
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonodisperse polysarcosine-based highly-loaded antibody-drug conjugates†
Warren
Viricel
 *a,
Guy
Fournet
b,
Sabine
Beaumel
c,
Emeline
Perrial
c,
Sébastien
Papot
de,
Charles
Dumontet
c and
Benoît
Joseph
b
*a,
Guy
Fournet
b,
Sabine
Beaumel
c,
Emeline
Perrial
c,
Sébastien
Papot
de,
Charles
Dumontet
c and
Benoît
Joseph
b
aMablink Bioscience SA, 14 rue Waldeck Rousseau, 69006 Lyon, France. E-mail: w.viricel@mablink.com
bUniversité de Lyon, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires, UMR CNRS 5246, 43 Boulevard du 11 Novembre 1918, 69622 Villeurbanne Cedex, France
cUniversité de Lyon, Centre de Recherche en Cancérologie de Lyon, INSERM 1052, CNRS 5286, 8 avenue Rockefeller, 69008 Lyon, France
dUniversité de Poitiers, Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP), UMR CNRS 7285, Groupe “Systèmes Moléculaires Programmés”, 4 rue Michel-Brunet, TSA 51106, 86073 Poitiers, France
eSeekyo SA, 4 rue Carol Heitz, 86000 Poitiers, France
First published on 6th March 2019
Abstract
Antibody-drug conjugates (ADCs) convey highly potent anticancer drugs to antigen-expressing tumor cells, thereby sparing healthy tissues throughout the body. Pharmacokinetics and tolerability of ADCs are predominantly influenced by the drug-antibody ratio (DAR) of the conjugates, which is to-date limited to a value of 3–4 drugs per antibody in ADCs under clinical investigations. Here, we report the synthesis of monodisperse (i.e. discrete) polysarcosine compounds and their use as a hydrophobicity masking entity for the construction of highly-loaded homogeneous β-glucuronidase-responsive antibody-drug conjugates (ADCs). The highly hydrophilic drug-linker platform described herein improves drug-loading, physicochemical properties, pharmacokinetics and in vivo antitumor efficacy of the resulting conjugates.
Introduction
Antibody-drug conjugates (ADCs) represent an emerging class of oncology therapeutics, with 4 ADCs already on the market and more than 80 currently under clinical evaluation for various cancer indications.1–3 Expected improvements in this field rely on the design of novel drug-linker technologies that strongly influence the physicochemical properties of the conjugates.4–6 Key parameters such as (i) plasmatic stability of the drug-linker,7,8 (ii) Drug-Antibody-Ratio (DAR),9,10 (iii) conjugation position on the antibody component,11,12 (iv) overall hydrophobicity13 and homogeneity14,15 of the conjugates dictates pharmacokinetics (PK) properties, efficacy and tolerability of ADCs.Within this framework, it has long been considered that a 2-to-4 cytotoxic payload per antibody ratio (DAR2–4) achieves the optimal balance among pharmacokinetics and in vivo potency.9,10 Higher DAR species are traditionally known to hamper the therapeutic efficacy of ADCs because of the increased overall hydrophobicity of the conjugate that is conferred by the excessive number of highly hydrophobic drug cargo. In light of this finding, a great emphasis on site-specific bioconjugation technologies aiming to deliver homogeneous DAR2 or DAR4 conjugates is observed in the field and began to translate into the clinic.16,17 These techniques require protein genetic re-engineering and/or the use of one or several coupling enzymes to graft the drug-linker payload to the antibody. As a consequence, their implementation is time-consuming, rather expensive and may prove to be difficult to transpose to large-scale production.
Recently, hydrophilic drug-linker architectures aiming to mask or minimize the apparent hydrophobicity of the payloads and overcome the DAR2–4 limitation have paved the way to a new generation of highly drug-loaded ADCs.18 These innovations enable improved physicochemical properties, excellent PK profiles, decreased non-specific uptake, protection against payload metabolism, superior efficacy in low-target expressing tumors and allow the use of moderately potent drugs as ADC payloads.2,13,19–22 Furthermore, this approach offers the possibility to obtain homogeneous ADCs without tricky site-specific conjugation technologies. Underlying all these observations, it is also admitted that a beneficial correlation exists between the overall hydrophilicity and tolerability of ADCs.13,23
In the present work we envisioned the use of polysarcosine (PSAR) as a hydrophobicity masking entity that would be embedded into an ADC drug-linker platform. Thus, we herein report a novel generation of strongly hydrophilic PSAR-containing β-glucuronidase-responsive self-immolative drug-linkers devoted to the preparation of highly-loaded homogeneous ADCs having improved physicochemical and pharmacological properties (Fig. 1). In this pilot study, we designed drug-linkers that include the potent monomethyl auristatin E (MMAE) cytotoxin, a glucuronide trigger,24 a self-immolative linker,25,26 an auto-hydrolyzable maleimide-based bioconjugation head27 and a PSAR unit. With this design, we anticipated that the presence of both PSAR and glucuronide hydrophilic moieties would allow the construction of homogenous DAR8 ADCs, programmed for releasing MMAE within targeted cancer cells upon intracellular β-glucuronidase activation. Most of the strategies that have been employed to increase drug-linker hydrophilicity rely on the introduction of polyethylene glycol (PEG),28–31 which is to-date the gold standard for improving physicochemical properties of therapeutic agents but is not exempt of several limitations (non-biodegradable backbone and reported cases of hypersensitivity or accelerated blood clearance).32,33 Other approaches use hydrophilic stealth polymer carriers as drug-linker platforms, thus providing ADCs reaching DAR10–20.21,34,35 The main drawback of these approaches is the extreme polydispersity of the final ADCs, arising from the polydisperse nature of the polymer-linker and the heterogeneous coupling procedure to the antibody.
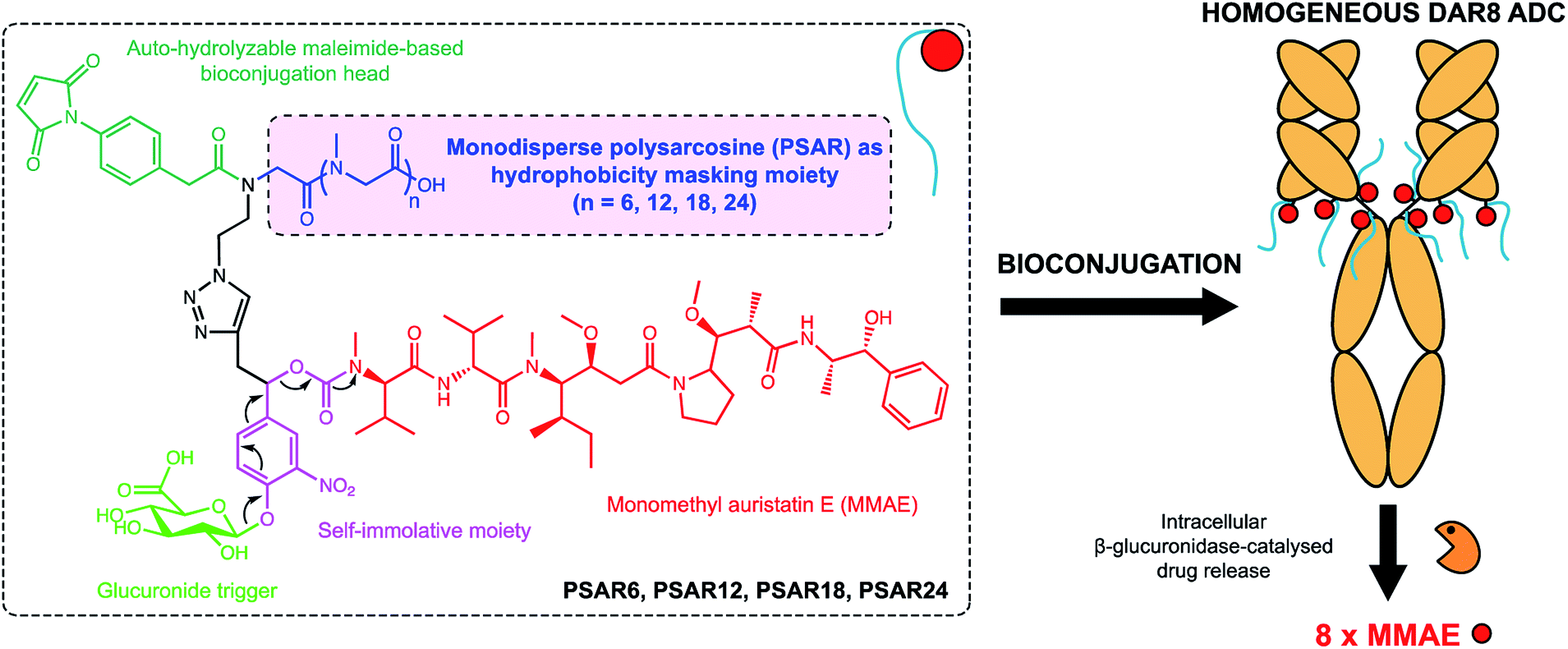 | ||
| Fig. 1 Chemical structure of the polysarcosine-based ADC drug-linker platform and schematic representation of homogeneous ADCs with a Drug-Antibody-Ratio (DAR) of 8. See ESI† for detailed chemical synthetic and bioconjugation procedures. | ||
Polysarcosine (PSAR) or poly(N-methylglycine), a polypeptoid based on the endogenous sarcosine aminoacid, is a rather underexplored biocompatible and biodegradable polymer that has been employed as an hydrophilic block in copolymers for nanosized drug delivery systems,36 as an antifouling polymer for surface modification37 and in fluorophore-conjugate constructs for imaging purposes.38 Recently PSAR was investigated for therapeutic protein conjugation purposes, conferring protease resistance to an interferon conjugate and increasing its circulation half-life.39
To date, only polydisperse polymeric PSAR is available, since it is prepared via a condensative ring-opening polymerization reaction.40 These polydisperse PSAR are suboptimal in the context of ADCs, where developing a drug-linker platform with absolute chemical homogeneity is highly preferable. Such a platform would provide chemically homogeneous ADCs sharing the exact same pharmacological properties (PK and efficacy), would be more straightforward to characterize and would allow greater control of the reproducibility of the manufacturing process.
Results and discussion
As a result, we decided to access monodisperse (i.e. discrete) PSAR oligomers by a submonomer solid-phase synthesis method that allowed a strict control over the repeat unit number (Scheme 1).41 Alternating acylation steps by bromoacetic acid and diisopropylcarbodiimide with nucleophilic displacement steps by methylamine afforded monodisperse polysarcosine oligomers with excellent purity (Schemes S1 and S2†). One of the major difficulties faced during this synthesis was the observation that an almost quantitative diketopiperazine formation occurred at the dimeric stage, despite the use of the sterically hindered 2-chlorotrityl solid support (Scheme S3†). This was avoided by adding the second and third sarcosine residues as a dipeptoid unit. The PSAR strands were then functionalized with an azide group for subsequent grafting of a cytotoxic payload and terminated with a maleimide reactive moiety for final coupling to the antibody.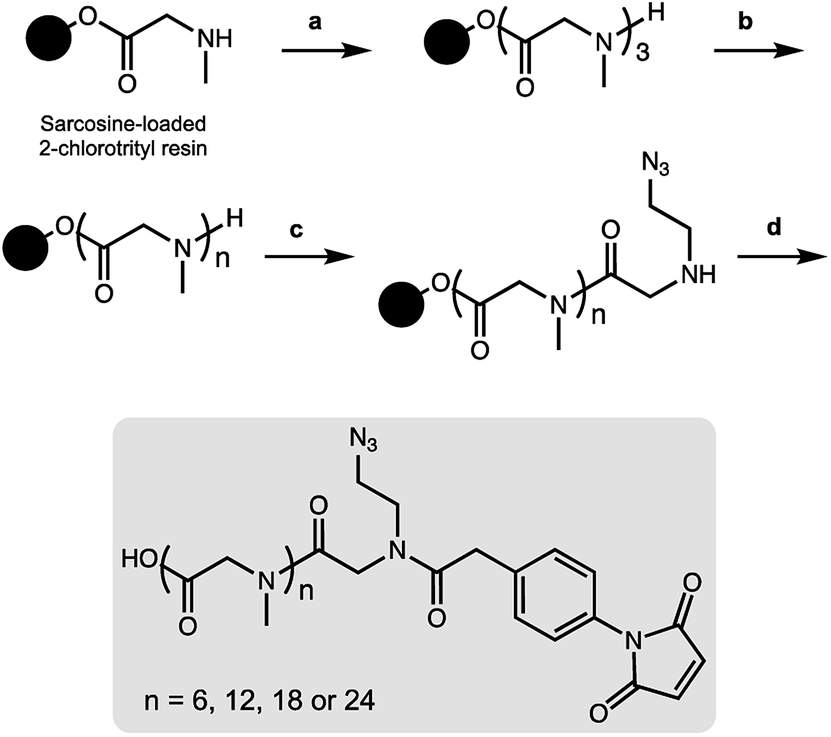 | ||
| Scheme 1 On-resin synthesis of monodisperse side-functionalized polysarcosine oligomers. (a) Fmoc-Sar-Sar-OH, HATU, DIPEA, DMF then piperidine/DMF. (b) Bromoacetic acid (BAA), diisopropylcarbodiimide (DIC), DMF then methylamine in water. This step is repeated until the desired oligomer length is obtained. (c) BAA, DIC, DMF then 2-azidoethan-1-amine, DMF. (d) 4-Maleimidophenylacetic acid, COMU, DIPEA, DMF then TFA, CH2Cl2. | ||
To support our proof-of-concept study, a family of PSAR-based drug-linkers was synthesized (Fig. 1, S5 and S6†). We used the previously described β-glucuronidase-sensitive linker associated with potent MMAE as the payload unit.24,25 With the aim to investigate its effects on physicochemical and pharmacological properties of the resulting ADCs, length of the PSAR hydrophobicity masking moiety was incremented from 6 to 24 monomer units. The drug-linker architecture was optimized in such a way that the hydrophobic payload was the closest as possible to the antibody and the shielding PSAR moiety was in an orthogonal orientation to the payload.13 An auto-hydrolyzable aryl-maleimide was used to prevent premature deconjugation of the linker-drug in plasma.27
Control drug-linkers were also synthesized (Fig. 2A and S4–S9†). PSAR12L aimed to assess the impact of linker architecture (orthogonal versus linear PSAR placement) whereas PSAR0 lacks PSAR hydrophobicity masking moiety. PEG12 incorporates PEG instead of PSAR in order to make a side-by-side comparison of the two hydrophobicity masking entities.
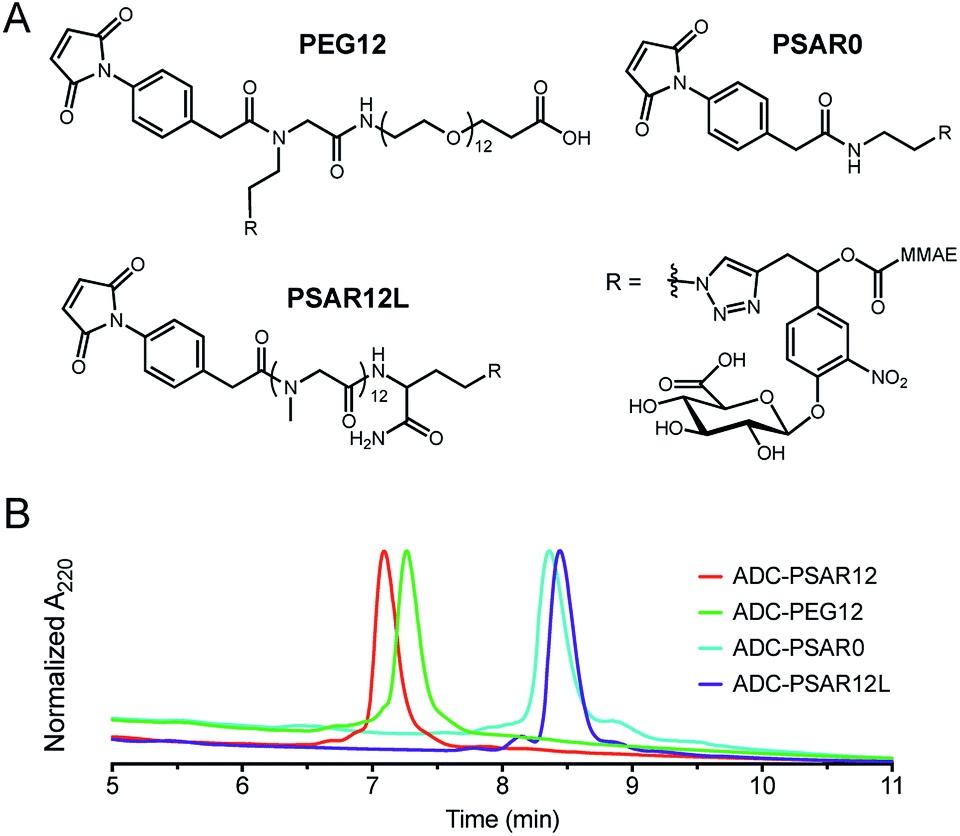 | ||
| Fig. 2 (A) Structures of the negative control drug-linkers used in the study. See ESI† for detailed chemical synthetic procedures. (B) Hydrophobic Interaction Chromatography (HIC) profiles of ADCs. | ||
Homogeneous DAR8 conjugates based on clinically validated antibody trastuzumab (anti-HER2 mAb) were produced (Fig. 2B and S11–S14†). mAb interchain disulfide bonds were totally reduced with excess tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and drug-linkers were coupled to the antibody (1.25 molar equivalent linker per mAb cysteine—30 min incubation time). The resulting ADCs were purified by buffer-exchange using Sephadex™ desalting columns and incubated 48 hours at 37 °C in PBS to promote complete maleimide hydrolysis.
We were pleased to observe that a significant reduction in hydrophobic interaction chromatography (HIC) retention time was observed for the ADC-PSAR12 conjugate, compared to its negative control ADC-PSAR0 (Fig. 2B and S13†). This result demonstrates that inclusion of PSAR in a parallel orientation to the drug unit was able to promote satisfactory hydrophobicity masking and stealth properties to the final conjugate. Inclusion of PSAR in a linear configuration between the mAb and the drug (ADC-PSAR12L) was detrimental to the masking of hydrophobicity, as already observed in previous studies.13 At equal length (n = 12 monomer units), PSAR provides slightly better shielding properties than PEG (ADC-PSAR12versusADC-PEG12). Albeit providing general distinctions between PSAR/PEG hydrophilic moieties or lack thereof, in silico log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D or logS predictions of the drug-linkers were unable to provide insight on the crucial impact of the linker architecture on the final ADC hydrophilicity (Fig. S10†). We also confirmed that the grafting of 8 drug-linkers, with or without hydrophobicity masking entity, had negligible impact on antigen-binding affinity compared to the native antibody (Fig. S15†).
D or logS predictions of the drug-linkers were unable to provide insight on the crucial impact of the linker architecture on the final ADC hydrophilicity (Fig. S10†). We also confirmed that the grafting of 8 drug-linkers, with or without hydrophobicity masking entity, had negligible impact on antigen-binding affinity compared to the native antibody (Fig. S15†).
To explore if inclusion of an orthogonal PSAR in the drug-linker structure would improve PK properties and antitumor activity, mice PK and xenograft studies were conducted with ADC-PSAR12 and ADC-PSAR0. Without inclusion of PSAR, a detrimental accelerated plasma clearance was observed, as anticipated for a DAR8 ADC based on a conventional drug-linker (Fig. 3A).9,10 PK profile was restored with the inclusion of the PSAR hydrophobicity masking moiety. Antitumor activity of these two ADCs in a BT-474 breast cancer model was directly correlated with their observed PK behavior (Fig. 3B and S16†). A single dose of ADC-PSAR12 at 3 mg kg−1 was curative whereas ADC-PSAR0 induced incomplete tumor regressions. At the same dose, the currently approved Trastuzumab-DM1 (Kadcyla®) was only able to promote a negligible tumor growth delay and no cures.
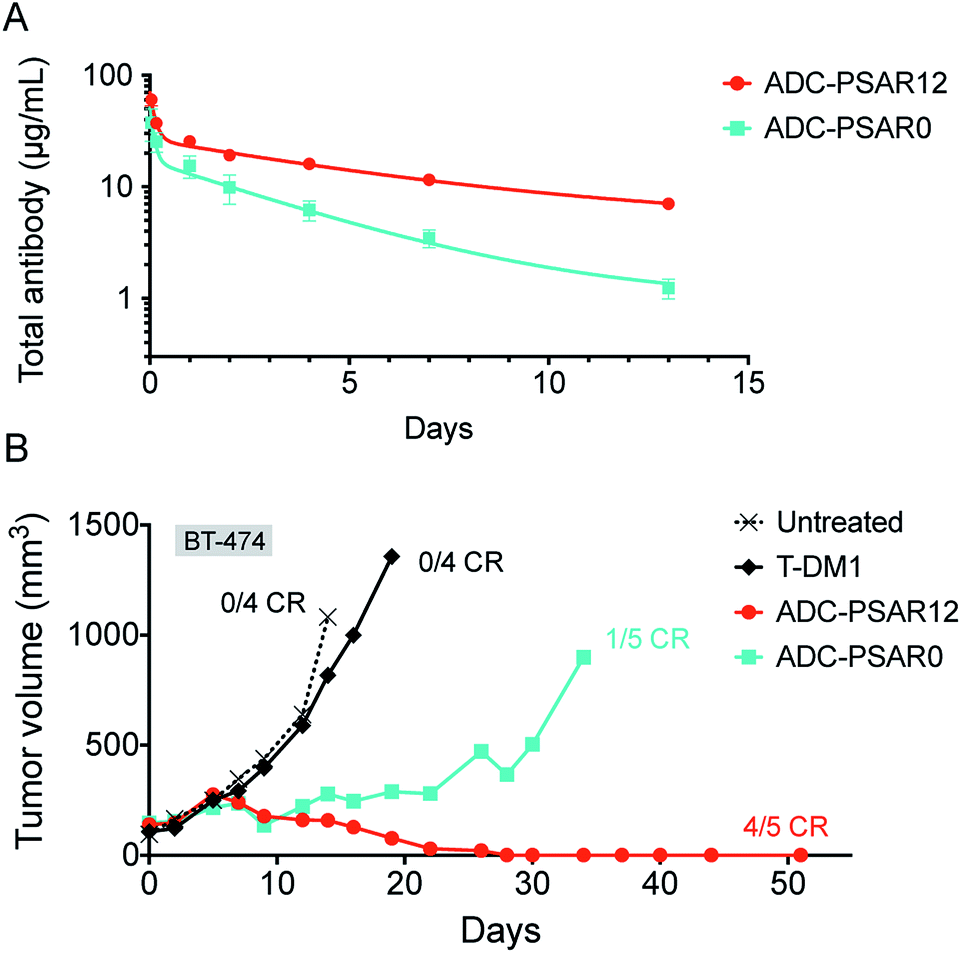 | ||
| Fig. 3 (A) Total ADC pharmacokinetic study in SCID mice after a single intravenous ADC dose of 3 mg kg−1. Clearance was 15.8 and 37.6 mL per day per kg respectively for ADC-PSAR12 and ADC-PSAR0, as calculated by two-compartmental model analysis. (B) Antitumor activity in SCID/BT-474 breast cancer model following a single intravenous ADC dose of 3 mg kg−1. No body-weight changes were observed during the study. CR = complete remission. | ||
In order to gain some insight on structure–activity relationship of the drug-linkers, more thorough in vivo investigations were conducted (Fig. 4). The impact of PSAR length on ADC PK properties was investigated in Sprague-Dawley rats, as it has been shown that magnitude of ADC PK differences are more important in rats than in mice.13 We observed that ADC exposure increased as a function of PSAR length up to 12 monomer residues, eventually reaching a point where further PSAR extension had little influence on the clearance rates of the conjugates (Fig. 4A, B and S17†). In accordance with their observed hydrophobicity (Fig. 2B), ADC-PSAR0 and ADC- PSAR12L showed unfavorable PK characteristics. These results clearly show that inclusion of a hydrophobicity masking entity in an orthogonal orientation to the drug is mandatory to efficiently restore PK properties. It was also observed that at equal length, PSAR more efficiently improve clearance rates when compared to PEG (respectively 38.9 and 47.3 mL per day per kg for ADC-PSAR12 and ADC-PEG12).
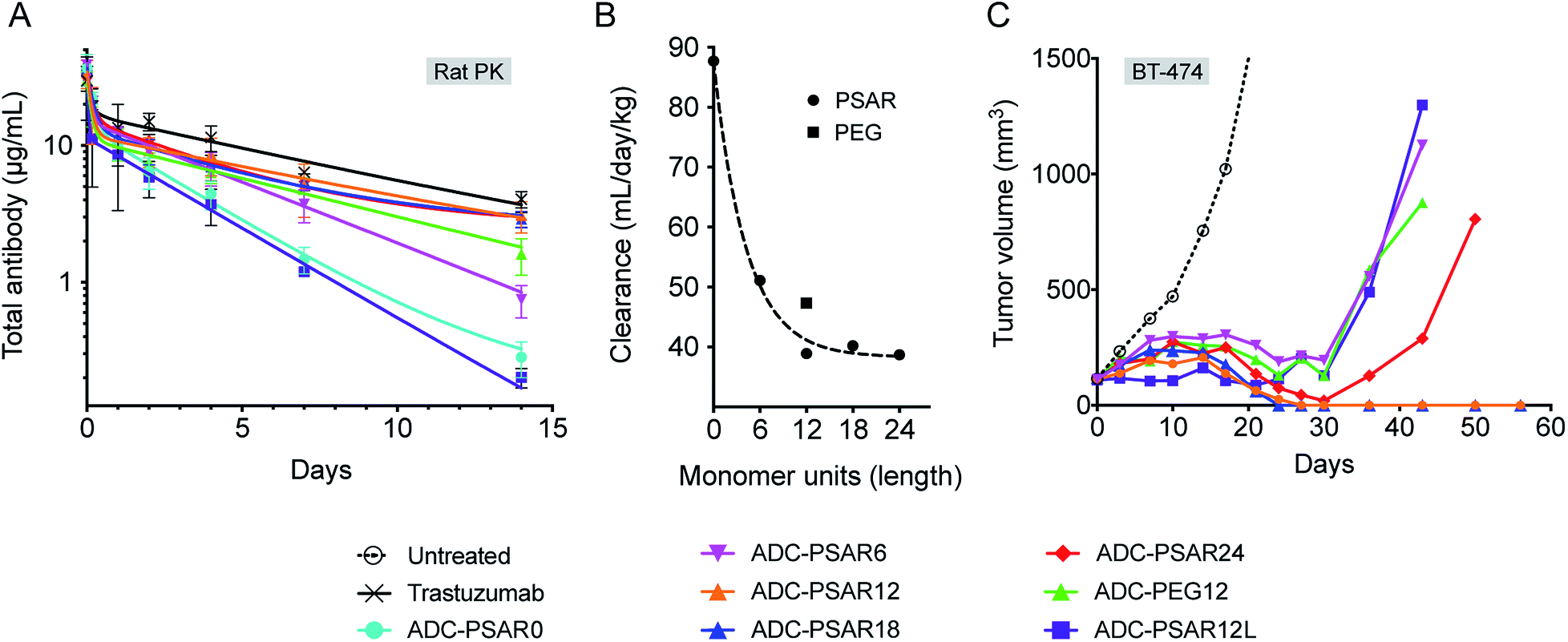 | ||
| Fig. 4 (A) Total ADC pharmacokinetic study in Sprague-Dawley rats after a single intravenous ADC dose of 3 mg kg−1 and (B) associated clearance rates (two-compartmental model analysis) as a function of the hydrophobicity masking entity length (monomer units). (C) Antitumor activity in SCID/BT-474 breast cancer model following a single intravenous ADC dose of 2.5 mg kg−1. No body-weight changes were observed during the study. | ||
Antitumor activity of these ADCs was investigated in the breast cancer model BT-474, at a single 2.5 mg kg−1 dose (Fig. 4C). Complete tumor regressions were only observed for conjugates having low clearance rates (ADC-PSAR12 and ADC-PSAR18), validating that a 12 monomer PSAR length appears to be an optimized value, at least for this MMAE-based drug-linker. In other experiments conducted in our labs, we observed that the optimized PSAR length is a payload-dependent value that can be more carefully optimized depending on the hydrophobic character and the number of payloads attached to one drug-linker. Interestingly we observed that the ADC-PSAR24 compound had a suboptimal activity, despite its excellent clearance profile. As already observed28 and because ADC hydrodynamic diameter increases as a function of the hydrophilic shielding entity size, it can be hypothesized that a 24 monomer PSAR length ultimately yields an overlarge and/or hindered conjugate. Such a conjugate may partially impede tumor penetration, interfere with internalization or β-glucuronidase digestion processes and ultimately lead to the observed loss of efficacy.
ADCs having unfavorable (ADC-PSAR12L) or suboptimal (ADC-PSAR6 and ADC-PEG12) clearance profiles were only able to promote delayed tumor growth at the tested dose (Fig. 4C). These results confirm the rational orthogonal placement of the PSAR hydrophobicity masking entity and show that, at equal length, PSAR more efficiently improve ADC antitumor activity when compared to PEG (ADC-PSAR12versusADC-PEG12).
Conclusions
In summary, we report the use of monodisperse polysarcosine as a hydrophobicity masking entity for the formulation of high drug-load ADCs having improved physicochemical properties, excellent pharmacokinetic characteristics and remarkable antitumor potencies. Owing to its simplicity, this straightforward approach allows the formulation of optimized plasma-stable homogeneous ADCs without the requirement of hard-to-implement bioconjugation technologies. Our approach can be tailored to obtain ADCs incorporating very hydrophobic payloads known for their aggregation propensity or highly-loaded ADCs bearing conventional or more moderately potent payloads with new mechanisms of action.1,2 Ongoing efforts focusing on the application of this platform to the preparation of homogeneous DAR8/16 ADCs based on differentiated payloads will be reported elsewhere.Conflicts of interest
Some authors are co-inventors on a patent application related to this work and are shareholders of Mablink Bioscience.Acknowledgements
The Cancéropôle Lyon Auvergne Rhône-Alpes (CLARA) and the Banque Publique d’Investissement (BPI) are acknowledged for financial support. The authors would like to thank the Antineo team for their contribution and insightful discussions as well as Dr Alexandra Berlioz-Barbier (Centre Commun de Spectrométrie de Masse, UMR CNRS 5246) for assistance with LC-MS analysis of antibody conjugates.Notes and references
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- Innovations for Next-Generation Antibody-Drug Conjugates, ed. M. Damelin, Humana Press, 2018th edn, 2018 Search PubMed.
- H. Wade, ADC Beacon [database], https://beacon-intelligence.com, 2018 Search PubMed.
- T. Rodrigues and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2018, 57, 2032–2034 CrossRef CAS PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- K. Tsuchikama and Z. An, Protein Cell, 2018, 9, 33–46 CrossRef CAS PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- R. P. Lyon, T. D. Bovee, S. O. Doronina, P. J. Burke, J. H. Hunter, H. D. Neff-LaFord, M. Jonas, M. E. Anderson, J. R. Setter and P. D. Senter, Nat. Biotechnol., 2015, 33, 733–735 CrossRef CAS PubMed.
- N. Gupta, J. Kancharla, S. Kaushik, A. Ansari, S. Hossain, R. Goyal, M. Pandey, J. Sivaccumar, S. Hussain, A. Sarkar, A. Sengupta, S. K. Mandal, M. Roy and S. Sengupta, Chem. Sci., 2017, 8, 2387–2395 RSC.
- P. Akkapeddi, S.-A. Azizi, A. M. Freedy, P. M. S. D. Cal, P. M. P. Gois and G. J. L. Bernardes, Chem. Sci., 2016, 7, 2954–2963 RSC.
- S. S. van Berkel and F. L. van Delft, Drug Discovery Today: Technol., 2018, 30, 3–10 CrossRef PubMed.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS PubMed.
- N. Bodyak and A. V. Yurkovetskiy, in Innovations for Next-Generation Antibody-Drug Conjugates, Humana Press, Cham, 2018, pp. 215–240 Search PubMed.
- S. V. Govindan, T. M. Cardillo, E. A. Rossi, P. Trisal, W. J. McBride, R. M. Sharkey and D. M. Goldenberg, Mol. Pharm., 2015, 12, 1836–1847 CrossRef CAS PubMed.
- Y. Ogitani, T. Aida, K. Hagihara, J. Yamaguchi, C. Ishii, N. Harada, M. Soma, H. Okamoto, M. Oitate, S. Arakawa, T. Hirai, R. Atsumi, T. Nakada, I. Hayakawa, Y. Abe and T. Agatsuma, Clin. Cancer Res., 2016, 22, 5097–5108 CrossRef CAS PubMed.
- A. V. Yurkovetskiy, M. Yin, N. Bodyak, C. A. Stevenson, J. D. Thomas, C. E. Hammond, L. Qin, B. Zhu, D. R. Gumerov, E. Ter-Ovanesyan, A. Uttard and T. B. Lowinger, Cancer Res., 2015, 75, 3365–3372 CrossRef CAS PubMed.
- D. Su, K. R. Kozak, J. Sadowsky, S.-F. Yu, A. Fourie-O'Donohue, C. Nelson, R. Vandlen, R. Ohri, L. Liu, C. Ng, J. He, H. Davis, J. Lau, G. Del Rosario, E. Cosino, J. dela Cruz-Chuh, Y. Ma, D. Zhang, M. Darwish, W. Cai, C. Chen, H. Zhou, J. Lu, Y. Liu, S. Kaur, K. Xu and T. H. Pillow, Bioconjugate Chem., 2018, 29, 1155–1167 CrossRef CAS PubMed.
- J. M. M. Verkade, M. A. Wijdeven, R. van Geel, B. M. G. Janssen, S. S. van Berkel and F. L. van Delft, Antibodies, 2018, 7, 12 CrossRef.
- T. Legigan, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski, A. Monvoisin, J.-M. Berjeaud, F. Guilhot and S. Papot, J. Med. Chem., 2012, 55, 4516–4520 CrossRef CAS PubMed.
- B. Renoux, F. Raes, T. Legigan, E. Péraudeau, B. Eddhif, P. Poinot, I. Tranoy-Opalinski, J. Alsarraf, O. Koniev, S. Kolodych, S. Lerondel, A. L. Pape, J. Clarhaut and S. Papot, Chem. Sci., 2017, 8, 3427–3433 RSC.
- A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
- R. J. Christie, R. Fleming, B. Bezabeh, R. Woods, S. Mao, J. Harper, A. Joseph, Q. Wang, Z.-Q. Xu, H. Wu, C. Gao and N. Dimasi, J. Controlled Release, 2015, 220, 660–670 CrossRef CAS PubMed.
- P. J. Burke, J. Z. Hamilton, S. C. Jeffrey, J. H. Hunter, S. O. Doronina, N. M. Okeley, J. B. Miyamoto, M. E. Anderson, I. J. Stone, M. L. Ulrich, J. K. Simmons, E. E. McKinney, P. D. Senter and R. P. Lyon, Mol. Cancer Ther., 2017, 16, 116–123 CrossRef CAS PubMed.
- M. R. Levengood, X. Zhang, J. H. Hunter, K. K. Emmerton, J. B. Miyamoto, T. S. Lewis and P. D. Senter, Angew. Chem., Int. Ed., 2017, 56, 733–737 CrossRef CAS PubMed.
- M. Pabst, W. McDowell, A. Manin, A. Kyle, N. Camper, E. De Juan, V. Parekh, F. Rudge, H. Makwana, T. Kantner, H. Parekh, A. Michelet, X. Sheng, G. Popa, C. Tucker, F. Khayrzad, D. Pollard, K. Kozakowska, R. Resende, A. Jenkins, F. Simoes, D. Morris, P. Williams, G. Badescu, M. P. Baker, M. Bird, M. Frigerio and A. Godwin, J. Controlled Release, 2017, 253, 160–164 CrossRef CAS.
- S.-J. Moon, S. V. Govindan, T. M. Cardillo, C. A. D'Souza, H. J. Hansen and D. M. Goldenberg, J. Med. Chem., 2008, 51, 6916–6926 CrossRef CAS PubMed.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed.
- Q. Yang and S. K. Lai, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 655–677 CAS.
- A. Yurkovetskiy, M. Yin, T. B. Lowinger, J. D. Thomas, C. E. Hammond, C. Stevenson, N. D. Bodyak, P. R. Conlon and D. R. Gumerov, WO2012171020 (A1), 2012.
- L. Zhang, Y. Fang, J. Kopeček and J. Yang, Eur. J. Pharm. Sci., 2017, 103, 36–46 CrossRef CAS PubMed.
- A. Birke, D. Huesmann, A. Kelsch, M. Weilbächer, J. Xie, M. Bros, T. Bopp, C. Becker, K. Landfester and M. Barz, Biomacromolecules, 2014, 15, 548–557 CrossRef CAS PubMed.
- K. H. A. Lau, C. Ren, T. S. Sileika, S. H. Park, I. Szleifer and P. B. Messersmith, Langmuir, 2012, 28, 16099–16107 CrossRef CAS PubMed.
- K. Sano, M. Ohashi, K. Kanazaki, A. Makino, N. Ding, J. Deguchi, Y. Kanada, M. Ono and H. Saji, Bioconjugate Chem., 2017, 28, 1024–1030 CrossRef CAS PubMed.
- Y. Hu, Y. Hou, H. Wang and H. Lu, Bioconjugate Chem., 2018, 29, 2232–2238 CrossRef CAS PubMed.
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Macromolecules, 2011, 44, 6746–6758 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00285e |
| This journal is © The Royal Society of Chemistry 2019 |