonine and tyrosine is underexplored in contrast to cysteine or lysine.11 Herein, we report Ni-catalyzed photoredox peptide C(sp2)–O cross-coupling for the late-stage diversification of linear peptides and macrocylization of peptidomimetics.
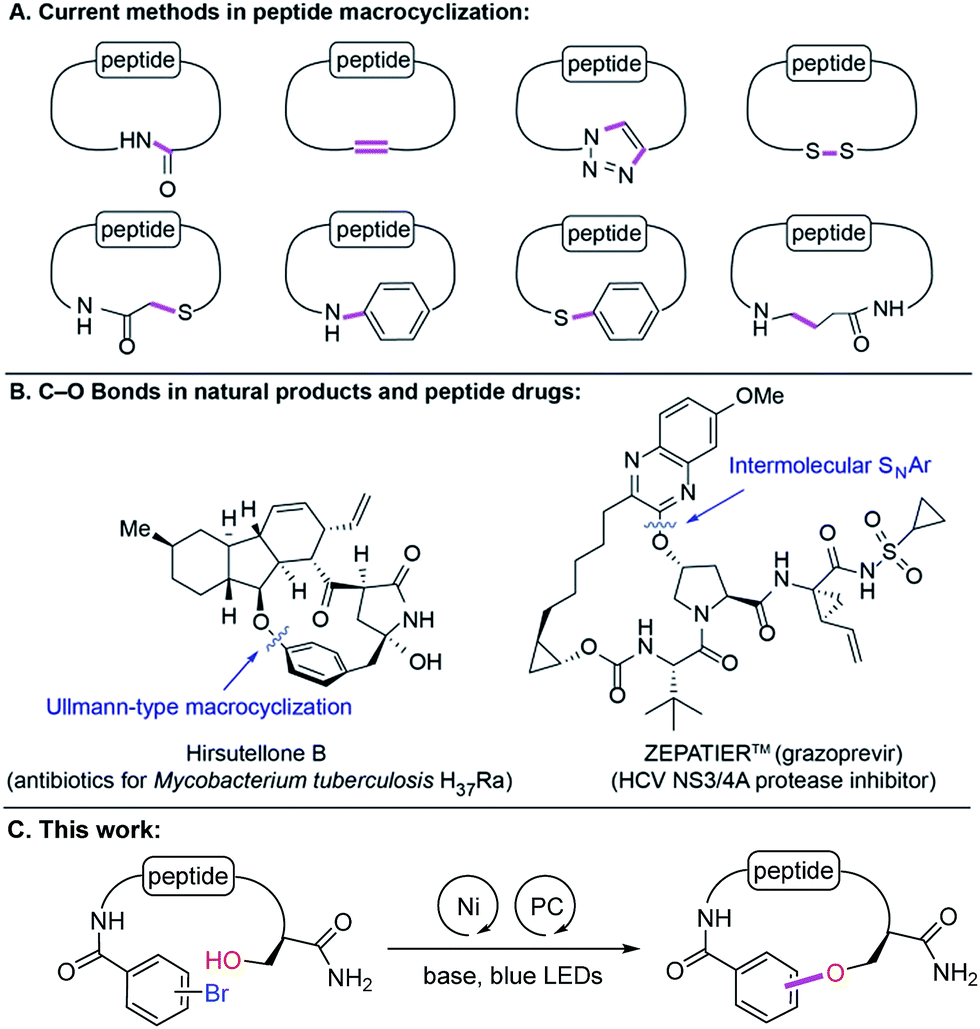 | ||
| Fig. 1 A) Existing synthetic methods in peptide macrocyclization. (B) Examples of C–O bond containing macrocyclic natural products and peptide drugs. (C) Ni-catalyzed photoredox C–O coupling of peptides. PC, photocatalyst. | ||
Results and discussion
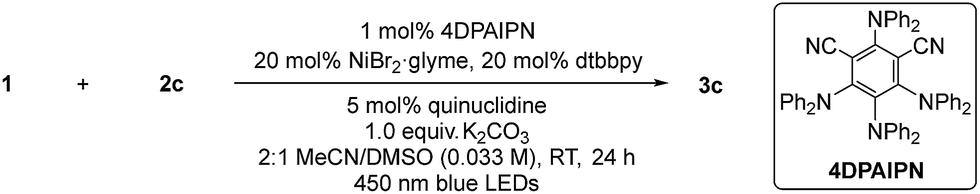
Seminal work from the MacMillan group demonstrated Ni-catalyzed C(sp2)–O coupling enabled by modulating oxidation states of nickel via photoredox catalysis.12 We envisioned that this approach could provide a general strategy to access C–O bonds in peptide post-synthetic modifications under mild conditions (Fig. 1C). Despite its undeniable advantages and generality, metallaphotoredox-mediated C–O coupling in peptides presents several challenges:13 (1) intrinsic poor nucleophilicity of alcohols, (2) presence of multiple epimerizable α-carbon centers in peptide backbones, (3) potential coordination of backbone amides to Ni(II),14 (4) chemoselectivity of C–O vs. C–N coupling, and (5) compatibility with various functional groups on side chains.Taking these potential challenges into account, we explored the coupling between dipeptides bearing an aryl bromide motif and the side chain of a protected serine derivative. During preliminary investigations, we observed slow oxidative addition of Ni(0) into an unactivated aryl bromide (e.g., bromophenyl alanine-containing dipeptides). We speculated that the presence of amide functionalities in peptide backbones, which could form stable Ni(II)-peptide complexes, may interfere with the productive Ni-catalytic cycle. To circumvent this issue, we designed a general strategy to incorporate the aryl bromide unit with an electron-withdrawing group by acylation of the N-terminus with bromobenzoyl chloride. We propose that the activated aryl bromide system would facilitate oxidative addition step and allow the equilibrium shifting towards the productive catalytic cycle. With this design, we initiated cross-coupling optimization between 1 and 2c (Fig. 2). Using conventional methods for optimization screening, 1,3-dicyano-2,4,5,6-tetrakis(diphenylamino)-benzene (4DPAIPN)15 was identified as the optimal photocatalyst. This organic dye was recently reported as one of the metal-free organic donor–acceptor photocatalysts15 and is substantially inexpensive compared to metal-based photocatalysts (e.g., Ir- or Ru-based catalysts). In parallel with optimization by iterative batch reactions,16 we utilized in-house capability of nano-scale high-throughput experimentation, which allows thorough screening for optimal conditions in a multireaction parameter platform.17 We simultaneously examined 240 combinations, and the best one was identified as 20 mol% NiBr2·glyme, 20 mol% 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbbpy), 1 equiv. K2CO3, 5 mol% 4DPAIPN, 5 mol% quinuclidine, N-methyl-2-pyrrolidone (NMP, 0.05 M).
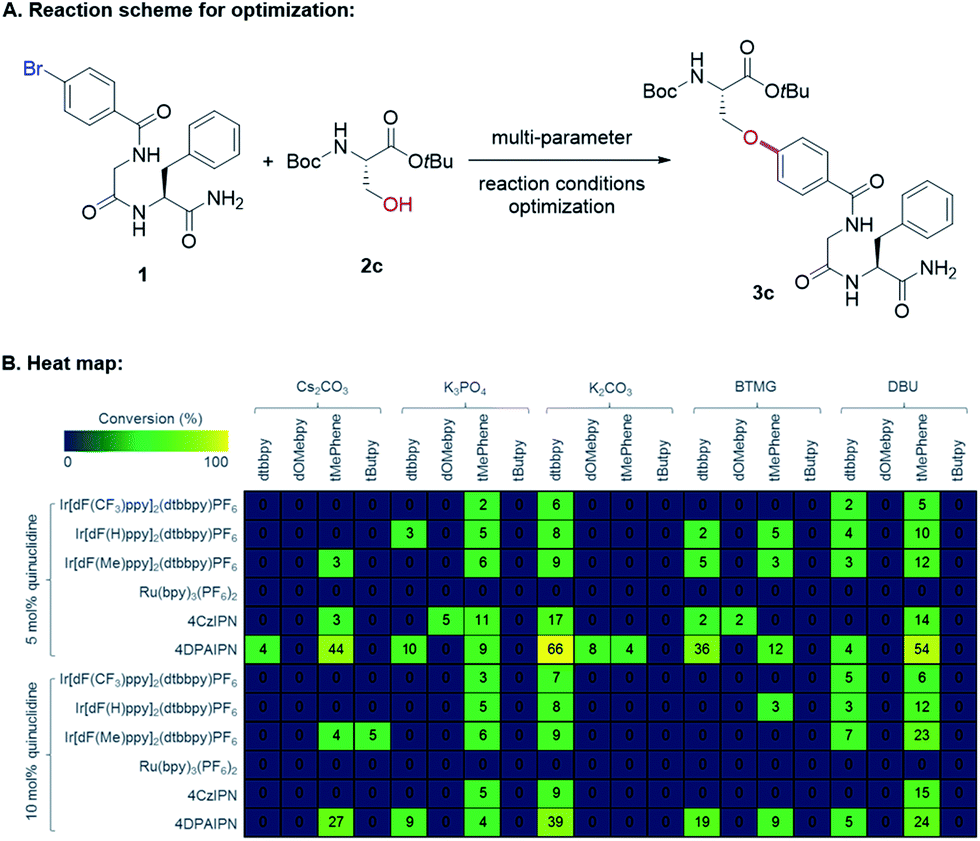 | ||
| Fig. 2 Multi-parameter reaction conditions optimization by nanomole-scale high-throughput experimentation. (A) Model reaction for peptide C–O cross-coupling. (B) Heat map showing data from 240 nano-scale reactions analyzed by UPLC-MS.16 | ||
Upon further exploration of reaction parameters, we validated the optimized conditions shown in the scheme of Table 1. When NiCl2·glyme was used as the nickel(II) source, substantial amounts of the corresponding aryl chloride of the starting material 1 was formed, thus lowering the yield (Table 1, entry 2). Considering slow oxidative addition of nickel to the aryl chloride,18 the aryl chlorides presumably inhibit the nickel catalytic cycle (Fig. S3†). A commonly used iridium-based photocatalyst (Ir[dF(CF3)ppy]2(dtbbpy)PF6)12 showed reduced reactivity (61%, entry 3) compared to 4DPAIPN (81%). One noticeable difference between the iridium catalyst and 4DPAIPN is that in its reduced form, 4DPAIPN is a better reductant than the Ir-catalyst (E1/2red[PC/PC−˙] = −1.52 V vs. E1/2red[IrIII/IrII] = −1.37 V), possibly facilitating single electron reduction of Ni(I) to Ni(0) to close the dual catalytic cycle.19 The major side-product was the phenol derivative, likely originating from the reaction of 1 with adventitious water present in the reaction mixture. Addition of water was detrimental to productivity of the desired C–O coupling (entry 4). In this case, the formation of the phenol side-product was not noticeable but there were substantial amounts of unreacted aryl bromide starting material, which may arise from quenching of the nickel catalyst by the excess water. Entry 5 clearly supports existence of a competing pathway where water can react with 1 to generate the corresponding phenol. Addition of molecular sieves did not significantly improve reaction efficiency, possibly due to much of the incident light being scattered off by the suspended particles (entry 6). Increasing photocatalyst loading was not beneficial (entry 7). In the absence of quinuclidine, the yield was diminished (81% to 64%), possibly because of the beneficial role of quinuclidine as an electron shuttle and/or reductant (entry 8).12,20
|
|
||
|---|---|---|
| Entry | Variations from above | Yielda (%) |
| a Yields were determined by 1H NMR spectroscopy against 1,3,5-trimethoxybenzene as an internal standard. b ArCl side-product was observed. c Yield of ArOH. | ||
| 1 | Conditions above | 81 |
| 2 | NiCl2·glyme instead of NiBr2·glyme | 61b |
| 3 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 instead of 4DPAIPN | 61 |
| 4 | 20 equiv. water added | 24 |
| 5 | 5 equiv. water instead of 2c | 43c |
| 6 | 4Å molecular sieves added | 77 |
| 7 | 5 mol% 4DPAIPN | 77 |
| 8 | No quinuclidine | 64 |
With optimized reaction conditions in hand, we examined the substrate scope of various alcohols in an intermolecular C–O coupling. Aliphatic alcohols successfully coupled with aryl bromide dipeptide 1, giving the desired products in 97% and 58% yield (Table 2, 3a and 3b), respectively. Surprisingly, a seemingly minor change in the ester protecting group of L-serine – switching from tert-butyl (2c) to methyl (2d) – led to diminished yield (81% to 53%). This may arise from instability of both starting material (2d) (hydrolysis by residual water to the corresponding acid) and product (3d) (β-elimination). The secondary alcohol from protected trans-4-hydroxy-L-proline (2e) provided the desired ether linkage in moderate yield (3e, 52%). 1,3-Propanediol (2f) was also examined to evaluate the formation of the corresponding di-ether. Using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1:2f, the major product was the mono-ether adduct (3f, 63% and 3f′, 25%). We then tested tolerance to functional groups commonly utilized in bioconjugation chemistry. Terminal alkyne 2g was tolerated (3g, 62%), whereas azide 2h was incompatible with the reaction conditions. Protected 6-amino-1-hexanol (2i) and α-D-galactopyranose (2j) can also undergo the desired coupling reactions with good efficiency (3i, 44% and 3j, 54%). We also explored the cross-coupling of hydroxyl-containing dipeptides (2k–2n). Serine-containing dipeptides were successfully coupled with 1 (3k, 60% and 3l, 38%). Reaction between 1 and the more sterically demanding threonine-containing peptide 2m afforded the desired alkyl aryl ether in moderate yield, likely due to poor solubility of 2m.
:1 ratio of 1:2f, the major product was the mono-ether adduct (3f, 63% and 3f′, 25%). We then tested tolerance to functional groups commonly utilized in bioconjugation chemistry. Terminal alkyne 2g was tolerated (3g, 62%), whereas azide 2h was incompatible with the reaction conditions. Protected 6-amino-1-hexanol (2i) and α-D-galactopyranose (2j) can also undergo the desired coupling reactions with good efficiency (3i, 44% and 3j, 54%). We also explored the cross-coupling of hydroxyl-containing dipeptides (2k–2n). Serine-containing dipeptides were successfully coupled with 1 (3k, 60% and 3l, 38%). Reaction between 1 and the more sterically demanding threonine-containing peptide 2m afforded the desired alkyl aryl ether in moderate yield, likely due to poor solubility of 2m.
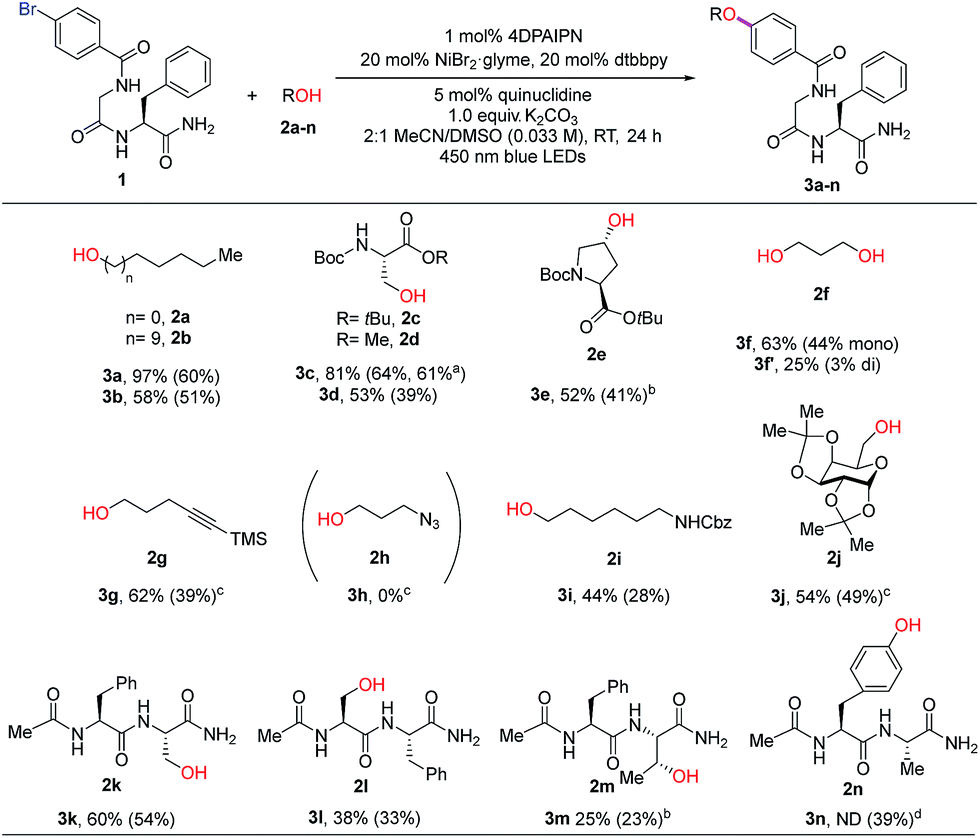
An unexpected but intriguing result is the successful coupling of tyrosine-containing dipeptide 2n, furnishing a C(sp2)–O–C(sp2) motif abundant in natural products and important glycopeptide antibiotics (e.g., vancomycin family). Compared to aliphatic alcohol substrates, there may be an alternative mechanistic pathway in play for tyrosine-based substrates (Fig. S4†). Considering the redox potential and bond dissociation free energy (BDFE) of the phenolic O–H bond being relatively high,22 the possibility of direct hydrogen atom transfer (HAT) mechanism is improbable. Alternatively, a transient phenoxy radical could be generated by either oxidation of phenolate or proton-coupled electron transfer (PCET), which is widely discussed in Tyr redox chemistry.23
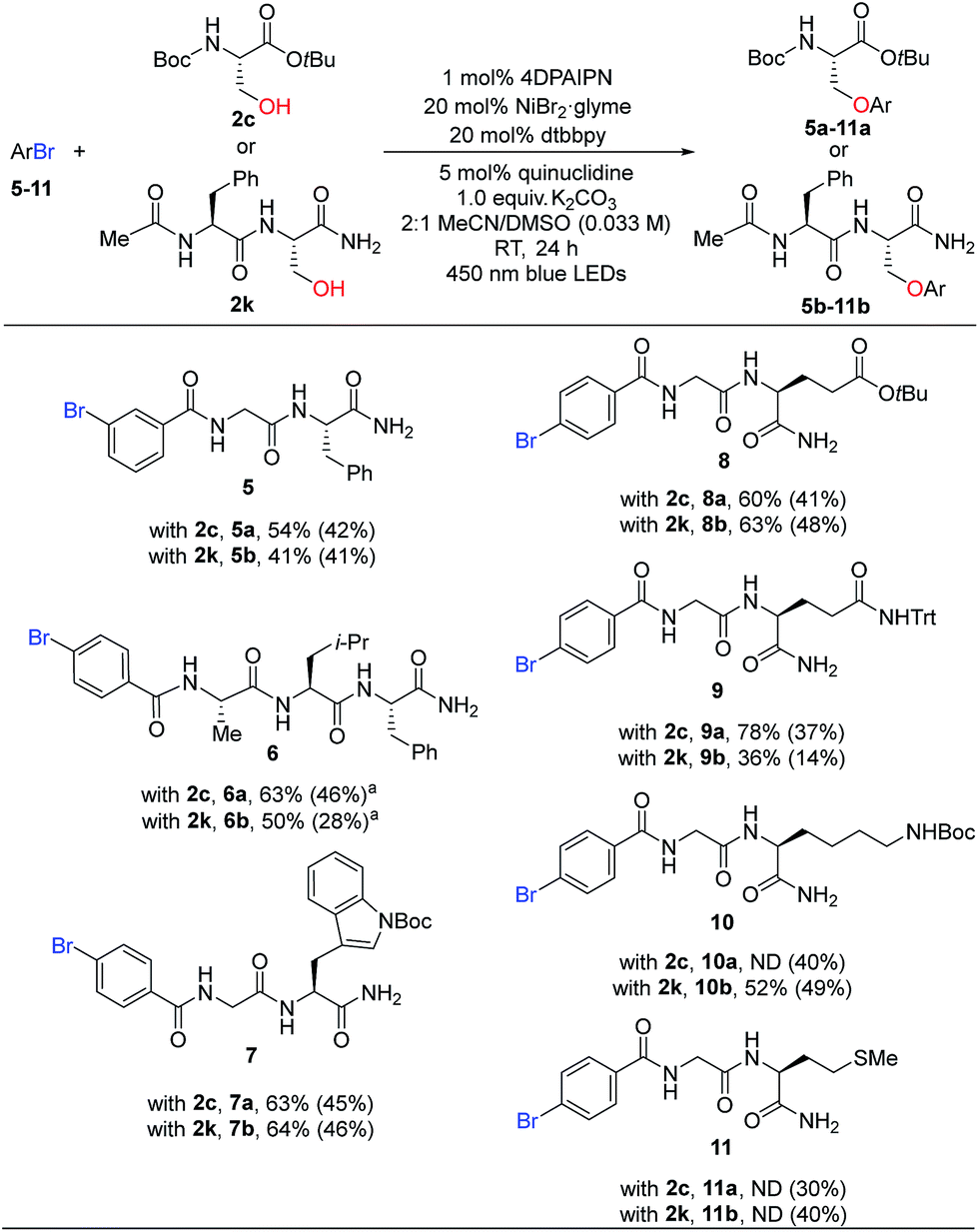
We then turned our attention to the substrate scope for the bromobenzoyl-capped peptides. By changing the substituent pattern of the bromide position from para (1) to meta (5), the reaction efficiency was moderately reduced for both coupling counterparts 2c and 2k (Table 3, 5a, 54% vs.3c, 81% and 5b, 41% vs.3k, 60%). Attenuated reactivity of 5 could be imputed to a slower oxidative addition of Ni(0) associated with the electron-withdrawing group at the meta position, in addition to greater sterics around the nickel coordination site compared to the para-substituted substrate. Tripeptide 6 was successfully coupled with either 2c or 2k to give desired products, 6a or 6b, respectively. Importantly, dipeptides bearing various functional groups (indole, carboxylic acid, amide, amine, thioethers from the side chains of Trp, Glu, Gln, Lys, Met) were also competent partners for peptide C–O coupling (7a–11a and 7b–11b).
| a 0.05 mmol ArBr scale. ND, not determined. |
|---|
|
To investigate peptide macrocyclization through C–O cross-coupling, we designed two model systems (Fig. 3A): (1) a tripeptide C-terminally modified with 4-amino-1-butanol (12) and (2) a pentapeptide containing the turn-inducer motif D-Pro–L-Pro (13).24 The former design affords conformational flexibility between the two reactive sites, while the latter has a higher propensity to form a β-hairpin that could promote substrate preorganization through intramolecular hydrogen bonds. To assess feasibility of such macrocyclization, we utilized conformational sampling to calculate accessible conformations of model system of linear substrates. A hundred thousand conformations were generated by using our in-house implementation of the distance geometry approach (Fig. S5†).25 The conformations were minimized by Merck Molecular Force Field (MMFFs)26 and clustered into 1500 conformations. The conformations within 2 kcal mol−1 compared to the lowest energy are exemplified in Fig. 3A. Linear precursor 12 appears to be highly flexible and disorganized while 13 is relatively well-organized with clear intramolecular hydrogen bonding interactions (yellow dashed lines in Fig. 3A, design 2). To our delight, Ni-catalyzed photoredox macrocyclization of 13 afforded the desired cyclized product in 67% isolated yield with two separable regioisomers (7:3 C–O/C–N connectivity) as confirmed by 1D and 2D NMR analyses.27 However, reactions attempted with 12 resulted primarily in starting material recovery, in addition to the formation of debrominated and phenol side-products (3% conversion to the cyclized product observed by UPLC-MS analysis). With the success in macrocyclization of peptide 13via C–O cross-coupling, we further examined substrates containing the D-Pro–L-Pro motif. Inversion of stereochemical configuration at the C-terminus was well tolerated with slightly diminished selectivity (14a, 67%, 6:4 C–O/C–N). Modification to methyl ester at the C-terminus allowed formation of the C–O macrocyclic peptide 15a in 42% yield. Interestingly, by eliminating the hydroxymethylene side chain (Ser to Gly), the C–N cross-coupling product was formed exclusively in good yield (16a, 41%). A pentapeptide containing Trp and Glu residues also underwent efficient macrocyclization (17a, 45%, 6:4 C–O/C–N). Macrocyclization of a longer peptide (7-mer) proceeded well, furnishing the desired C–O coupling product to give the 26-membered macrocyclic peptide 18a (27%).
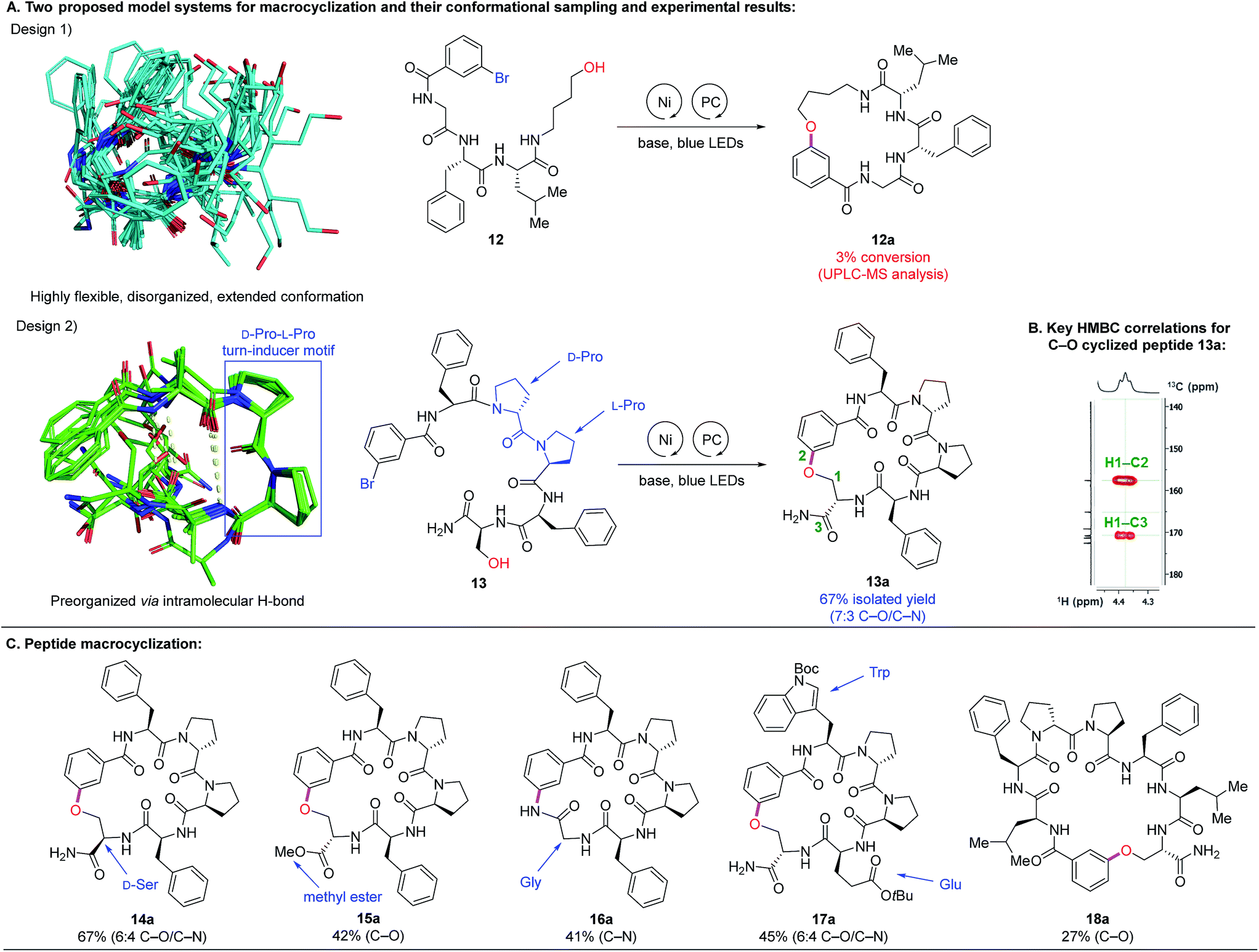 | ||
| Fig. 3 Macrocyclization of peptides with a β-hairpin motif (design 2) via Ni-catalyzed photoredox C–O coupling (modeling and experimental results). (A) Two proposed designs for macrocyclization. (B) Key 1H–13C HMBC correlations confirming C (sp2)–O bond formation for 13a. (C) Examples of macrocyclic peptides. Isolated yields are reported after purification using preparative reverse-phase HPLC. General reaction conditions: 0.05 mmol scale, 1 equiv. K2CO3, 10 mol% 4DPAIPN, 5 mol% quinuclidine, 20 mol% NiBr2·glyme, 20 mol% dtbbpy, 2:1 MeCN/DMSO (0.005 M), RT, 24 h, 450 nm blue LEDs using MSD photoreactor. | ||
Conclusions
We have demonstrated that a challenging peptide C–O cross-coupling can be achieved by Ni-catalyzed photoredox catalysis. Successful intermolecular etherification was reported between various alcohols and bromobenzoyl-capped peptides with high chemoselectivity and good yields. Strategic structural designs with the aid of computational modeling enabled side chain-to-tail macrocyclization. This work expands chemical space to broaden the applicability of cyclic peptides as powerful ligands and potential leads for challenging targets. Furthermore, we plan to utilize such synthetic methodology to advance macrocyclic peptides and elucidate their structure-cell permeability relationships. Specifically, efforts towards the direct comparison of cell permeability and stability of C–O vs. C–N or C–S containing macrocyclic peptides are ongoing. Results will be reported in due course.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Postdoctoral Research Program of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. We thank Prof. David W. C. MacMillan and his group for helpful discussions. We also thank Valerie Shurleff and Brandon Vara for helpful discussions and reviewing this paper. We greatly acknowledge Miroslawa Darlak, Spencer McMinn, Lisa Nogle, Mark Pietrafitta, David Smith for purification of the final compounds. We also thank Adam Beard and Shishi Lin for HRMS and HTE supports, respectively.References
- For select reviews, see: (a) P. G. Dougherty, Z. Qian and D. Pei, Biochem. J., 2017, 474, 1109 CrossRef CAS PubMed; (b) D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533 CrossRef CAS; (c) N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454 CrossRef CAS; (d) J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700 CrossRef CAS PubMed.
- (a) L. J. Walport, R. Obexer and H. Suga, Curr. Opin. Biotechnol., 2017, 48, 242 CrossRef CAS PubMed; (b) L. R. Malins, Curr. Opin. Chem. Biol., 2018, 46, 25 CrossRef CAS PubMed.
- (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed; (b) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961 CrossRef CAS.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509 CrossRef CAS PubMed.
- (a) T. Passioura and H. Suga, Chem.–Eur. J., 2013, 19, 6530 CrossRef CAS PubMed; (b) K. Ito, T. Passioura and H. Suga, Molecules, 2013, 18, 3502 CrossRef CAS PubMed.
- B. A. Hopkins, G. F. Smith and N. Sciammetta, Org. Lett., 2016, 18, 4072 CrossRef CAS PubMed.
- (a) K. Kubota, P. Dai, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 3128 CrossRef CAS PubMed; (b) A. J. Rojas, C. Zhang, E. V. Vinogradova, N. H. Buchwald, J. Reilly, B. L. Pentelute and S. L. Buchwald, Chem. Sci., 2017, 8, 4257 RSC.
- J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2017, 56, 728 CrossRef PubMed.
- Only few examples exist for the synthesis of C(sp2)–O bond containing macrocyclic natural products or peptide drugs and they are shown in Fig. 1B. (a) H. Uchiro, R. Kato, Y. Arai, M. Hasegawa and Y. Kobayakawa, Org. Lett., 2011, 13, 6268 CrossRef CAS PubMed; (b) J. Kuethe, Y.-L. Zhong, N. Yasuda, G. Beutner, K. Linn, M. Kim, B. Marcune, S. D. Dreher, G. Humphrey and T. Pei, Org. Lett., 2013, 15, 4174 CrossRef CAS PubMed.
- (a) D. L. Boger and D. Yohannes, J. Org. Chem., 1991, 56, 1763 CrossRef CAS; (b) D. L. Boger, Y. Nomoto and B. R. Teegarden, J. Org. Chem., 1993, 58, 1425 CrossRef CAS; (c) Q. Cai, B. Zou and D. Ma, Angew. Chem., Int. Ed., 2006, 45, 1276 CrossRef CAS PubMed; (d) D. A. Evans, M. R. Wood, B. W. Trotter, T. I. Richardson, J. C. Barrow and J. L. Katz, Angew. Chem., Int. Ed., 1998, 37, 2700 CrossRef CAS; (e) A. V. R. Ram, K. L. Reddy, A. S. Ram, T. V. S. K. Vittal, M. M. Reddy and P. L. Pathi, Tetrahedron Lett., 1996, 37, 3023 CrossRef; (f) R. Beugelmans, G. P. Singh, M. Bois-Choussy, J. Chastanet and J. Zhu, J. Org. Chem., 1994, 59, 5535 CrossRef CAS; (g) D. P. Fairlie, J. D. A. Tyndall, R. C. Reid, A. K. Wong, G. Abbenante, M. J. Scanlon, D. R. March, D. A. Bergman, C. L. L. Chai and B. A. Burkett, J. Med. Chem., 2000, 43, 1271 CrossRef CAS; (h) K. V. Lawson, T. E. Rose and P. G. Harran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3753 CrossRef PubMed.
- (a) J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863 CrossRef CAS; (b) C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed; (c) X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Bioconjugate Chem., 2012, 23, 500 CrossRef CAS PubMed; (d) H. Chen, R. Huang, Z. Li, W. Zhu, J. Chen, Y. Zhan and B. Jiang, Org. Biomol. Chem., 2017, 15, 7339 RSC; (e) G. Lautrette, F. Touti, H. G. Lee, P. Dai and B. L. Pentelute, J. Am. Chem. Soc., 2016, 138, 8340 CrossRef CAS PubMed.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed.
- While there is an increasing number of reports for peptide modifications by photoredox catalytic reactions, only a few examples were reported using Ni/photoredox dual-catalyzed reactions, (a) B. A. Vara, X. Li, S. Berritt, C. R. Walters, E. J. Petersson and G. A. Molander, Chem. Sci., 2018, 9, 336 RSC; select examples for photoredox catalytic reactions of peptides, (b) S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205 CrossRef CAS PubMed; (c) Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, J. Am. Chem. Soc., 2018, 140, 6797 CrossRef CAS PubMed; (d) N. Ichiishi, J. P. Caldwell, M. Lin, W. Zhong, X. Zhu, E. Streckfuss, H.-Y. Kim, C. A. Parish and S. W. Krska, Chem. Sci., 2018, 9, 4168 RSC.
- (a) H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385 CrossRef CAS; (b) J. Schapp, K. Haas, K. Sünkel and W. Beck, Eur. J. Inorg. Chem., 2003, 2003, 3745 CrossRef; (c) M. K. Kim and A. E. Martell, J. Am. Chem. Soc., 1967, 89, 5138 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS.
- Details can be found in ESI.†.
- A. B. Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49 CrossRef PubMed.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319 CrossRef CAS.
- While this may be true, the redox potentials of photocatalysts’ excited state would also play roles in reaction kinetics (E1/2red[PC*/P−˙] = +1.10 V vs. E1/2red[*IrIII/IrII] = +1.21 V). The photoexcited *IrIII being a stronger oxidant would facilitate SET oxidation step from Ni(II) to Ni(III) in the catalytic cycle. E1/2 = A half-reaction redox potential in volts versus the saturated calomel electrode (SCE) in acetonitrile, PC = 4DPAIPN, Ir = Ir[dF(CF3)ppy]2(dtbbpy)PF6. The values are from ref. 15 and M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
- (a) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532 CrossRef CAS PubMed; (b) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369 CrossRef CAS PubMed.
- C. C. Le, M. K. Wismer, Z.-C. Shi, R. Zhang, D. V. Conway, G. Li, P. Vachal, I. W. Davies and D. W. C. MacMillan, ACS Cent. Sci., 2017, 3, 647 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961 CrossRef CAS PubMed.
- (a) J. J. Warren, J. R. Winkler and H. B. Gray, FEBS Lett., 2012, 586, 596 CrossRef CAS PubMed; (b) B. W. Berry, M. C. Martínez-Rivera and C. Tommos, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9739 CrossRef CAS PubMed; (c) C. Li and M. Z. Hoffman, J. Phys. Chem. B, 1999, 103, 6653 CrossRef CAS.
- M. Favre, K. Moehle, L. Jiang, B. Pfeiffer and J. A. Robinson, J. Am. Chem. Soc., 1999, 121, 2679 CrossRef CAS.
- G. M. Crippen and T. F. Havel, Distance Geometry and Molecular Conformation, Wiley, New York, 1988 Search PubMed.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 730 CrossRef CAS.
- The C–N coupling product is from the reaction between aryl bromide and the carboxamide at the C-terminus.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00694j |
| ‡ Present address: Analytical Research and Development, Pfizer Inc., Andover, Massachusetts 01810, USA. |
| This journal is © The Royal Society of Chemistry 2019 |