
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantioselective synthesis of perfluoroalkyl-substituted β-lactones via a concerted asynchronous [2 + 2] cycloaddition: a synthetic and computational study†
Diego-Javier
Barrios Antúnez
a,
Mark D.
Greenhalgh
 a,
Alexander C.
Brueckner
b,
Daniel M.
Walden
b,
Pilar
Elías-Rodríguez
a,
Patrick
Roberts
a,
Benjamin G.
Young
c,
Thomas H.
West
a,
Alexandra M. Z.
Slawin
a,
Paul
Ha-Yeon Cheong
*b and
Andrew D.
Smith
*a
a,
Alexander C.
Brueckner
b,
Daniel M.
Walden
b,
Pilar
Elías-Rodríguez
a,
Patrick
Roberts
a,
Benjamin G.
Young
c,
Thomas H.
West
a,
Alexandra M. Z.
Slawin
a,
Paul
Ha-Yeon Cheong
*b and
Andrew D.
Smith
*a
aEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
bDepartment of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, Oregon 97333, USA. E-mail: cheongh@oregonstate.edu
cDepartment of Chemistry, Physics, and Engineering, Biola University, 315 Lim Center, La Mirada, California 90639, USA
First published on 29th April 2019
Abstract
The enantioselective preparation of a range of perfluoroalkyl-substituted β-lactones through an isothiourea (HyperBTM) catalysed reaction using symmetric anhydrides as ammonium enolate precursors and perfluoroalkylketones (RF = CF3, C2F5, C4F9) is reported. Following optimisation, high diastereo- and enantioselectivity was observed for β-lactone formation using C2F5- and C4F9-substituted ketones at room temperature (26 examples, up to >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr and >99:1 er), whilst −78 °C was necessary for optimal dr and er with CF3-substituted ketones (11 examples, up to >95:5 dr and >99:1 er). Derivatisation of the β-lactones through ring-opening, as well as a two-step conversion to give perfluoroalkyl-substituted oxetanes, is demonstrated without loss of stereochemical integrity. Density functional theory computations, alongside 13C natural abundance KIE studies, have been used to probe the reaction mechanism with a concerted asynchronous [2 + 2]-cycloaddition pathway favoured over a stepwise aldol–lactonisation process.
:5 dr and >99:1 er), whilst −78 °C was necessary for optimal dr and er with CF3-substituted ketones (11 examples, up to >95:5 dr and >99:1 er). Derivatisation of the β-lactones through ring-opening, as well as a two-step conversion to give perfluoroalkyl-substituted oxetanes, is demonstrated without loss of stereochemical integrity. Density functional theory computations, alongside 13C natural abundance KIE studies, have been used to probe the reaction mechanism with a concerted asynchronous [2 + 2]-cycloaddition pathway favoured over a stepwise aldol–lactonisation process.
1. Introduction
The enantioselective preparation of β-lactones remains a significant goal in chemical synthesis due to the synthetic versatility of β-lactones and their presence in numerous natural products and bioactive compounds.1 Accordingly, a range of approaches has been reported for the synthesis of enantioenriched β-lactones, including substrate-, chiral auxiliary- and catalyst-controlled processes. Although enantioselective Lewis acid catalysis has proven a successful approach,2 the majority of catalytic methods involve the use of chiral Lewis basic catalysts.3Lewis base-catalysed β-lactone synthesis is generally proposed to proceed by generation of a chiral enolate 1 which reacts with an electrophilic carbonyl-containing substrate to give a β-lactone product 2 (Scheme 1a). In principle, this reaction could take place through either a [2 + 2] cycloaddition, or a stepwise aldol–lactonisation process. Most methods have focussed on using highly-reactive ketenes as the enolate precursor, in ketene dimerisations or through reaction with aldehydes.4–9 Although N-heterocyclic carbenes (NHCs) and phosphines have been used in these processes,4,5 the majority of examples have reported the use of tertiary amine catalysts – in particular Cinchona alkaloid derivatives.7,8a–d,f–i,9
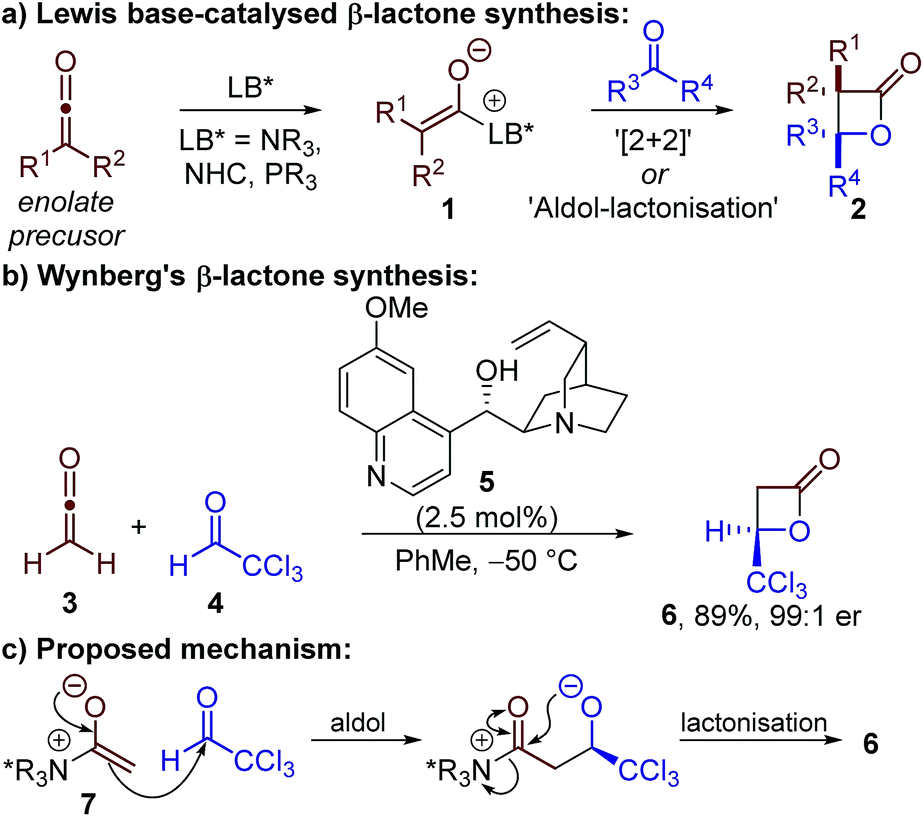 | ||
| Scheme 1 Synthesis and previously proposed mechanism for the formation of β-lactones using Lewis base catalysis. | ||
Building on the work of Borrmann and Wegler,6 a series of seminal studies by Wynberg in the 1980s demonstrated that quinidine 5 could catalyse the highly enantioselective formal [2 + 2] cycloaddition between ketene 3 and chloral 4 (Scheme 1b).7 It was proposed the reaction proceeded by addition of quinidine 5 to ketene 3 to give a C(1)-ammonium enolate intermediate 7,3b followed by reaction with chloral 4via an aldol–lactonisation process (Scheme 1c). This stepwise mechanism was preferred over the thermally-forbidden concerted [2 + 2] cycloaddition, however no experimental evidence was provided to support this assertion. The scope and applicability of this transformation has been expanded by Calter, Romo, Nelson and Fu, amongst others, to allow the use of di- or monosubstituted ketenes (isolated or generated in situ) with a broader range of aldehydes.8 The power of these synthetic methods has been exemplified through their application in a number of elegant total syntheses.9 Despite these significant advances, no experimental or computational studies of the reaction mechanism using tertiary amine catalysts have been published, with the reaction usually assumed to proceed by a stepwise aldol–lactonisation pathway.
The limitation of using highly reactive ketenes in these reactions was originally addressed by Romo, who introduced the use of carboxylic acids as bench-stable ammonium enolate precursors (Scheme 2a).10 A modified Mukaiyama reagent 9 was used for in situ functionalisation of carboxylic acid substrates bearing pendant aldehydes or ketones 8, to promote ammonium enolate formation and subsequent intramolecular formal [2 + 2] cycloaddition. We expanded this general approach to allow the use of alternative bench-stable carboxylic acid derivatives, such as anhydrides 11, in intermolecular formal [2 + 2] cycloadditions (Scheme 2b).11 Reaction of the catalytically-generated chiral ammonium enolate intermediates with aldimines 12 was used for the diastereo- and enantioselective synthesis of β-lactams 14; however analogous methods to generate β-lactones have not been developed.
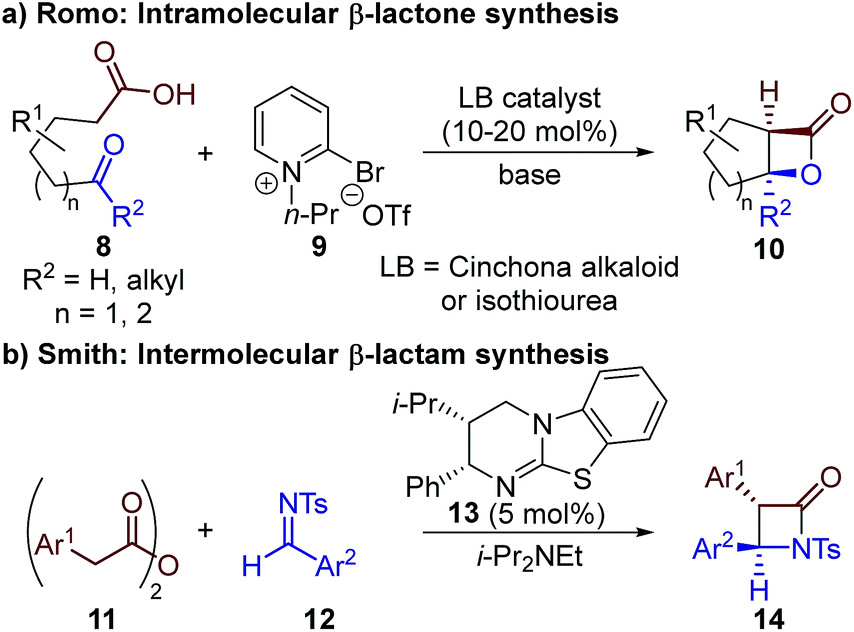 | ||
| Scheme 2 Previous applications of bench-stable carboxylic acid derivatives as ammonium enolate precursors in formal [2 + 2] cycloaddition reactions. | ||
It is widely acknowledged that the chemical and physical properties of small molecules can be significantly affected by the incorporation of fluorine and perfluorinated groups.12 Many drugs and drug candidates contain perfluorinated groups, making simple and robust fluorination methods an area of significant interest. Numerous methods have been reported for the enantioselective introduction of trifluoromethyl groups; however analogous methods for the introduction of longer chain perfluoroalkyl groups at stereogenic centres remains underdeveloped.13 To address this limitation, in previous work we reported the use of perfluoroalkyl-substituted ketones for the synthesis of β-lactones using NHC redox catalysis.14 Although highly enantioselective, this method was restricted to the synthesis of α-alkyl-substituted β-lactones, which proved unstable to isolation by column chromatography and therefore required in situ derivatisation to give acyclic products.
Herein we report the catalytic enantioselective synthesis of perfluoroalkyl-substituted β-lactones from bench-stable anhydrides and perfluoroalkyl-substituted ketones using isothiourea catalysis (Scheme 3). This method precludes the requirement of using highly unstable ketene starting materials. Derivatisation of the enantioenriched β-lactones into a range of products, including perfluoroalkyl-substituted oxetanes, is reported in good yield and with no loss in enantiopurity. Natural abundance 13C kinetic isotope effect (KIE) experiments in tandem with computational analyses were utilised to investigate the reaction mechanism, with results indicating the operation of a concerted asynchronous [2 + 2] cycloaddition.
 | ||
| Scheme 3 This work: enantioselective synthesis of highly-substituted perfluoroalkyl-substituted β-lactones. | ||
2. Results and discussion
2.1. Reaction optimisation
Initial studies for the synthesis of perfluoroalkyl-substituted β-lactones used the isothiourea catalyst HyperBTM 13, perfluorobutyl-substituted ketone 18, and a range of ammonium enolate precursors 15–17 in CH2Cl2 at room temperature (Table 1). First, the use of in situ generated mixed anhydrides was investigated. Functionalisation of phenylacetic acid with pivaloyl chloride to provide mixed anhydride 15 provided access to perfluorobutyl-substituted β-lactone 21 in 51% yield and promising stereoselectivity (92:8 dr, 89:11 er; entry 1). Co-elution of the β-lactone product 21 with pivalic anhydride complicated product isolation and therefore alternative enolate precursors were investigated. The use of para-methoxybenzoic mixed anhydride 16 simplified product isolation and provided β-lactone 21 with improved diastereoselectivity and enantioselectivity (entry 2). The use of bench-stable phenylacetic anhydride 17 gave β-lactone 21 in an improved yield of 61%, and with excellent diastereo- and enantioselectivity (entry 3). The use of alternative isothiourea catalysts, solvents, or reaction temperatures was not beneficial,15 however, increasing the equivalents of anhydride 17 provided β-lactone 21 in improved yield (entry 4). The catalyst loading could be reduced to just 1 mol% without compromising the yield or stereoselectivity of the transformation (entries 5 and 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Anhydride (equiv.) | Ketone (RF) | Yield (%) | dra | erb |
| a Determined by 1H NMR spectroscopic analysis of the crude reaction product mixture. b Determined by HPLC analysis using a chiral support. c Mixed anhydride formed in situ from phenylacetic acid and the appropriate acid chloride. d 5 mol% 13 used. e 1 mol% 13 used. f Reaction conducted at −78 °C. | |||||
| 1 | R = t-Buc | 18 (C4F9) | 51 | 92:8 |
89:11 |
| 15 (1.25) | |||||
| 2 | R = 4-MeO-C6H4c | 18 (C4F9) | 51 | 95:5 |
96:4 |
| 16 (1.25) | |||||
| 3 | R = CH2Ph | 18 (C4F9) | 61 | >95:5 |
97:3 |
| 17 (1.25) | |||||
| 4 | 17 (2.5) | 18 (C4F9) | 69 | >95:5 |
96:4 |
| 5d | 17 (2.5) | 18 (C4F9) | 74 | >95:5 |
97:3 |
| 6e | 17 (2.5) | 18 (C4F9) | 75 | >95:5 |
97:3 |
| 7d | 17 (2.5) | 19 (C2F5) | 75 | >95:5 |
97:3 |
| 8d | 17 (2.5) | 20 (CF3) | 88 | 88:12 |
77:23 |
| 9d,f | 17 (2.5) | 20 (CF3) | 95 | 90:10 |
95:5 |

Variation of the perfluoroalkyl substituent on the ketone was investigated next. The developed methodology proved equally applicable at room temperature when using perfluoroethyl-substituted ketone 19, with β-lactone 22 obtained in 75% yield, >95:5 dr and 97:3 er (entry 7). Reducing the size of the perfluoroalkyl group led to a decrease in stereoselectivity, with trifluoroacetophenone derivative 20 providing β-lactone 23 in 88:12 dr and 77:23 er at room temperature (entry 8). Reducing the reaction temperature to −78 °C provided access to β-lactone 23 in excellent yield (95%), similar diastereoselectivity (90:10) but with significantly improved enantioselectivity (95:5; entry 9).16 In contrast to our previous work with α-alkyl-substituted β-lactones,14 all three α-aryl-substituted β-lactones (21–23) were easy to isolate and were found to be thermally stable up to 140 °C.17
2.2. Reaction scope and limitations
:3 er), indicating this reaction temperature to be optimal when using trifluoromethyl-substituted ketones.

The electronic nature of the aromatic substituent was then investigated. The introduction of electron-withdrawing groups (positive Hammett sigma constants)19 was tolerated, with p-CF3 and m-OMe-substituted β-lactone products 27 and 28 obtained in excellent yield and with excellent diastereoselectivity and good enantioselectivity. The use of ketones bearing electron-neutral or weakly electron-donating groups (negative Hammett sigma constants)19 were also tolerated, with 29 and 30 obtained with good to excellent diastereo- and enantioselectivity, albeit in slightly reduced yield (∼50%). The introduction of a more strongly electron-donating para-methyl substituent led to only trace amounts of β-lactone 31 in the perfluorobutyl-substituted ketone series. In contrast, using the analogous trifluoromethyl-substituted ketone led to the formation of β-lactone 32 in high yield (78%) and with excellent enantioselectivity (99:1 er). The system was further challenged by the introduction of a highly electron-donating para-methoxy group. β-Lactone 33 was produced with excellent stereoselectivity (>95:5 dr, 99:1 er), albeit in a relatively low yield (22%), which was attributable to lower conversion of the ketone starting material. The use of heteroaromatic ketones was also reasonably successful, with thienyl-substituted β-lactone 34 obtained in 43% yield, with high diastereoselectivity, but reduced enantioselectivity (>95:5 dr, 87:13 er). Finally, the reaction scope was extended to include alkyl-substituted ketones, with the use of trifluoroacetone providing access to β-lactone 35 in an excellent 91% yield and with high diastereo- and enantioselectivity (95:5 dr, 93:7 er).
The generality of the procedure was further probed by applying a subset of anhydrides for the formation of β-lactones using perfluoroethyl-substituted ketone 19 (Table 4) and trifluoromethyl-substituted ketone 20 (Table 5). The results obtained using perfluoroethyl-substituted ketone 19 feature similar trends as when using perfluorobutyl-substituted ketone 18. β-Lactones 47–51 were obtained with excellent diastereo- and enantioselectivity when using arylacetic anhydride derivatives,21 and slightly reduced stereocontrol when using (E)-pent-3-enoic anhydride (52). In all cases excellent yields were obtained, with the exception of an electron-withdrawing group on the aryl acetic anhydride, which again resulted in a slight drop in yield (50:65%).
The same set of anhydrides was also applied in the synthesis of β-lactones with trifluoromethyl-substituted ketone 20 at −78 °C (Table 5). All 3-aryl-substituted β-lactones 53–56 were obtained with similar diastereoselectivity (∼90:10 dr),22 however alkenyl-substituted β-lactone 58 was obtained with relatively low diastereocontrol (69:31 dr). In this series, high to excellent enantioselectivities were obtained in each case, including for the formation of 3-alkenyl-substituted β-lactone 58, which is in contrast to the other series in which a drop in enantioselectivity was observed when using this anhydride (see Tables 3 and 4, 45 and 52).
| a dr determined by 1H NMR spectroscopic analysis of the crude reaction product mixture. er determined by HPLC analysis using a chiral support. |
|---|

|
| a dr determined by 1H NMR spectroscopic analysis of the crude reaction product mixture. er determined by HPLC analysis using a chiral support. |
|---|

|
| a dr determined by 1H NMR spectroscopic analysis of the crude reaction product mixture. er determined by HPLC analysis using a chiral support. |
|---|

|
:11 er). The addition of sodium methoxide in dichloromethane at −78 °C however allowed access to ester 62 in 93% yield with no loss in enantiopurity (98:2 er). Finally, the arylbromide functionality of β-lactone 21 was exploited for a Suzuki–Miyaura cross-coupling with 2-indolylboronic acid derivative 63 to give β-lactone 64 with no loss in diastereo- or enantiopurity. Although β-lactone 64 was only isolated in 35% yield, this derivatisation demonstrates the appreciable stability of these β-lactones under basic aqueous conditions, even at high reaction temperatures.
 | ||
| Scheme 4 Derivatisations of β-lactone 21. Conditions: (i) 21 (1 equiv.), NH3 (5 equiv.), 1,4-dioxane (0.1 M), r.t., 16 h; (ii) 21 (1 equiv.), BnNH2 (5 equiv.), CH2Cl2 (0.1 M), r.t., 16 h; (iii) 21 (1 equiv.), morpholine (5 equiv.), CH2Cl2 (0.1 M), r.t., 16 h; (iv) 21 (1 equiv.), NaOMe (5 equiv.), MeOH/CH2Cl2 (1:1, 0.1 M), −78 °C, 2 h; (v) 21 (1 equiv.), N-Boc-5-methoxyindole-2-boronic acid 63 (1 equiv.), Pd(PPh3)4 (0.1 equiv.), Na2CO3 (3 equiv.), DMF/H2O (9:1, 0.06 M), 85 °C, 4 h. | ||
Finally, derivatisation of the β-lactone products to give perfluoroalkyl-substituted oxetanes was investigated (Table 6). Oxetanes are of significant interest in medicinal chemistry as bioisosteres for gem-dimethyl and carbonyl groups, and also in synthetic chemistry as versatile reactive intermediates.24 Reduction of the β-lactones using i-Bu2AlH (DIBAL) at −78 °C provided access to diols 65–69 in moderate yield and with no loss in diastereo- or enantiopurity. Treatment of these diols with sodium hydride, followed by 2,4,6-triisopropylbenzenesulfonyl chloride, provided perfluoroalkyl-substituted oxetanes 70–74 in excellent yield and in highly enantioenriched form. The protocol proved to be general with comparable results obtained when using β-lactones bearing perfluorobutyl, perfluoroethyl or trifluoromethyl substituents and various functionalised aromatic groups.
| a dr determined by 1H NMR spectroscopic analysis of the crude reaction product mixture. er determined by HPLC analysis using a chiral support. |
|---|

|
2.3. Mechanism
Our initial mechanistic objective was to identify the nature of the potential energy surface (PES) for the lactonisation step of the isothiourea-catalysed formation of perfluoroalkyl-substituted β-lactones.15 Phenylacetic anhydride 17, 4′-bromo-2,2,2-trifluoroacetophenone 20 and HyperBTM 13 were used as model substrates to examine the PES. We began by computing transition states (TSs) connecting HyperBTM-enolate complex D to product–catalyst complex F using two widely applied density functional theory (DFT) methods, M06-2X29 and PBE30 (Fig. 1a).
 | ||
| Fig. 1 (a) Possible mechanistic pathways for the formation of intermediate F; Ar = 4-bromophenyl. (b) Potential energy surface (PES) contours generated with parallel distance scans in the vicinity of the β-lactone forming transition state bond lengths. Blue regions are lower in energy while red regions are higher in energy. Structure D is not a stationary point on the PES but represents a relatively lower energy acylated catalyst–ketone complex directly “downhill” from the first saddle point. Contour images generated and rendered with Matplotlib.34 (c) Computed concerted asynchronous [2 + 2] and stepwise aldol TSs using PBE and M06-2X, respectively. The Newman projection looks down the forming C–C bond. Distances are in Ångströms. Structure images made with CYLView.35 | ||
Surprisingly, the two DFT methods revealed different reaction mechanisms. M06-2X predicted a stepwise process in which TS-Aldol (Fig. 1) was located where only the β-lactone C–C bond was formed. A subsequent ring-closing lactonisation TS (TS-Lact) was located that forms the C–O bond, ultimately giving the product–catalyst complex F. Attempts to locate a concerted TS with M06-2X were unsuccessful. Interestingly, PBE gave the concerted asynchronous TS-[2 + 2],31,32 and no stepwise transition structures could be located.
We computed the respective PESs around the located transition structures (Fig. 1b) in order to clarify the differences between the predicted mechanisms for each method. The PESs were computed by fixing the C–C bond distance and varying the C–O bond distance of the forming β-lactone. The M06-2X surface indeed revealed a stepwise process involving two first-order saddle-points (TS-Aldol and TS-Lact) and a zwitterionic intermediate (Int). In contrast, the PBE surface showed a single first-order saddle point connecting the enolate–ketone complex D directly to the product–catalyst complex F. As these two popular theoretical methods revealed differing mechanistic pathways, we turned to the use of coordinated kinetic isotope effect experiments and computations to elucidate the active mechanism.33
:1 ratio of reagents and give just a single product (as a single stereoisomer). The formal cycloaddition using phenylacetic anhydride 17 and 4′-bromo-2,2,2-trifluoroacetophenone 20 was therefore further optimised to fulfil these criteria. By conducting the reaction at −90 °C in anhydrous CH2Cl2 under inert conditions, the reaction stoichiometry could be reduced to 1:1, with β-lactone 23 isolated in 92% yield and 90:10 dr (Scheme 5a). Notably, the major diastereoisomer 23 was isolated in highly enantioenriched form (97:3 er), while the minor diastereoisomer 75 was isolated in a 72:28 er. To investigate the origin of this observation, a 97:3 sample of β-lactones 23 (97:3 er) and 75 (72:28 er) was treated with i-Pr2NEt in CH2Cl2 (Scheme 5b). After 2 h, the diastereomeric ratio had readjusted to 90:10 (96:4 er for both diastereoisomers), with no further change observed upon extended exposure times. This is consistent with base-mediated epimerisation at C(3), and allowed the configuration of the minor diastereoisomer 75 isolated from the catalytic reaction to be unambiguously assigned as (3R,4S).37 This stereoisomer is most likely generated by C(3)-epimerisation of the major (3S,4S)-stereoisomer, and indicates that the kinetically-derived diastereomeric ratio in the catalytic reaction is likely to be significantly higher than 90:10. The optimised system is therefore proposed to provide ∼90–95% selectivity for initial generation of the (3S,4S)-stereoisomer, which was considered to be sufficient for reliable assessment of the 13C KIEs at natural abundance.
 | ||
| Scheme 5 Reaction optimisation for KIE at natural abundance analysis. | ||
Determination of 13C KIEs for each carbon of the β-lactone 23 ring was approached by performing reactions to low conversion (∼5%) and analysing the 13C isotopic composition of β-lactone product 23.38,39 The isotopic composition of each sample was compared to a sample of β-lactone 23 obtained from a reaction taken to completion. In each case, the signal from the meta-carbons of the 4-bromophenyl (Ar) substituent was used as an internal standard, with clear baseline separation and the assumption of negligible isotope effect at this position. Unfortunately, due to incomplete chromatographic separation of the diastereoisomers, the 13C KIE for C(4) of β-lactone 23 could not be reliably measured due to partial overlap of the signals arising from each diastereoisomer, which appear as quartets (J = 33 Hz) from 13C–19F coupling with the CF3 substituent. 13C KIEs were therefore only calculated for C(2) and C(3) (Fig. 2a). Three independent experiments were performed, with each sample analysed five times by quantitative 13C NMR spectroscopy.15 Computed KIEs (Fig. 2b) are reported using the Bigeleisen–Mayer method40 with the meta-carbons of the 4-bromophenyl substituent set to unity and thermal corrections computed at −90 °C to match experimental conditions (see ESI†). Computation of all predicted KIEs were automated by use of the Onyx isotope effect program.41
 | ||
| Fig. 2 Experimentally and computationally-determined 13C KIEs. (a) 13C KIEs measured at −90 °C in three independent reactions, with standard deviations from five measurements given in parentheses; (b) computed KIEs are reported with the Bigeleisen–Mayer method with frequencies derived from M06-2X and PBE methods at −90 °C. M06-2X predicts deprotonation as rate-limiting, therefore the reported KIEs are derived from TS-C. PBE predicts cycloaddition as rate-limiting, and the associated KIEs are derived from TS-[2 + 2]. | ||
The experimental KIEs at natural abundance reveal a small, normal isotope effect at C(2), and essentially no isotope effect at C(3) (Fig. 2a).42 For PBE/6-31G(d),43 the predicted KIEs at C(2) and C(3) of the rate-limiting concerted asynchronous TS-[2 + 2] are in excellent agreement with the experimental values. Interestingly, at −90 °C, M06-2X predicted deprotonation (TS-C, Fig. 2b right) as the rate-limiting step, whereas at room temperature, the TS-Aldol was rate-limiting (see ESI†). It is important to note that computations using the M06-2X level of theory did not match the experimental KIEs, regardless of the rate-limiting step. Based on these results, we propose a concerted asynchronous [2 + 2] cycloaddition as rate-limiting in the HyperBTM-catalysed synthesis of perfluoroalkyl-substituted β-lactones reported in this manuscript.
The computed catalytic cycle with PBE at −90 °C is summarised in Fig. 3, as this method correctly predicted the experimental KIE results (vide supra). Addition of HyperBTM to phenylacetic anhydride 17 proceeds, viaTS-A (ΔG‡ = 16.4 kcal mol−1), to give the acylated-HyperBTM and phenylacetate ion pair B (ΔG = 6.7 kcal mol−1), without location of a tetrahedral intermediate.48 Deprotonation of the acyl group by phenylacetate (TS-C, ΔG‡ = 9.5 kcal mol−1) yields (Z)-enolate D (ΔG = 4.6 kcal mol−1).49 Intermediate D is the key reactive species, with subsequent rate-limiting concerted asynchronous [2 + 2] cycloaddition with ketone 20 (TS-[2 + 2]-(Re,Si), ΔG‡ = 20.8 kcal mol−1) leading to tetrahedral intermediate F (ΔG = 17.8 kcal mol−1). Catalyst turnover occurs through cleavage of the catalyst–lactone N–C bond, releasing product and initiating a new catalytic cycle (TS-G, ΔG‡ = 20.3 kcal mol−1). All acylated HyperBTM intermediates and transition states in the catalytic cycle exhibit a stabilising and rigidifying 1,5-S⋯O interaction50,51 (dashed orange line, Fig. 3).
 | ||
| Fig. 3 (Top) Proposed catalytic cycle; Ar = 4-bromophenyl. (Bottom) Computed reaction coordinate using PBE/6-311++G(2df,p)/PCM(DCM)//PBE/6-31G(d)/PCM(DCM) with thermal corrections computed at −90 °C. | ||
Computations overpredict both enantioselectivity  and diastereoselectivity
and diastereoselectivity  compared to experiments. These deviations may arise in part from the previously discussed epimerisation at the C(3) position. Alternative theoretical methods (e.g. B3LYP) and reaction mechanisms that would lead to the formation of racemic β-lactone (e.g. cycloaddition between in situ generated ketene and ketone 20) were investigated. However, all alternative mechanisms were significantly disfavoured (ΔG‡ > 30.7 kcal mol−1) compared to the HyperBTM-catalysed concerted asynchronous [2 + 2] mechanism, and other quantum mechanical methods gave similar predicted selectivities.15
compared to experiments. These deviations may arise in part from the previously discussed epimerisation at the C(3) position. Alternative theoretical methods (e.g. B3LYP) and reaction mechanisms that would lead to the formation of racemic β-lactone (e.g. cycloaddition between in situ generated ketene and ketone 20) were investigated. However, all alternative mechanisms were significantly disfavoured (ΔG‡ > 30.7 kcal mol−1) compared to the HyperBTM-catalysed concerted asynchronous [2 + 2] mechanism, and other quantum mechanical methods gave similar predicted selectivities.15
Conclusions
In conclusion, the isothiourea-catalysed reaction between symmetric anhydrides and perfluoroalkyl ketones (RF = CF3, C2F5, C4F9) has been demonstrated for the enantioselective generation of a range of isolable perfluoroalkyl-substituted β-lactones with excellent stereocontrol (37 examples, up to >95:5 dr and >99:1 er). Derivatisation of the β-lactones was achieved through ring-opening processes, in addition to conversion to perfluoroalkyl-substituted oxetanes through a simple two-step process. Quantum mechanical computations were used to elucidate the mechanism of this process. Interestingly, two widely applied density functional theory (DFT) methods gave different mechanistic pathways. PESs were used to verify the opposing mechanisms, wherein M06-2X predicted a stepwise aldol/lactonisation process and PBE predicted a concerted asynchronous [2 + 2] process. Experimental and computed 13C natural abundance KIE studies were juxtaposed to elucidate the active mechanism. The experimental and PBE-predicted KIE values were in excellent agreement, suggesting the reaction proceeds through a concerted asynchronous [2 + 2] mechanism.52
Conflicts of interest
There are no conflicts to declare.Author contributions
D.-J. B. A., M. D. G., P. E.-R., P. R. and T. H. W. contributed to the synthetic work. A. M. Z. S. obtained X-ray crystal structures. A. C. B., D. M. W. and B. G. Y. performed the computational work. M. D. G., A. C. B. and D. M. W. wrote the manuscript. A. D. S. and P. H. Y. C. supervised the project and the writing of the manuscript.Acknowledgements
The research leading to these results (D.-J. B. A., T. H. W., A. D. S.) has received funding from the ERC under the European Union's Seventh Framework Programme (FP7/2007-2013)/E.R.C. Grant Agreement No. 279850. A. D. S. thanks the Royal Society for a Wolfson Research Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University. P. H. Y. C. is the Bert and Emelyn Christensen professor of OSU, and gratefully acknowledges financial support from the Vicki & Patrick F. Stone family. P. H. Y. C., A. C. B., D. M. W. and B. G. Y. gratefully acknowledge support from the National Science Foundation (NSF CHE-1352663). P. E.-R. thanks the Spanish government for a FPU Fellowship and the University of Seville (V Plan Propio de Investigación) for financial support.Notes and references
- (a) Y. Wang, R. L. Tennyson and D. Romo, Heterocycles, 2004, 64, 605–658 CrossRef CAS; (b) A. Pommier and J.-M. Pons, Synthesis, 1995, 7, 729–744 CrossRef; (c) C. Lowe and J. C. Vederas, Org. Prep. Proced. Int., 1995, 27, 305–346 CrossRef CAS; (d) H. W. Yang and D. Romo, Tetrahedron, 1999, 55, 6403–6434 CrossRef CAS; (e) T. Böttcher and S. A. Sieber, Med. Chem. Commun., 2012, 3, 408–417 RSC; (f) A. Pommier and J.-M. Pons, Synthesis, 1993, 5, 441–459 CrossRef.
- (a) Y. Tamai, H. Yoshiwara, M. Someya, J. Fukumoto and S. Miyano, J. Chem. Soc., Chem. Commun., 1994, 2281–2282 RSC; (b) B. W. Dymock, P. J. Kocienski and J.-M. Pons, Chem. Commun., 1996, 1053–1054 RSC; (c) H. W. Yang and D. Romo, Tetrahedron Lett., 1998, 39, 2877–2880 CrossRef CAS; (d) S. G. Nelson, T. J. Peelen and Z. Wan, J. Am. Chem. Soc., 1999, 121, 9742–9743 CrossRef CAS; (e) S. G. Nelson and Z. Wan, Org. Lett., 2000, 2, 1883–1886 CrossRef CAS PubMed; (f) D. A. Evans and J. M. Janey, Org. Lett., 2001, 3, 2125–2128 CrossRef CAS PubMed; (g) S. G. Nelson, C. Zhu and X. Shen, J. Am. Chem. Soc., 2004, 126, 14–15 CrossRef CAS PubMed; (h) R. E. Forslund, J. Cain, J. Colyer and M. P. Doyle, Adv. Synth. Catal., 2005, 347, 87–92 CrossRef CAS; (i) V. Gnanadesikan and E. J. Corey, Org. Lett., 2006, 8, 4943–4945 CrossRef CAS PubMed; (j) T. Kull and R. Peters, Adv. Synth. Catal., 2007, 349, 1647–1652 CrossRef CAS; (k) T. Kull and R. Peters, Angew. Chem., Int. Ed., 2008, 47, 5461–5464 CrossRef CAS PubMed; (l) X. Hao, X. Liu, W. Li, F. Tan, Y. Chu, X. Zhao, L. Lin and X. Feng, Org. Lett., 2014, 16, 134–137 CrossRef CAS PubMed.
- For reviews see: (a) J. Douglas, L. C. Morrill, E. Richmond and A. D. Smith, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, ed. N. Nishiwaki, Wiley, Hoboken, 2014, ch. 3, pp. 89–114 Search PubMed; (b) K. N. Van, L. C. Morrill, A. D. Smith and D. Romo, in Lewis Base Catalysis in Organic Synthesis, ed. E. Vedejs and S. E. Denmark, Wiley-VCH, Weinheim, 2016, ch. 13, vol. 2, pp. 527–653 Search PubMed.
- (a) C. Burstein, S. Tschan, X. Xie and F. Glorius, Synthesis, 2006, 14, 2418–2439 Search PubMed; (b) L. He, H. Lv, Y.-R. Zhang and S. Ye, J. Org. Chem., 2008, 73, 8101–8103 CrossRef CAS PubMed; (c) X.-N. Wang, P.-L. Shao, H. Lv and S. Ye, Org. Lett., 2009, 11, 4029–4031 CrossRef CAS PubMed; (d) X.-N. Wang, Y.-Y. Zhang and S. Ye, Adv. Synth. Catal., 2010, 352, 1892–1895 CrossRef CAS; (e) M. Zhang, D. Wei, Y. Wang, S. Li, J. Liu, Y. Zhu and M. Tang, Org. Biomol. Chem., 2014, 12, 6374–6383 RSC; (f) J. Douglas, J. E. Taylor, G. Churchill, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2013, 78, 3925–3938 CrossRef CAS PubMed; (g) J. J. Douglas, G. Churchill, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem.–Eur. J., 2015, 21, 16354–16358 CrossRef CAS PubMed; for an example of NHC redox catalysis starting from an aldehyde see: (h) J. Mo, R. Yang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 50–53 CrossRef CAS PubMed.
- M. Mondal, A. A. Ibrahim, K. A. Wheeler and N. J. Kerrigan, Org. Lett., 2010, 12, 1664–1667 CrossRef CAS PubMed.
- (a) D. Borrmann and R. Wegler, Chem. Ber., 1966, 99, 1245–1251 CrossRef CAS; (b) D. Borrmann and R. Wegler, Chem. Ber., 1967, 100, 1575–1579 CrossRef CAS.
- (a) H. Wynberg and E. G. J. Staring, J. Am. Chem. Soc., 1982, 104, 166–168 CrossRef CAS; (b) H. Wynberg and E. G. J. Staring, J. Org. Chem., 1985, 50, 1977–1979 CrossRef CAS; (c) H. Wynberg and E. G. J. Staring, J. Chem. Soc., Chem. Commun., 1984, 1181–1182 RSC.
- (a) M. A. Calter, J. Org. Chem., 1996, 61, 8006–8007 CrossRef CAS PubMed; (b) R. Tennyson and D. Romo, J. Org. Chem., 2000, 65, 7248–7252 CrossRef CAS PubMed; (c) M. A. Calter, R. K. Orr and W. Song, Org. Lett., 2003, 5, 4745–4748 CrossRef CAS PubMed; (d) C. Zhu, X. Shen and S. G. Nelson, J. Am. Chem. Soc., 2004, 126, 5352–5353 CrossRef CAS PubMed; (e) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 6358–6360 CrossRef CAS PubMed; (f) M. A. Calter, O. A. Tretyak and C. Flaschenriem, Org. Lett., 2005, 7, 1809–1812 CrossRef CAS PubMed; (g) V. C. Purohit, R. D. Richardson, J. W. Smith and D. Romo, J. Org. Chem., 2006, 71, 4549–4558 CrossRef CAS PubMed; (h) A. Armstrong, S. P. Geldart, C. R. Jenner and J. N. Scutt, J. Org. Chem., 2007, 72, 8091–8094 CrossRef CAS PubMed; (i) A. A. Ibrahim, D. Nalla, M. Van Raaphorst and N. J. Kerrigan, J. Am. Chem. Soc., 2012, 134, 2942–2945 CrossRef CAS PubMed.
- (a) M. E. Green, J. C. Rech and P. E. Floreancig, Angew. Chem., Int. Ed., 2008, 47, 7317–7320 CrossRef CAS PubMed; (b) X. Jiang, C. Fu and S. Ma, Chem.–Eur. J., 2008, 14, 9656–9664 CrossRef CAS PubMed; (c) G. Liu and D. Romo, Org. Lett., 2009, 11, 1143–1146 CrossRef CAS PubMed; (d) B. Chandra, D. Fu and S. G. Nelson, Angew. Chem., Int. Ed., 2010, 49, 2591–2594 CrossRef CAS PubMed; (e) T. R. Vargo, J. S. Hale and S. G. Nelson, Angew. Chem., Int. Ed., 2010, 49, 8678–8681 CrossRef CAS PubMed; (f) S. Wan, F. Wu, J. C. Rech, M. E. Green, R. Balachandran, W. S. Horne, B. W. Day and P. E. Floreancig, J. Am. Chem. Soc., 2011, 133, 16668–16679 CrossRef CAS PubMed; (g) M. Yoshino, K. Eto, K. Takahashi, J. Ishihara and S. Hatakeyama, Org. Biomol. Chem., 2012, 10, 8164–8174 RSC.
- (a) G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945–7946 CrossRef CAS PubMed; (b) S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835–2838 CrossRef CAS PubMed; (c) H. Henry-Riyad, C. Lee, V. C. Purohit and D. Romo, Org. Lett., 2006, 8, 4363–4366 CrossRef CAS PubMed; (d) G. Ma, H. Nguyen and D. Romo, Org. Lett., 2007, 9, 2143–2146 CrossRef CAS PubMed; (e) V. C. Purohit, A. S. Matla and D. Romo, J. Am. Chem. Soc., 2008, 130, 10478–10479 CrossRef CAS PubMed; (f) C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed., 2010, 49, 9479–9483 CrossRef CAS PubMed; (g) K. A. Morris, K. M. Arendt, S. H. Oh and D. Romo, Org. Lett., 2010, 12, 3764–3767 CrossRef CAS PubMed; (h) C. A. Leverett, V. C. Purohit, A. G. Johnson, R. L. Davis, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2012, 134, 13348–13356 CrossRef CAS PubMed; (i) W. Kong and D. Romo, J. Org. Chem., 2017, 82, 13161–13170 CrossRef CAS PubMed.
- (a) S. R. Smith, J. Douglas, H. Prevet, P. Shapland, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1626–1639 CrossRef CAS PubMed; (b) L. C. Morrill, S. M. Smith, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1640–1655 CrossRef CAS PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119–6146 CrossRef CAS PubMed; (b) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455–529 CrossRef CAS PubMed; (c) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- A. T. Davies, A. M. Z. Slawin and A. D. Smith, Chem.–Eur. J., 2015, 21, 18944–18948 CrossRef CAS PubMed.
- See the ESI† for more details.
- The absolute configurations of products 21–23 were assigned by analogy to products 60, 47 and 53, respectively, which were unambiguously assigned by single crystal X-ray crystallographic analysis.
- Heating samples of 21–23 in d6-DMSO at 140 °C resulted in slow decomposition to a mixture of unidentified products, with compounds 21–23 displaying the following half-lives: 21: t½ = 80 h; 22: t½ = 50 h; 23: t½ = 16 h. See page S51 of the ESI† for more details.
- Although the synthesis of β-lactone 21 was successful using 1 mol% catalyst loading (Table 1, entry 6), lower yields of β-lactone were obtained in some cases when using different anhydrides and ketones. A catalyst loading of 5 mol% was therefore chosen as standard for investigating the substrate scope.
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS.
- (a) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed; (b) C. Simal, T. Lebl, A. M. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653–3657 CrossRef CAS PubMed; (c) J. N. Arokianathar, A. B. Frost, A. M. Z. Slawin, D. Stead and A. D. Smith, ACS Catal., 2018, 8, 1153–1160 CrossRef CAS.
- Crystallographic data for (3S,4S)-47 (CCDC 1886142†).
- Crystallographic data for (3S,4S)-53 (CCDC 1886144†).
- Crystallographic data for (2S,3S)-60 (CCDC 1886143†).
- J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed.
- (a) X. Wang and K. N. Houk, J. Am. Chem. Soc., 1990, 112, 1754–1756 CrossRef CAS; (b) E. T. Seidl and H. F. Schaefer III, J. Am. Chem. Soc., 1991, 113, 5195–5200 CrossRef CAS; (c) D. V. Deubel, J. Phys. Chem. A, 2002, 106, 431–437 CrossRef CAS; (d) D. A. Singleton, Y. Wang, H. W. Yang and D. Romo, Angew. Chem., Int. Ed., 2002, 41, 1572–1575 CrossRef CAS; (e) B. R. Ussing, C. Hang and D. A. Singleton, J. Am. Chem. Soc., 2006, 128, 7594–7607 CrossRef CAS PubMed; (f) C. M. Rasik, Y. J. Hong, D. J. Tantillo and K. M. Brown, Org. Lett., 2014, 16, 5168–5171 CrossRef CAS PubMed.
- S. J. Ryan, A. Stasch, M. N. Paddon-Row and D. W. Lupton, J. Org. Chem., 2012, 77, 1113–1124 CrossRef CAS PubMed.
- R. C. Johnston, D. T. Cohen, C. C. Eichman, K. A. Scheidt and P. H.-Y. Cheong, Chem. Sci., 2014, 5, 1974–1982 RSC.
- S. R. Hare and D. J. Tantillo, Chem. Sci., 2017, 8, 1442–1449 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 145–241 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- This phenomenon is not unknown and has been reported in computational studies of Claisen rearrangements, where B3LYP predicts a stepwise pathway, whilst mPW1K predicts a concerted pericyclic TS, see ref 25e.
- M06-2X has been previously shown to predict a concerted TS in theoretical studies on the mechanism of NHC-catalysed β-lactone formation from ketenes and aldehydes, see ref 4e.
- G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706–1725 CrossRef CAS PubMed.
- J. D. Hunter, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- (a) D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9358 CrossRef CAS; (b) A. J. DelMonte, J. Haller, K. N. Houk, K. B. Sharpless, D. A. Singleton, T. Strassner and A. A. Thomas, J. Am. Chem. Soc., 1997, 119, 9907–9908 CrossRef CAS; (c) D. E. Frantz and D. A. Singleton, J. Am. Chem. Soc., 2000, 122, 3288–3395 CrossRef CAS; (d) D. A. Singleton, C. Hang, M. J. Szymanski, M. P. Meyer, A. G. Leach, K. T. Kuwata, J. S. Chen, A. Greer, C. S. Foote and K. N. Houk, J. Am. Chem. Soc., 2003, 125, 1319–1328 CrossRef CAS PubMed; (e) J. K. Lee, A. D. Bain and P. J. Berti, J. Am. Chem. Soc., 2004, 126, 3769–3776 CrossRef CAS PubMed; (f) D. A. Singleton and Z. Wang, J. Am. Chem. Soc., 2005, 127, 6679–6685 CrossRef CAS PubMed; (g) S. E. Denmark, S. M. Pham, R. A. Stavenger, X. Su, K.-T. Wong and Y. Nishigaichi, J. Org. Chem., 2006, 71, 3904–3922 CrossRef CAS PubMed; (h) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694–4697 CrossRef CAS PubMed.
- This configuration at C(3) is opposite to that expected for products formed under the control of the isothiourea catalyst, indicating that this stereoisomer is mostly likely generated through C(3)-epimerisation of the major (3S,4S)-stereoisomer. See pages S46–S50 of the ESI† for more details.
- For examples of 13C KIEs at natural abundance measured at low conversion, see ref. 25d and 36c, g, h.
- For adapted or alternative methods for measuring 13C KIEs at natural abundance see: (a) A. Pabis, R. Kamiński, G. Ciepielowski, S. Jankowski and P. Paneth, J. Org. Chem., 2011, 76, 8033–8035 CrossRef CAS PubMed; (b) K. A. Manning, B. Sathyamoorthy, A. Eletsky, T. Szyperski and A. S. Murkin, J. Am. Chem. Soc., 2012, 134, 20589–20592 CrossRef CAS PubMed; (c) S. Xiang and M. P. Meyer, J. Am. Chem. Soc., 2014, 136, 5832–5835 CrossRef CAS PubMed; (d) E. E. Kwan, Y. Park, H. A. Besser, T. L. Anderson and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 43–46 CrossRef CAS PubMed.
- J. Bigeleisen and M. G. Mayer, J. Chem. Phys., 1947, 15, 261–267 CrossRef CAS.
- A. C. Brueckner, S. L. Cevallos, O. M. Ogba, D. M. Walden, M. P. Meyer, D. J. O'Leary and P. H.-Y. Cheong, Onyx, version 1.0, Oregon State University, Corvallis, OR, USA, 2016 Search PubMed.
- The absence of a significant 13C KIE at C(3) contrasts the seminal isotope effect studies on the Lewis acid-catalysed cycloaddition of ketenes and aldehydes by Singleton and Romo (ref. 25d), in which a normal KIE at the C(3) position was observed and a stepwise mechanism proposed. In the current study, KIEs consistent with those reported in ref. 25d were predicted by M06-2X at room temperature for a step-wise mechanism (see ESI† for more details).
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford CT, 2009, see ESI† for full citation.
- See ESI† for M06-2X reaction coordinate diagram.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- (a) S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem.–Eur. J., 2005, 11, 4751–4757 CrossRef CAS PubMed; (b) J. F. Marlier, Acc. Chem. Res., 2001, 34, 283–290 CrossRef CAS PubMed.
- Deprotonation by i-Pr2NEt was computed, but gave a much higher barrier of 32.9 kcal mol−1. See ESI† for structural details.
- (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed; (b) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed.
- (a) V. B. Birman, X. Li and Z. Han, Org. Lett., 2007, 9, 37–40 CrossRef CAS PubMed; (b) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef CAS PubMed; (c) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef CAS PubMed; (d) E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC; (e) T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnston, P. H.-Y. Cheong, G. C. Lloyd-Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366–4375 CrossRef CAS PubMed; (f) M. D. Greenhalgh, S. M. Smith, D. M. Walden, J. E. Taylor, Z. Brice, E. R. T. Robinson, C. Fallan, D. B. Cordes, A. M. Z. Slawin, H. C. Richardson, M. A. Grove, P. H.-Y. Cheong and A. D. Smith, Angew. Chem., Int. Ed., 2018, 57, 3200–3206 CrossRef CAS PubMed.
- The research data underpinning this publication can be found at DOI: https://doi.org/10.17630/07f4bb41-b486-4be0-87de-517ae9eb8d4d.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterisation data (mpt, NMR, IR, HRMS, [α]D, HPLC), traces (1H, 13C, 19F NMR, HPLC), X-ray crystallographic data (CCDC 1886142, CCDC 1886143 and CCDC 1886144), coordinates, thermal corrections and energies of all computed structures. CCDC 1886142–1886144. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00390h |
| This journal is © The Royal Society of Chemistry 2019 |