DOI:
10.1039/C9SC00244H
(Edge Article)
Chem. Sci., 2019,
10, 4550-4559
Breaking the 1,2-HOPO barrier with a cyclen backbone for more efficient sensitization of Eu(III) luminescence and unprecedented two-photon excitation properties†
Received
16th January 2019
, Accepted 27th March 2019
First published on 28th March 2019
Abstract
A cyclen backbone was utilized to study the effect of backbone rigidity on Eu(III) luminescence sensitization using a 1,2-HOPO derivative and 2-thenoyltrifluoroacetonate (TTA) as chromophores. The restriction of molecular movement of Eu-Cy-HOPO brought about by the increased rigidity provided a tightly packed coordination environment for the octadentate Eu(III) center which resulted in the highest overall quantum yield (30.2%) and sensitization efficiency (64.6%) among 1,2-HOPO sensitized Eu(III) complexes. Eu-Cy-HOPO is also the first 1,2-HOPO-based lanthanide complex to emit Eu(III) luminescence under two-photon excitation.
Introduction
Trivalent lanthanide (Ln(III)) ions exhibit unique luminescence properties, such as fingerprint spectral profiles, sharp emission bands and long luminescence lifetimes and thus are widely considered as potential replacements of organic chromophores and transition metal complexes especially in biological applications in which autofluorescence from biological entities often interferes with fluorescent signals due to their similar emission lifetimes and/or emission profile.1–3 However, optimizing lanthanide luminescence requires delicate molecular design to fulfill vital elements during the sensitization process. The f–f transitions of Ln(III) ions are Laporte forbidden and therefore have intrinsically low absorption coefficients (ε ≈ 1–10 M−1 cm−1). Indirect excitation (sensitization) of Ln(III) is achieved by an excited energy transfer process from a light-harvesting chromophore and the energy transfer efficiency is principally governed by two main energy transfer mechanisms dependent on the chromophore-Ln(III) distance. The Förster mechanism is a ‘through space’ interaction that requires the donor's emission spectrum to overlap with the acceptor's excitation spectrum with a r−6 distance-dependence.4,5 Alternatively, the Dexter mechanism requires a physical orbital overlap between the donor and acceptor for a concerted electron exchange and is dependent on e(−2r/L).5 Secondly, as the 4f electrons of Ln(III) are well-shielded, interaction between Ln(III) ions and ligands is mainly ionic, resulting in flexible coordination numbers from 8–12 depending on steric factors. It has therefore become prevalent to design multidentate chromophoric ligands to obtain good luminescence properties.
In 2006, Raymond's group extended Picard's work6 on using a bidentate 1,2-hydroxypyridinonate (1,2-HOPO) chromophore to sensitize Ln(III) by modifying it into a tetradentate ligand consisting of two 6-amide derivatives of 1,2-HOPO to form an ML2 complex and dramatically improved the luminescence properties of the Eu(III) complex: from Φ = 0.3% of Picard's group to 21.5%.7 Then, they designed the first example of an octadentate ligand with four units of the 1,2-HOPO derivative connected through an N,N,N′,N′-tetrakis-(2-aminoethyl)-ethane-1,2-diamine (H(2,2)) to form an ML complex with improved aqueous stability but exhibited weak luminescence (Φ = 3.6%) due to the presence of one water molecule in the inner coordination sphere of Eu(III).8 Replacing the branched tetrapodal skeleton of H(2,2) with a linear spermine-based (3,4,3-LI) offered sufficient protection of the Eu(III) center from water molecules coordination and directly resulted in an increased radiative decay rate and decreased non-radiative decay rate; however, the quantum yield was only 7.0% as the linear backbone contributed to poorer sensitization.9 Later, they found out that isolating the same (3,4,3-LI) Eu(III) complex prior to luminescence measurements would allow the 1,2-HOPO derivative units to fully coordinate with Eu(III) and lead to a slower non-radiative decay rate and higher sensitization efficiency, achieving a quantum yield of 15.6%.10 In 2015, a systematic study on derivatives based on the H(2,2) skeleton was carried out on investigating how the change of central chain length and the length between two bridgehead tertiary nitrogen atoms would affect the photophysical properties of Eu(III).11 The authors concluded that with a shorter length, the steric constraints would lead to coordination of a water molecule whereas a longer length would give highly luminescent complexes (Φ = 19.6%). Most recently, the highest quantum yield of an Eu(III) complex sensitized by the 6-amide 1,2-HOPO derivative was obtained (Φ = 23.9%) with a tetradentate ligand with two methylene groups between the two chelating chromophores, and such a geometry allows better wrapping of the Eu(III), resulting in better sensitization efficiency.16 Nonetheless, a systematic relationship between the coordination geometry and photophysical properties remains elusive despite more than a decade's thorough work on a selected system. Table 1 selectively summarizes the work of Raymond's group.
Table 1 Summary of literature data of Eu(III) complexes sensitized by 1,2-HOPO derivatives done by Raymond's groupa
| Compound |
ε (M−1 cm−1) |
τ
H2O (ms) |
q
|
Φ
LnL
|
Φ
LnLn
|
η
sens
|
Ref. |
|
All measurements were done at pH 7.4.
|
| [Eu(5LIO-1,2-HOPO)2]− |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 250@333 nm 250@333 nm |
0.727 |
0 |
21.5% |
43.0% |
49.0% |
7
|
| [Sm(5LIO-1,2-HOPO)2]− |
19200@331 nm |
0.013 |
— |
0.44% |
— |
— |
12
|
| Eu(5LINMe-1,2,-HOPO)2 |
18750@332 nm |
0.728 |
0 |
0.173 |
44.2% |
39.1% |
8
|
| [Eu(H(2,2)-1,2-HOPO)]− |
18200@341 nm |
0.48 |
1 |
3.6% |
17.8% |
20.2% |
13
|
| [Eu(o-Phen-1,2-HOPO)2]− |
21020@342 nm |
0.536 |
0 |
6.2% |
36.5% |
17.0% |
14
|
| [Eu(5LI-1,2-HOPO)2]− |
18800@331 nm |
0.737 |
0 |
20.7% |
49.0% |
42.0% |
12
|
| [Eu(5LI-1,2-HOPO)2]− |
19400@331 nm |
0.011 |
— |
0.44% |
— |
— |
12
|
| [Eu(3,4,3-LI(1,2,-HOPO))]− (in situ) |
17700@315 nm |
0.805 |
0 |
0.7% |
43.2% |
16.2% |
9
|
| [Sm(3,4,3-LI(1,2,-HOPO))]− (in situ) |
17850@316 nm |
0.017 |
— |
0.2% |
— |
— |
15
|
| [Eu(3,4,3-LI(1,2,-HOPO))]− (isolated) |
— |
0.814 |
0 |
15.6% |
46.9% |
39.7% |
10
|
| [Sm(3,4,3-LI(1,2,-HOPO))]− (isolated) |
— |
0.019 |
— |
0.41% |
— |
— |
10
|
| [Eu(H(17O5,2)-1,2-HOPO)]− |
15000@336 nm |
0.704 |
0 |
19.6% |
52.2% |
37.5% |
11
|
| [Eu(2LI-1,2-HOPO)2]− |
21600@338 nm |
0.578 |
0 |
23.9% |
42.0% |
51.0% |
16
|
| [Sm(2LI-1,2-HOPO)2]− |
21600@334 nm |
0.017 |
0 |
0.4% |
— |
— |
16
|
1,4,7,10-Tetraazacyclododecane (cyclen)-based chelators are vastly common amongst Ln(III) and transition metals for various applications, especially the carboxylate derivative DO3A (and its derivatives) which gives octadentate complexes with exceptional stability.17–19 In this work, we intend to utilize the 12-memebered ring as a macrocyclic backbone and investigate how it would influence the molecular arrangement and hence the luminescence properties of the complexes compared to tetrapodal and linear backbones. We expect the relatively rigid cyclen ring would restrict the movement of the tetradentate 1,2-HOPO units20 and reduce the rate of non-radiative deactivation while simultaneously increasing the energy transfer efficiency by limiting the average Eu(III)-1,2-HOPO separation. To further confirm the effect of the macrocyclic backbone, we also designed an analogous chelate with 2-thenoylfluoroacetonate (TTA), a known efficient sensitizer for Eu(III) luminescence,21 as comparison.
While 1,2-HOPO-based and cyclen-based Ln(III) complexes are often water-soluble, the application of luminescent Ln(III) complexes in a biological context, in general, is often hindered by the high-energy, tissue-damaging energy required for exciting the chromophore during antenna effect. The invention of femtosecond-pulsed laser sources has made multi-photon absorption – a non-linear optical process in which two or more photons with a combined amount of energy equal to the ΔE of a single-photon absorption process are absorbed almost simultaneously by a molecule affording a convenient solution by significantly shifting the excitation wavelength near or beyond the red region.22,23 However, as the selection rules for single-photon, two-photon and three-photon excitation are different, chromophores with a high ε does not guarantee a high two-photon absorption cross section (σ2). Highly absorptive dyes such as fluorescein and rhodamine 6G, with absorption maxima at ca. 500 nm and 530 nm respectively, have δ values of 8.0 and 9.2 GM (1 GM = 10−40 cm4 s photon−1) at 950 nm.24 Following a systematic study, Albota et al. suggested a design rationale for chromophores with a high σ2: ‘π-conjugated molecules with large changes of quadrupole moment upon excitation’25 and organic chromophores with d > 5000 GM have been gradually developed.26–29 Ln(III) complexes containing chromophores with σ2 from 0.37 to 775 GM have also been reported.21,30,31 As a result, a suitable balance between electronic density gradient and water-solubility should be attained when designing the structure of chromophores for two-photon biological applications.
Results and discussion
Synthesis of ligands and complexes
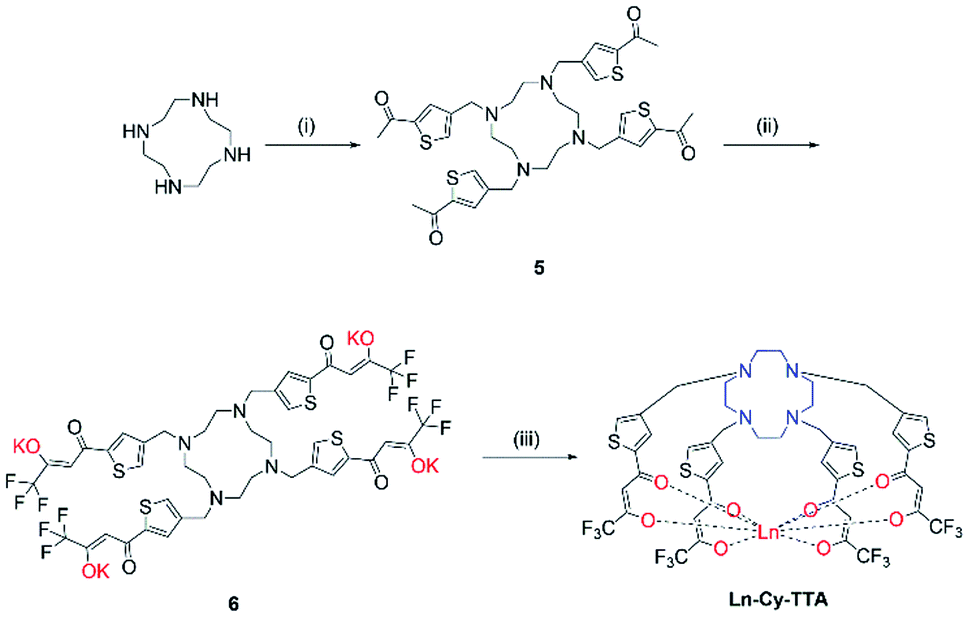
The syntheses of ligand 4 is presented in Scheme 1. Originally, we tried reacting cyclen with phthalimide- or Boc-protected bromoethylamine to give a protected tetra-amine cyclen derivative, but both reaction routes gave very low yields due to the reactivity of the bromo group and side reactions from nucleophilic substitution. By using tosylaziridine in a ring-opening, zwitterion-forming reaction, the protected derivative was obtained in a good yield after recrystallization with acetonitrile and benzene. Deprotection with acetic acid and hydrobromic acid gave 1 readily for reaction with the protected 1,2-HOPO derivative to give 3, which was purified by semi-preparative HPLC. The octadentate ligand 4 was obtained by recrystallization with methanol and diethyl ether after deprotection. Complexation with Ln(III) trichloride hexahydrate was performed in methanol in the presence of pyridine at 55 °C for 8 hours (Fig. 1).
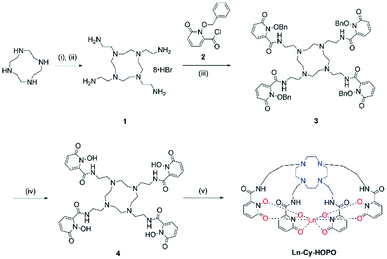 |
| | Scheme 1 Synthetic route of Ln-Cy-HOPO. (i) tosylaziridine, MeCN/benzene; (ii) AcOH/HBr; (iii) DIPEA, THF; (iv) AcOH/HCl; (v) LnCl3·6H2O, pyridine, MeOH, 55 °C, 8 h. | |
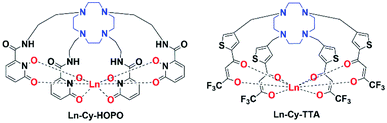 |
| | Fig. 1 Structures of europium(III) complexes Ln-Cy-HOPO and Ln-Cy-TTA (Ln = Eu/Sm/Gd). | |
Chloromethylation of 2-acetylthiophene was performed successfully with aluminum chloride as Lewis acid and strict control over the reaction time and stoichiometry and was subsequently reacted with cyclen to give 5. The substitution reaction was performed at room temperature for two days since a lot of side products – reaction of ketone with amine and acetylthiophene should be well-controlled, too, to avoid over- alkylation. Compound 6 was obtained by reacting 5 with ethyl trifluoroacetate with potassium bis(trimethylsilyl)amide and complexation was carried out with Eu(III) trichloride hexahydrate in methanol at 60 °C overnight (Scheme 2).
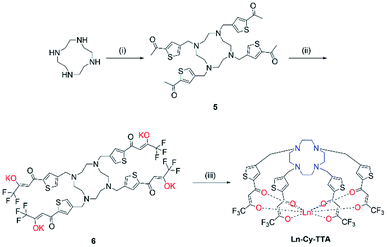 |
| | Scheme 2 Synthetic route of Ln-Cy-TTA. (i) 4-Chloromethyl-2-acetylthiophene, K2CO3, ACN, RT, 48 h; (ii) ethyl trifluoroacetate, KHMDS, −78 °C, RT; (iii) LnCl3·6H2O, MeOH, 60 °C, 12 h. | |
Photophysical properties
UV-vis absorption spectroscopy.
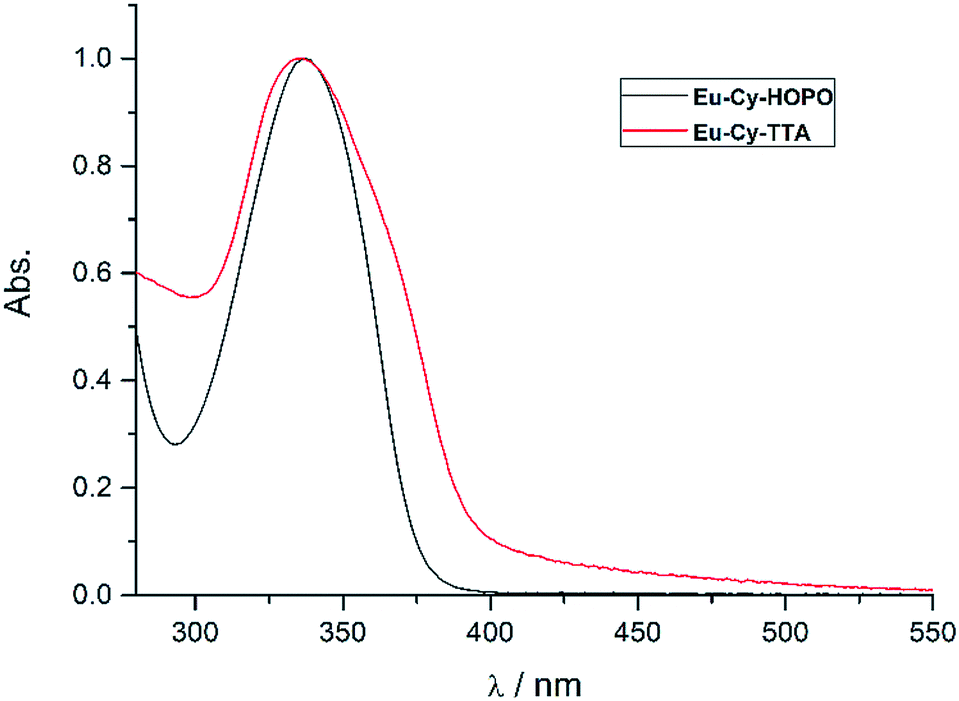
Fig. 2 shows the UV-vis absorption properties of Eu-Cy-HOPO and Eu-Cy-TTA in water (pH 5.5) and aqueous solution (3% DMSO) respectively. Both absorption spectra only show one absorption band with maxima at 337 nm (ε350 nm = 12100 M−1 cm−1) and 336 nm (ε350 nm = 22690 M−1 cm−1), assigned as the π–π* transitions of the chromophores. The molar absorption coefficients of are lower than values of reported 1,2-HOPO- and TTA-based compounds, due to the crowdedness between the chromophores brought about by the rigid cyclen backbone.
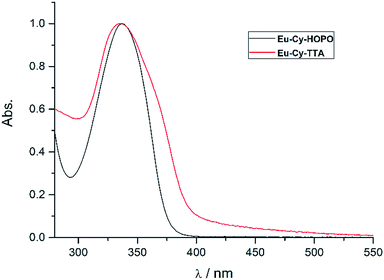 |
| | Fig. 2 Normalized UV-vis absorption spectra of Eu-Cy-HOPO and Eu-Cy-TTA. | |
Luminescence properties of Eu-Cy-HOPO and Eu-Cy-TTA.
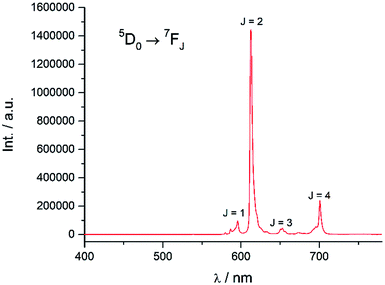
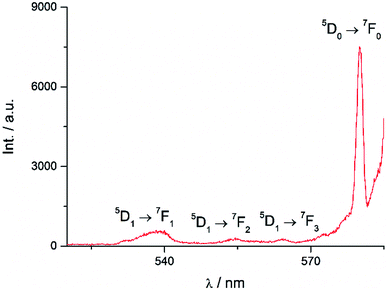
Excitation at 350 nm resulted in the characteristic Eu(III) emission profile with the 5D0 → 7FJ (J = 1–4) transitions clearly observed (Fig. 3). Fig. 4 offers a higher magnification into the 5D0 → 7F0 transition, which is often very weak in due to its forbidden nature, as well as the 5D1 → 7FJ transitions, indicating the involvement of the higher excited state in sensitization.
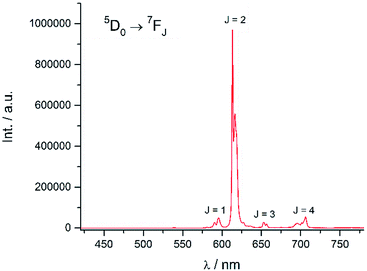 |
| | Fig. 3 Emission spectrum of Eu-Cy-HOPO in water (λex = 350 nm). | |
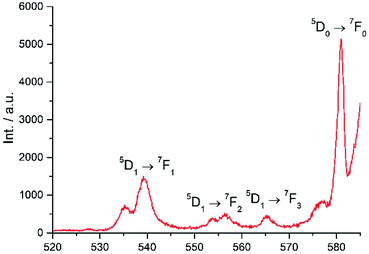 |
| | Fig. 4 Higher magnification of partial emission spectrum of Eu-Cy-HOPO in Fig. 3. | |
Energy transfer from the 1,2-HOPO unit is efficient as residual ligand fluorescence was not observed. The high intensity of the 5D0 → 7F2 hypersensitive transition relative to the other transitions, quantified by an asymmetry ratio of 14, reveals a large extent of deviation from a centrosymmetric geometry of the Eu(III) center,32 corroborating with the narrow octadentate structure optimized using the RM1 model by the LUMPAC software package (Fig. 5).33,34
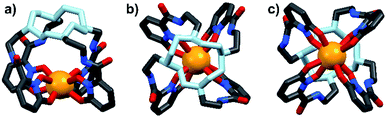 |
| | Fig. 5 Optimized structure of Eu-Cy-HOPO. View from side (a); above cyclen backbone (b); view from below Eu(III) center (c). | |
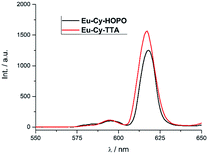
The luminescence lifetime of the 5D0 → 7F2 transition was measured to be a respectable 0.784 ms. The number of coordinated water molecule in the inner coordination sphere (q) of Eu(III) was determined to be 0 using both Parker's35 and Horrocks'36 equation respectively, which is similar to the octadentate complexes of Raymond's group. The luminescence lifetime was also measured in methanol and methanol-d4 and the number of coordinated methanol molecule (m) is determined to be 0.37 An overall quantum yield of 30.2% was measured relative to quinine sulfate; this value is the highest amongst Eu(III) complexes sensitized by a 1,2-HOPO chromophore thus far. The sensitization efficiency in water was calculated to be 64.6%, which is noticeably higher compared to the ceiling of 50% of Raymond's group in 0.1 M TRIS buffer (Table 1). These parameters indicated efficient luminescence sensitization which is attributed to two factors from our molecular design: (1) sufficient protection by the four 1,2-HOPO units prevented the coordination of solvent molecules which quenches the excited energy of Eu(III) by vibrational overtones of O–H oscillators35 and; (2) rigid macrocyclic backbone restricting the movement of the 1,2-HOPO unit and maintaining a close distance between the chromophore and Eu(III) since energy transfer mechanisms are highly distance-dependent.
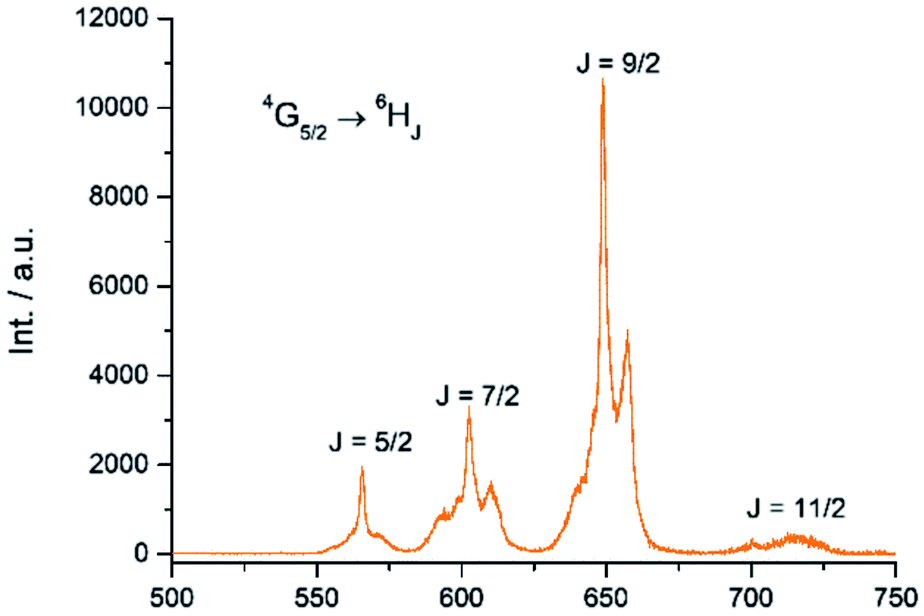
The Sm(III) analog, Sm-Cy-HOPO, was synthesized as chromophores that could sensitize Eu(III) luminescence could often sensitize Sm(III) luminescence as well (vide infra). As seen in Fig. 6, certain 4G5/2 → 6HJ transitions (J = 5/2–11/2) in the visible region could be observed by exciting the ligand at 350 nm. The luminescence lifetime of the most intense 4G5/2 → 6H9/2 transition was measured to be 16 μs in water and the q value was determined to be 0 and 0.5 by Kimura's38 and Hakala's39 equation respectively (Table 2). While it is impractical for half a water molecule to be coordinated, this value reflects that the Sm(III) is not as well secluded from water molecules by the macrocyclic ligand as the Eu(III) counterpart due to the Sm(III)'s slightly larger ionic radius (Fig. S41†) and this is also supported by a larger m value. Furthermore, as the energy gap between the emitting state and the next lower energy level of Sm(III) is quite small (vide infra), Sm(III) complexes suffers an intrinsic disadvantage of having low luminescent quantum yields and thus the 0.4% determined for Sm-Cy-HOPO is not surprising. The 5D0 → 7FJ transitions (J = 0–4) and some of the 5D1 → 7FJ (J = 0–3) could be clearly observed when Eu-Cy-TTA was excited at 350 nm in aqueous solution (Fig. 8). Like Eu-Cy-HOPO, the 5D0 → 7F2 transition is more intense than other transitions, yet the coordination environment is expected to be slightly different since the asymmetry ratio is 11.5 and the splitting of the hypersensitive transition is not the same.40 The luminescence lifetime measured in aqueous solution (3% DMSO) was best-fitted with a bi-exponential decay (0.968 and 0.377 ms), indicating the presence of two radiatively decaying species. The shorter-lived species is believed to be due to coordinated water molecules, however, the presence of DMSO renders the calculation of q value inaccurate. Alternatively, the m value41,42 – representing the number of coordinated methanol molecules – was determined to be 2. It is also worth mentioning that Eu-Cy-TTA exhibited a mono-exponential decay in methanol with a much shorter lifetime (0.463 ms), suggesting the complex is more vulnerable to methanol coordination than water molecules. Nonetheless, in aqueous solution, a considerably decent overall quantum yield of 21.7% was recorded for Eu-Cy-TTA despite the co-existence of the hydrated species as indicated by the bi-exponential lifetime. Such an interesting observation could be explained by our proposed cage-like structure – supported by Sparkle optimization – such arrangement of chromophore creates much higher steric hindrance among the four TTA molecules around the Eu(III) compared to the smaller 1,2-HOPO units, leading to a less tight structure than Eu-Cy-HOPO, thus allowing space for the infiltration of solvent molecules. An optimized structure was obtained from LUMPAC with RM1 model (Fig. 7).
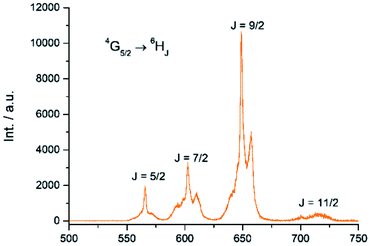 |
| | Fig. 6 Emission spectrum of Sm-Cy-HOPO in water (λex = 350 nm). | |
Table 2 Photophysical data of Ln-Cy-HOPO and Eu-Cy-TTA in water (pH 5.5) and methanola
|
|
Eu-Cy-HOPO
|
Sm-Cy-HOPO
|
Eu-Cy-TTA
|
|
Estimated error in τ and Φ are 10% and 15% respectively.
|
|
ε (M−1 cm−1) |
12110@337 nm |
15360@337 nm |
22690@336 nm |
|
τ
H2O (ms) |
0.784 |
0.018 |
0.968, 0.377 |
|
τ
D2O (ms) |
1.06 |
0.113 |
— |
|
q
|
0 |
0.5 |
— |
|
Φ
LnL
|
30.2% |
0.4% |
21.7% |
|
τ
MeOH (ms) |
0.825 |
0.025 |
0.463 |
|
τ
MeOD (ms) |
0.962 |
0.104 |
0.876 |
|
m
41,42
|
0 |
1.5 |
2 |
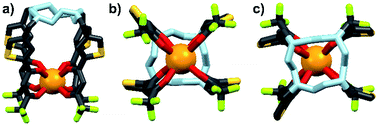 |
| | Fig. 7 Optimized structure of Eu-Cy-TTA. View from side (a); view from above cyclen backbone (b); view from below Eu(III) center (c). | |
 |
| | Fig. 8 Emission spectrum of Eu-Cy-TTA in 3% DMSO aqueous solution (λex = 350 nm). | |
The sensitization efficiency was calculated by eqn (1)–(3) (Table 3). The intrinsic quantum yield (ΦLnLn) for Eu-Cy-HOPO was calculated to be 46.8%, in agreement with literature values whereas that of Eu-Cy-TTA is higher at 52.9%. Nonetheless, the overall quantum yield of the latter is indeed lower per the above data, resulting in a lower sensitization efficiency (ηsens). The intrinsic quantum yield is defined as the quantum yield obtained from direction 4f–4f excitation whereas the overall quantum yield takes the sensitization process into account. In other words, ηsens is a parameter to evaluate the extent of excited energy lost prior to reaching the Eu(III)'s accepting state; quenching of the Eu(III)'s excited state, such as by overtones of O–H oscillators, is irrelevant to the sensitization. The lower ηsens of Eu-Cy-TTA is attributed to the steric demand of the larger TTA molecule, resulting in a longer Eu(III)-chromophore distance (6.1 Å of Eu-Cy-HOPOvs. 7.1 Å of Eu-Cy-TTA as measured from their optimized structures) and less efficient energy transfer via distance-dependent energy transfer mechanisms, as indicated by the slower rate of radiative deactivation (τrad).
Table 3 Calculated Eu(III) parameters of Eu-Cy-HOPO and Eu-Cy-TTA in water and aqueous solution respectively
|
|
Eu-Cy-HOPO
|
Eu-Cy-TTA
|
|
Φ
LnL
|
30.2% |
21.7% |
|
τ
rad (ms) |
1.675 |
1.830 |
|
k
rad (s−1) |
597 |
546 |
|
k
nr (s−1) |
678 |
487 |
|
Φ
LnLn
|
46.8% |
52.9% |
|
η
sens
|
64.6% |
41.1% |
Energy transfer pathway.
To study the antenna effect, the Gd(III) counterpart was synthesized to probe the triplet state of the chelated chromophore. Since the 4f electrons are very well-shielded, the ionic radii of Gd(III) and Eu(III) are very similar and hence it is commonly accepted that the coordination environments are comparable. Furthermore, the excited states of Gd(III) are situated beyond 30000 cm−1,43 so energy transfer is often impractical. At low temperature (77 K), reverse intersystem crossing is hindered, and the excited energy would have a higher tendency to relax from the triplet excited state to give phosphoresce. At room temperature, the emission spectrum of Gd-Cy-HOPO showed very weak ligand fluorescence with two emission maxima at 408 and 434 nm. After cooling Gd-Cy-HOPO at 77 K, a new, broad emission band with peak maximum at 503 nm was observed, and the emission band is assigned as ligand phosphorescence since the emission lifetime was determined to be 6.87 ms. For Gd-Cy-TTA, there was negligible emission at two band maxima at 510 nm and 534 nm appeared with biexponential lifetimes of 1.4 ms and 6.4 μs and 1.2 ms and 7.4 μs respectively. The triplet excited state of 1,2-HOPO is determined to be at ca. 19900 cm−1 and that of TTA, taken as average of the two peak maxima, is ca. 19200 cm−1 (Fig. S4 and S6†). The 5D0 accepting state of Eu(III) is at ca. 17200 cm−1,44 and the energy gap between the respective triplet states and the accepting state(s) falls within the ideal 2500 to 4000 cm−1 range for efficient energy transfer while preventing thermally-promoted back energy transfer.45 The higher excited state 5D1, at ca. 19200 cm−1, despite its proximity with the triplet energy levels, is also involved as shown in Fig. 4 and 9. Furthermore, the exceptionally long phosphorescence lifetime – implying a low non-radiative deactivation rate of the excited triplet state – provides a stable and long-lived excited state for energy transfer to take place, resulting in efficient luminescence sensitization. On the other hand, the 4G5/2 accepting state of Sm(III) is located at ca. 17860 cm−1,46 and is therefore expected to be the recipient of the excited energies from HOPO and TTA. While the energy gap between the emitting state and the next lower energy state of Sm(III) (ΔE(4G5/2→6F11/2) = ca. 7500 cm−1) does not resonate with oscillator overtones, a closer examination of the next energy level (ΔE(4G5/2→6F9/2) = ca. 8700 cm−1) reveals a close match between the second C–H overtone (ca. 8700 cm−1), leading to efficient non-radiative deactivation (Fig. 10). This result is consistent with Doffek et al.'s finding regarding how the smallest energy gap is not ‘universally relevant’, especially in Sm(III) contexts.47 Consequently, luminescent quantum yields of organo-Sm(III) complexes are generally expected to be low due to the abundant C–H oscillators in proximity, and this also explains why only the sensitization barrier of Eu(III) luminescence could be broken by a change to the cyclen backbone but not Sm(III)'s, with the quantum yield of Sm-Cy-HOPO (0.4%) the same as those reported by Raymond's group.
 |
| | Fig. 9 Magnification of partial emission spectrum of Eu-Cy-TTA in Fig. 8. | |
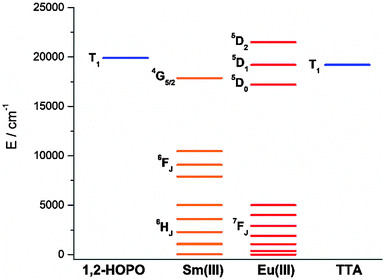 |
| | Fig. 10 Energy level diagram depicting the energy levels of Eu(III), Sm(III) and the triplet states of 1,2-HOPO and TTA determined from Gd-Cy-HOPO and Gd-Cy-TTA. | |
Two-photon absorption and excitation.
Under two-photon excitation at 700 nm with an ultrafast Ti:Sapphire laser, Eu(III) luminescence spectra were recorded for Eu-Cy-HOPO and Eu-Cy-TTA in DMSO (Fig. 11). The emission profile is typical of Eu(III) and the intense 5D0 → 7F2 transitions relative to the 5D0 → 7F1 transition resemble those in Fig. 3 and 8, suggesting the same emitting species compared to single-photon excitation. The two-photon excitation mode was confirmed by the dependence of luminescence intensity on incident power (Fig. S35 and S37†). The two-photon absorption cross sections (σ2) were determined against fluorescein chromophores are not structurally constructed with a large quadrupole moment upon photo-excitation, i.e. the expected to play any role in the two-photon absorption process as it is spatially distant from and has minimal influence on the electronic environment of both the chromophore and the Eu(III). Nevertheless, the unprecedented observation of Eu(III) luminescence via two-photon excitation of a 1,2-HOPO-based chromophore is an encouraging result to develop Eu-Cy-HOPO for two-photon optical microscopy given its excellent water-solubility.
 |
| | Fig. 11 Emission spectra of Eu-Cy-HOPO and Eu-Cy-TTA under two-photon excitation in DMSO (λex = 700 nm). | |
MTT assay and in vitro imaging.
Eu-Cy-HOPO exhibited low cytotoxicity as its IC50 value was determined to be 600 μM by MTT assay in HeLa cells (Fig. S39†), and its cellular uptake behavior was evaluated by fluorescent microscopy and multiphoton confocal microscopy in HeLa cells, too. Fig. 12 shows the uptake of Eu-Cy-HOPO by HeLa cells after 3 hours of incubation as indicated by the red luminescence under 380 nm excitation. Multiphoton excitation at 760 nm by a femtosecond pulsed laser under a confocal microscope also gave red luminescence, and the localization of Eu-Cy-HOPO in the lysosomes was confirmed by co-staining with LysoTracker® (Fig. 13). Due to the much lower luminescent quantum yield of Sm-Cy-HOPO, a higher incubation concentration (40 μM) and longer incubation time (24 hours) was required for orange-red luminescence to be observed via multiphoton excitation at 780 nm (Fig. S40†). On the other hand, precipitation was observed for Eu-Cy-TTA in aqueous solutions at concentrations used for in vitro studies, therefore no studies were performed.
 |
| | Fig. 12 Bright field (left) and fluorescent microscopy image (middle) and overlaid image of Eu-Cy-HOPO (2 μM) in HeLa cells after 3 hours of incubation (λex = 380 nm, BP filter 550–650 nm). | |
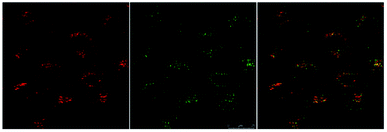 |
| | Fig. 13 Multi-photon confocal microscopy images of Eu-Cy-HOPO (4 μM) after 3 hours of incubation (λex = 760 nm) (left), LysoTracker® (λex = 488 nm) (middle) and overlaid image (right). | |
Conclusions
In this work, a 1,2-HOPO derivative was incorporated into a rigid cyclen backbone and the overall quantum yield of the resulting Eu(III) complex Eu-Cy-HOPO was determined to be 30.2%, with a sensitization efficiency of 64.6%, both the highest thus far amongst 1,2-HOPO-based Eu(III) complexes. The rigidity of the backbone restricts the movement of the pendant chromophores to a higher extent than the linear and branched backbones reported in literature, hence leading to less non-radiative energy loss and a closer Eu(III)-chromophore distance for more efficient energy transfer. A TTA analog, Eu-Cy-TTA also gave decent luminescent properties as a result, but the steric hindrance among the TTA units allowed space for solvent molecules to exploit and penetrate the inner coordination sphere of the Eu(III). Eu(III) luminescence was also unprecedentedly observed under two-photon excitation of the 1,2-HOPO-based chromophore by a femtosecond laser (and in two-photon confocal microscopy), displaying emission profile and lifetimes near-identical to the single-photon excitation process. In addition, co-staining experiments with LysoTracker® confirmed the localization of Eu-Cy-HOPO in lysosomes in vitro.
Experimental
Materials and methods
Unless noted otherwise, all chemicals were of reagent grade and were purchased from Sigma-Aldrich or Acros Organics and used without further purification. Moisture-sensitive synthetic procedures were performed under a nitrogen atmosphere using standard Schlenk techniques. Davisil silica gel (40–63 μm) was obtained from Grace Davison. Analytical reagent grade solvents were used, and acetonitrile was dried with sodium hydride and distilled under nitrogen. 1H, 13C and 19F NMR spectra were recorded on a Bruker Ultrashield 400 Plus NMR spectrometer (at 400 MHz, 100 MHz and 376 MHz respectively) or a Bruker Ultrashield 600 Plus NMR spectrometer (at 600 MHz and 150 MHz respectively). The 1H and 13C NMR chemical shifts were referenced to solvent residual peaks. Mass spectra, reported as m/z, was obtained either on a Micromass Q-TOF 2 mass spectrometer or on an Agilent Technologies 6540 UHD Accurate-Mass Q-TOF LC/MS system or on a Bruker UltrafleXtreme Matrix Assisted Laser Ionization (MALDI) Mass Spectrometer. Analytical high performance liquid chromatography (HPLC) was performed on Waters 1525 series apparatus with PDA detector. The method used on this system is as follows: Atlantis T3 column (4.6 × 250 mm), mobile phase of water (with 0.05% TFA) with 10% of ACN was increased to 100% ACN within 15 min, then maintained at 100% ACN for 5 min and re-equilibrated for 5 min. Reverse-phase semi-preparative purification was performed on Waters 2535 series apparatus with PDA detection and Fraction Collector III. The method used on this system is as follows: Atlantis T3 column (19 × 250 mm), mobile phase of water (with 0.05% TFA) with 30% methanol was gradient increased to 100% methanol within 20 min, then the system was re-equilibrated for 4 min. Inductively coupled plasma - optical emission spectrometry (ICP-OES) was performed on an Agilent 700 Series system, with 6 points standards (0.5–20 ppm) of Eu, Sm and Gd in 2% of HNO3 for the determination of metal content. Fourier Transform Infrared (FT-IR) spectra were recorded on a Nicolet iS 50 FT-IR spectrometer with a KBr pellet.
Photophysical measurements
Milli-Q water (18.2 MΩ cm at 25 °C) was used for aqueous measurements; methanol used were of CHROMASOLV®Plus grade from Sigma-Aldrich, deuterated water and methanol used were from Cambridge Isotope; all were used without further purification. Solution samples of ca. 0.1 absorbances at 350 nm were prepared for visible photoluminescence measurements (Eu-Cy-TTA is insoluble in pure water, hence is first dissolved in DMSO and diluted with Milli-Q water.). Measurements were prepared in the unit of absorbance instead of concentration as the absorbances at 350 nm are slightly different for the two complexes. Separate samples were used for (1) UV-vis, emission and excitation scans; (2) luminescence lifetime measurements and (3) quantum yield measurements.
All room temperature solution measurements were done in a quartz cuvette (Starna) of 1 cm path length. UV-vis spectra were recorded with an HP UV-8453 spectrophotometer. Room temperature photoluminescence measurements data obtained with Edinburgh Instruments FLSP920 spectrophotometer equipped with a Xe900 continuous xenon lamp (450 W), xenon flashlamp (60 W) and a Hamamatsu R928P thermoelectrically cooled at −20 °C. Low temperature (77 K) measurements were measured on FLSP920 using an EPR dewar from Edinburgh Instruments. Samples were dissolved in ethanol–methanol mixture (v/v = 4:1), inserted into an EPR quartz sample rod and cooled with liquid nitrogen. Emission spectra were recorded at 30 min intervals until the intensity and emission profiles remained constant (∼2 hours) and the spectra were taken as final. Visible emission spectra obtained were corrected for spectral responses.
Luminescence lifetimes of visible emissions were measured with FLSP290 and fitted with Origin. Luminescence quantum yields were measured relative to quinine sulfate in 0.1 M sulfuric acid (λex = 350 nm, Φ = 0.577). All photophysical measurements were averages of triplicate.
The intrinsic quantum yield of the complex was also calculated using the below equations to gain more insight into the sensitization processes:48
| | 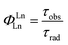 | (2) |
| | 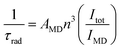 | (3) |
| | 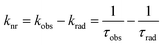 | (4) |
The overall quantum yield (ΦLnL) is the product of intrinsic quantum yield (ΦLnLn) and sensitization efficiency (ηsens). The reciprocal of the radiative lifetime (1/τrad) could be calculated by eqn (3), where AMD denotes the spontaneous emission probability of the magnetic dipole transition (5D0 → 7F0 for Eu(III)) which is a constant equal to 14.65 s−1, n is the refractive index of the medium and Itot and IMD are the integrated intensities of the total 5D0 → 7FJ transitions and the magnetic dipole transition respectively. The rate of non-radiative decay could be determined by eqn (4).
Determination of two-photon absorption cross section
For two-photon experiments, the 700 nm pump source was from an optical parametric amplifier (TOPAS-C) of a femtosecond mode-locked Ti:Sapphire laser system (Coherent Micra and Legend-Elite output beam ∼100 fs duration and 100 Hz repetition rate). The laser was focused to spot size ∼100 μm via an f = 30 cm lens onto the sample. The emitting light was collected with a right angle configuration into a 0.3 m spectrograph and detected by a liquid nitrogen-cooled CCD detector. A power meter was used to monitor the stability of the pump source and its intensity was controlled by using a variable ND filter. For two photon absorption cross-section measurements, the theoretical framework and experimental protocol for the two-photon cross-section measurement have been outlined by Webb and Xu.49 In this approach, the two-photon excitation ratios of the reference and sample systems are given by:| | 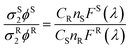 | (5) |
where ϕ is the quantum yield, C is the concentration, n the refractive index, and F(λ) is the integrated photoluminescent spectrum. In our measurements, we have ensured that the excitation flux and the excitation wavelengths are the same for both the sample and the reference. The two-photon absorption cross-sections σ2 of compounds were determined using fluorescein as a reference.24 Note the σ2 values are underestimation of the actual values because only the Eu Δ = J bands of 1, 2, 3 were used in the calculations due to the interference of the laser excitation at 700 nm which is in the ΔJ = 4 band of the Eu transition.
Cell imaging studies
For single-photon microscopy, images were obtained by a Carl Zeiss AxioObserver Z1 fluorescent microscope using a UV light source. For multi-photon microscopy, images were collected by a Leica TCS SP8 spectral confocal microscope equipped with a Ti:Sapphire laser. Living HeLa cells were used.
Synthesis
Compound 1.
Cyclen (1 g, 5.8 mmol, 1.0 equiv.) was dissolved in acetonitrile (10 mL) and benzene (10 mL), then tosylaziridine (5.5 g, 27.9 mmol, 4.8 equiv.) was added and the mixture was reacted at 60 °C for 3 days. After filtration and washing with acetonitrile, the tosylated intermediate was obtained as a white solid and used for the next step directly without any further purification (4.0 g, 4.2 mmol, yield: 71%). 1H NMR (400 MHz, DMSO) δ 7.61 (t, J = 10.7 Hz, 8H), 7.34 (d, J = 7.6 Hz, 8H), 2.72 (t, J = 6.5 Hz, 8H), 2.33 (d, J = 7.6 Hz, 12H), 2.32–2.20 (m, 24H). 13C NMR (100 MHz, DMSO) δ 143.00, 138.14, 130.05, 128.79, 54.12, 52.55, 40.80, 21.40. m/z (ESI-MS+) 961.3864 ([M + H]+ calculated: 961.3809). Acetic acid (7 mL) and hydrobromic acid (5 mL) were added to dissolve the intermediate (1.0 g, 1.0 mmol, 1.0 equiv.) and the mixture was reacted at 100 °C for 3 days. After cooling down to room temperature, the reaction mixture was filtered and washed with acetic acid and the filter cake was dried at 55 °C in oven to obtain a white solid (300 mg, 0.3 mmol, yield 29%).
Compound 3.
Compound 1 (114 mg, 0.11 mmol, 1.0 equiv.) and 2 (242 mg, 0.92 mmol, 8.0 equiv., synthesized according to literature10) were dissolved in dry THF (10 mL) and NMM (N-methylmorpholine) (184 mg, 1.8 mmol, 16.0 equiv.) was added at room temperature. HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate) (350 mg, 0.92 mmol, 8.4 equiv.) was added at 4 °C and the reaction mixture was allowed to react at room temperature overnight before quenching with water (50 mL). The organic solvent was evaporated and the aqueous layer was discarded. The residual oil adhered to the wall of flask was washed with water (30 mL) again and purified by preparative HPLC to obtain the product (80 mg, 0.064 mmol, yield: 55.5%). 1H NMR (400 MHz, CD3OD) δ 7.88–7.04 (m, 24H), 6.72 (d, J = 9.0 Hz, 4H), 6.41 (d, J = 6.5 Hz, 1H), 5.27 (s, 8H), 3.54 (s, 8H), 3.21–2.78 (m, 24H). 13C NMR (100 MHz, CD3OD) δ 161.79, 159.16, 142.56, 139.16, 133.63, 129.93, 129.30, 128.51, 123.15, 106.17, 79.00, 52.10, 48.6, 34.30. m/z (ESI-MS+) 1253 ([M + H]+ calculated: 1253).
Compound 4.
Compound 3 (80 mg, 0.064 mmol, 1.0 equiv.) was dissolved in acetic acid (3 mL) and hydrochloric acid (3 mL) and the reaction mixture was stirred at room temperature for 3 days. The solution was then concentrated and dissolved in methanol (0.5 mL). The product was precipitated by slow addition of diethyl ether and collected by centrifugation in the form of an HCl salt (65 mg, 0.063 mmol, yield: 98%). 1H NMR (400 MHz, D2O) δ 7.44 (t, J = 7.9 Hz, 4H), 6.69 (d, J = 8.9 Hz, 4H), 6.61 (d, J = 6.7 Hz, 4H), 3.63 (s, 8H), 3.30 (s, 16H), 3.20 (s, 8H). 13C NMR (100 MHz, D2O) δ 162.79, 159.85, 139.56, 139.03, 121.14, 108.86, 52.14, 49.00, 35.05. m/z (ESI-MS+) 893.3911 ([M + H]+ calculated: 893.3906).
Ln-Cy-HOPO
.
Compound 4 (15 mg, 0.014 mmol, 1.0 equiv.) was dissolved in methanol (1 mL) and a methanol (1 mL) solution of LnCl3·6H2O (0.015 mmol, 1.05 equiv.) was added. The pH of the solution was adjusted to 7.5 by pyridine. The solution was stirred at 55 °C for 12 hours. The product was precipitated by slow addition of diethyl ether (5 mL), and the solids were collected by centrifugation, washed twice with diethyl ether (5 mL x 2) and dried as white solids (∼80% yields). Eu-Cy-HOPO (12.5 mg, yield: 83%): m/z (ESI-MS+) 1043.2893 ([M + 2H]+ calculated: 1043.2883); calcd Eu content 14.6% (C40H49EuN12O12), found 13.9%. Sm-Cy-HOPO (12.9 mg, yield: 86%): m/z (ESI-MS+) 1042.2863 ([M+2H]+ calculated: 1042.2865); calcd Sm content 14.5% (C40H49SmN12O12), found 13.3%. Gd-Cy-HOPO (11.8 mg, yield: 78%): m/z (ESI-MS+) 1048.2914 ([M + 2H]+ calculated: 1048.2916); calcd Gd content 15.0% (C40H49GdN12O12), found 14.3%.
Compound 5.
Cyclen (202 mg, 1.16 mmol 1.0 equiv.) was dissolved in dried acetonitrile (4 mL). Potassium carbonate (660 mg, 4.78 mmol, 4.1 equiv.) and 2-acetyl-4-chloromethyl-thiophene (806 mg, 4.62 mmol, 4.0 equiv.) were added into the solution and the reaction mixture was stirred at room temperature for 24 hours, before increasing the temperature to 40 °C and allowed to react for another 2 days. The reaction mixture was then cooled, filtered and dried under vacuum. The product was purified by column chromatography on silica gel (CHCl3:EtOH, 100:1–10:1) to obtain compound 5 as a light yellow oil (550 mg, 0.76 mmol) with a 65% yield. 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 16.5 Hz, 8H), 3.42 (s, 8H), 2.63 (s, 16H), 2.42 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 190.65, 144.22, 141.91, 133.45, 130.96, 54.63, 52.93, 26.80. m/z (ESI-MS+) 725.2363 ([M + H]+ calculated: 725.2324).
Compound 6.
Compound 5 (51.6 mg, 0.07 mmol, 1.0 equiv.) was dissolved in dried THF (4 mL) and cooled to −78 °C. Potassium bis(trimethylsilyl)amide (KHMDS) (1.0 M in THF, 0.43 mL, 0.42 mmol, 6.0 equiv.) was dropped slowly into the reaction mixture and stirred for 20 minutes. Ethyl trifluoroacetate (60.7 mg, 0.43 mmol, 6.1 equiv.) was then added and the reaction was continually stirred at −78 °C for 2 more hours before stirring overnight at room temperature. Petroleum ether (5 mL) was added to the solution and stirred for 20 minutes to give precipitates, which were collected by centrifugation and further washed with dichloromethane twice (13 mL × 2). The product was obtained as an orange solid after drying under vacuum (60 mg, 0.05 mmol, yield: 67%). 1H NMR (400 MHz, DMSO) δ 7.85–7.35 (m, 8H), 6.09–5.73 (m, 4H), 3.93–3.3 (s, 4H), 3.58–3.42 (m, 4H), 2.65–2.61 (m, 8H), 2.48–2.42 (m, 8H). 19F NMR (376 MHz, DMSO) δ −73.49 (6F), −7.72 (6F). m/z (ESI-MS+) 1109.1645 ([M − 4K + 5H]+ calculated: 1109.1620).
Ln-Cy-TTA
.
Compound 6 (12 mg, 0.0095 mmol, 1.0 equiv.) was dissolved in methanol (1 mL) and a methanol solution (1 mL) of LnCl3·6H2O (0.01 mmol, 1.1 equiv.) was added. The reaction mixture was stirred for 16 hours at 50 °C. The product was obtained by precipitating with water (6 mL), centrifuged and washed with water twice more (6 mL × 2). A dried yellow/orange solid was obtained as the final complex (∼80% yields) with a counter ion of potassium. Eu-Cy-TTA (10 mg, yield: 81%): m/z (ESI-MS+) 1259.0579 ([M − K + 2H]+ calculated: 1259.0585); calcd Eu content 11.7% (C44H36F12EuKN4O8S4), found 12.1%. Gd-Cy-TTA (10 mg, yield: 81%): m/z (ESI-MS+) 1264.0605 ([M − K + 2H]+ calculated: 1264.0619); calcd Gd content 12.1% ((C44H36F12GdKN4O8S4), found 12.5%.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Mr Ho-Yin Wong for his help in assisting with some of the synthesis and also for rendering the graphics and designing the graphical artwork. We also thank Dr Pui-Kin So for his help in the mass spectrometry measurements. W.-T. W. gratefully acknowledges financial support from the Basic Research Program of Shenzhen (JCYJ20160531184120814). G.-L. L. gratefully acknowledges financial support from Natural Science Foundation of China (NSFC, 21875201) and financial support from Research Grants Council of Hong Kong (PolyU 153013/17P). W.-M. K. gratefully acknowledges financial support from Research Grants Council of Hong Kong (PolyU/153020/17P). All authors also gratefully acknowledge the State Key Laboratory of Chemical Biology and Drug Discovery, The Hong Kong Polytechnic University ((a) University Research Facility in Chemical and Environmental Analysis (UCEA); (b) University Research Facility in Life Sciences (ULS)).
Notes and references
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- E. G. Moore, A. P. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939 RSC.
- T. Forster, Naturwissenschaften, 1946, 33, 166–175 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- C. Tedeschi, J. Azéma, H. Gornitzka, P. Tisnès and C. Picard, Dalton Trans., 2003, 1738–1745, 10.1039/b300159h.
- E. G. Moore, J. Xu, C. J. Jocher, E. J. Werner and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 10648–10649 CrossRef CAS PubMed.
- E. G. Moore, C. J. Jocher, J. Xu, E. J. Werner and K. N. Raymond, Inorg. Chem., 2007, 46, 5468–5470 CrossRef CAS PubMed.
- R. J. Abergel, A. D'Aleo, C. N. Leung, D. K. Shuh and K. N. Raymond, Inorg. Chem., 2009, 48, 10868–10870 CrossRef CAS PubMed.
- L. J. Daumann, D. S. Tatum, B. E. Snyder, C. Ni, G. L. Law, E. I. Solomon and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 2816–2819 CrossRef CAS PubMed.
- A. D'Aleo, E. G. Moore, J. Xu, L. J. Daumann and K. N. Raymond, Inorg. Chem., 2015, 54, 6807–6820 CrossRef PubMed.
- E. G. Moore, J. Xu, C. J. Jocher, I. Castro-Rodriguez and K. N. Raymond, Inorg. Chem., 2008, 47, 3105–3118 CrossRef CAS PubMed.
- M. Seitz, E. G. Moore, A. J. Ingram, G. Muller and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 15468–15470 CrossRef CAS PubMed.
- A. D'Aleo, J. Xu, E. G. Moore, C. J. Jocher and K. N. Raymond, Inorg. Chem., 2008, 47, 6109–6111 CrossRef PubMed.
- M. Sturzbecher-Hoehne, C. N. Leung, A. D'Aleo, B. Kullgren, A. L. Prigent, D. K. Shuh, K. N. Raymond and R. J. Abergel, Dalton Trans., 2011, 40, 8340–8346 RSC.
- L. J. Daumann, D. S. Tatum, C. M. Andolina, J. I. Pacold, A. D'Aleo, G. L. Law, J. Xu and K. N. Raymond, Inorg. Chem., 2016, 55, 114–124 CrossRef CAS PubMed.
- D. Parker and J. A. Williams, J. Chem. Soc., Dalton Trans., 1996, 18, 3613–3628 RSC.
- R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS.
- L. Dai, W.-S. Lo, J. Zhang and G.-L. Law, Asian J. Org. Chem., 2017, 6, 1845–1850 CrossRef CAS.
- N. Chatterton, Y. Bretonniere, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2005, 44, 7595–7598 CrossRef CAS PubMed.
- W. S. Lo, W. M. Kwok, G. L. Law, C. T. Yeung, C. T. Chan, H. L. Yeung, H. K. Kong, C. H. Chen, M. B. Murphy, K. L. Wong and W. T. Wong, Inorg. Chem., 2011, 50, 5309–5311 CrossRef CAS PubMed.
- C. Wu, W. Zipfel, J. B. Shear, R. M. Williams and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10763–10768 CrossRef.
- F. Kielar, A. Congreve, G. L. Law, E. J. New, D. Parker, K. L. Wong, P. Castreno and J. de Mendoza, Chem. Commun., 2008, 2435–2437, 10.1039/b803864c.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS PubMed.
- M. Albota, D. Beljonne, J. L. Bredas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Rockel, M. Rumi, G. Subramaniam, W. W. Webb, X. L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- S. J. Chung, M. Rumi, V. Alain, S. Barlow, J. W. Perry and S. R. Marder, J. Am. Chem. Soc., 2005, 127, 10844–10845 CrossRef CAS PubMed.
- A. M. Marin, J. P. Telo, D. Collado, F. Najera, E. Perez-Inestrosa and U. Pischel, Chemistry, 2018, 24, 2929–2935 CrossRef CAS PubMed.
- O. Mongin, L. Porres, M. Charlot, C. Katan and M. Blanchard-Desce, Chemistry, 2007, 13, 1481–1498 CrossRef CAS PubMed.
- B. Sadowski, H. Kita, M. Grzybowski, K. Kamada and D. T. Gryko, J. Org. Chem., 2017, 82, 7254–7264 CrossRef CAS PubMed.
- G. F. White, K. L. Litvinenko, S. R. Meech, D. L. Andrews and A. J. Thomson, Photochem. Photobiol. Sci., 2004, 3, 47–55 RSC.
- A. D'Aleo, A. Picot, P. L. Baldeck, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10269–10279 CrossRef PubMed.
- P. A. Tanner, Chem. Soc. Rev., 2013, 42, 5090–5101 RSC.
- R. O. Freire, G. B. Rocha and A. M. Simas, J. Braz. Chem. Soc., 2009, 20, 1638–1645 CrossRef CAS.
- J. D. Dutra, T. D. Bispo and R. O. Freire, J. Comput. Chem., 2014, 35, 772–775 CrossRef CAS PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504, 10.1039/a808692c.
- R. M. Supkowski and W. D. Horrocks, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- W. S. Lo, J. Zhang, W. T. Wong and G. L. Law, Inorg. Chem., 2015, 54, 3725–3727 CrossRef CAS PubMed.
- T. Kimura and Y. Kato, J. Alloys Compd., 1995, 225, 284–287 CrossRef CAS.
- H. Hakala, P. Liitti, J. Peuralahti, J. Karvinen, V.-M. Mukkala and J. Hovinen, J. Lumin., 2005, 113, 17–26 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- R. C. Holz, C. A. Chang and W. D. Horrocks, Inorg. Chem., 1991, 30, 3270–3275 CrossRef CAS.
- A. P. Bassett, S. W. Magennis, P. B. Glover, D. J. Lewis, N. Spencer, S. Parsons, R. M. Williams, L. De Cola and Z. Pikramenou, J. Am. Chem. Soc., 2004, 126, 9413–9424 CrossRef CAS PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4443–4446 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4450–4455 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef CAS.
- C. Doffek, J. Wahsner, E. Kreidt and M. Seitz, Inorg. Chem., 2014, 53, 3263–3265 CrossRef CAS PubMed.
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346–1353 RSC.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00244h |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Yanjuan
Gu
ab,
Yanjuan
Gu