
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular genetic design of multi-domain functional amyloids: insights into self-assembly and functional properties†
Mengkui
Cui‡
abc,
Qi
Qi‡
a,
Thomas
Gurry‡
 d,
Tianxin
Zhao
abc,
Bolin
An
a,
Jiahua
Pu
a,
Xinrui
Gui
ce,
Allen A.
Cheng
f,
Siyu
Zhang
a,
Dongmin
Xun
a,
Michele
Becce
fgh,
Francesco
Briatico-Vangosa
g,
Cong
Liu
e,
Timothy K.
Lu
f and
Chao
Zhong
*a
d,
Tianxin
Zhao
abc,
Bolin
An
a,
Jiahua
Pu
a,
Xinrui
Gui
ce,
Allen A.
Cheng
f,
Siyu
Zhang
a,
Dongmin
Xun
a,
Michele
Becce
fgh,
Francesco
Briatico-Vangosa
g,
Cong
Liu
e,
Timothy K.
Lu
f and
Chao
Zhong
*a
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 200120, China. E-mail: zhongchao@shanghaitech.edu.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cShanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
dDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139-4307, USA
eInterdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
fDepartment of Electrical Engineering and Computer Science, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139-4307, USA
gDipartimento di Chimica Materiali e Ingegneria Chimica G. Natta, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy
hDepartment of Materials, Imperial College London, London SW7 2AZ, UK
First published on 15th February 2019
Abstract
Engineering functional amyloids through a modular genetic strategy represents new opportunities for creating multifunctional molecular materials with tailored structures and performance. Despite important advances, how fusion modules affect the self-assembly and functional properties of amyloids remains elusive. Here, using Escherichia coli curli as a model system, we systematically studied the effect of flanking domains on the structures, assembly kinetics and functions of amyloids. The designed amyloids were composed of E. coli biofilm protein CsgA (as amyloidogenic cores) and one or two flanking domains, consisting of chitin-binding domains (CBDs) from Bacillus circulans chitinase, and/or mussel foot proteins (Mfps). Incorporation of fusion domains did not disrupt the typical β-sheet structures, but indeed affected assembly rate, morphology, and stiffness of resultant fibrils. Consequently, the CsgA-fusion fibrils, particularly those containing three domains, were much shorter than the CsgA-only fibrils. Furthermore, the stiffness of the resultant fibrils was heavily affected by the structural feature of fusion domains, with β-sheet-containing domains tending to increase the Young's modulus while random coil domains decreasing the Young's modulus. In addition, fibrils containing CBD domains showed higher chitin-binding activity compared to their CBD-free counterparts. The CBD-CsgA-Mfp3 construct exhibited significantly lower binding activity than Mfp5-CsgA-CBD due to inappropriate folding of the CBD domain in the former construct, in agreement with results based upon molecular dynamics modeling. Our study provides new insights into the assembly and functional properties of designer amyloid proteins with increasing complex domain structures and lays the foundation for the future design of functional amyloid-based structures and molecular materials.
Natural protein molecules are typically composed of several protein domains organized in a modular manner. The sequences and arrangement of domains within single protein molecules and within the protein complexes into which they are subsequently assembled endow natural molecular materials with well-defined and tailored multifunctionalities that are usually absent from chemically synthesized molecules and polymeric materials. For example, the complex domains of extracellular matrix (ECM) proteins provide diverse functionalities in mediating cell and tissue behaviors, ranging from providing structural support for organs and tissues to acting as substrates for cell migration.1–3 In contrast, existing artificial self-assembling ECM proteins, though they form network that morphologically resembles natural ECM fibrous networks, can hardly recapitulate all the functional features of natural ECMs. The engineering of artificial protein-based molecular materials that mimic or even outperform their natural counterparts may become feasible by rationally designing tailored molecular sequences using a bottom-up modular genetic strategy.4–6 Such a research strategy represents new opportunities for engineering multifunctional molecular materials with predictable structures and customizable performance.7
Although originally identified as pathological hallmarks of diverse neurological disorders including Alzheimer's, Parkinson's, and Huntington's diseases,8,9 amyloids of diverse sequences have been recently identified in several organisms for physiological applications.10 One distinctive feature of functional amyloids is that the amyloidogenic core of assembled structures does not significantly diverge from that of the original amyloid backbone structure when domains with appropriate size and conformation are present.11 Such attributes, along with the intrinsic self-assembly and outstanding material properties of amyloids, have prompted an increasing amount of research in producing functional amyloid-based molecular materials based on a modular genetic design for diverse biomaterials and nanotechnologies.10,12–17 However, genetically modified amyloids created through this strategy do not always self-assemble into “normal” structures with predictable functions.6,18 For example, Forman et al. revealed that the conformation and position of proteins displayed on the same amyloidogenic cores significantly changed the morphology of the assembled fibrils.18 The increasing development and application of such self-assembling molecular materials, therefore, call for deeper understanding of how genetically fused domains affect the stability, morphology, assembly kinetics, mechanical and functional properties of structural amyloids. Such knowledge, though at present unfortunately incomplete, will be particularly important for advancing future amyloid-based nanotechnologies and materials.
Here, using E. coli curli as a model system, we investigated how flanking domains affect the morphology, assembly kinetics, stiffness, and functional properties of amyloids by rationally engineering functional amyloid proteins with variable domain structures and arrangement. We constructed functional CsgA-fusion proteins consisting of an amyloid core CsgA (a major amyloid protein associated with E. coli biofilm formation) and one or two functional protein domains, fused at either or both the C or N terminus of CsgA. The functional domains applied in this study include mussel foot proteins from Mytilus galloprovincialis (Mfp3 and Mfp5), which are interfacial adhesive proteins associated with mussel adhesive plaque,19,20 and chitin-binding domain (CBD) from Bacillus circulans chitinase,21 a functional domain that can specifically recognize and bind to chitin (Fig. 1b). In the designs, CsgA served as the backbone domain that mainly drove the self-assembly of constructed molecules, while CBD and/or Mfps acted as flanking domains that contributed to functional properties of assembled structures. The CsgA protein is an ideal amyloid model system for this study, as it has conserved amyloid sequences that are known to tolerate chemical elaboration,22–24 and the resultant CsgA fusion proteins have been demonstrated with emerging functional properties, for example, underwater adhesion.5 Using curli as a model system, our study here aims at promoting better understanding of fibril assembly, mechanical and functional properties of multi-domain functional amyloids, as well as providing new insights into rational design of functional amyloid fibrils.
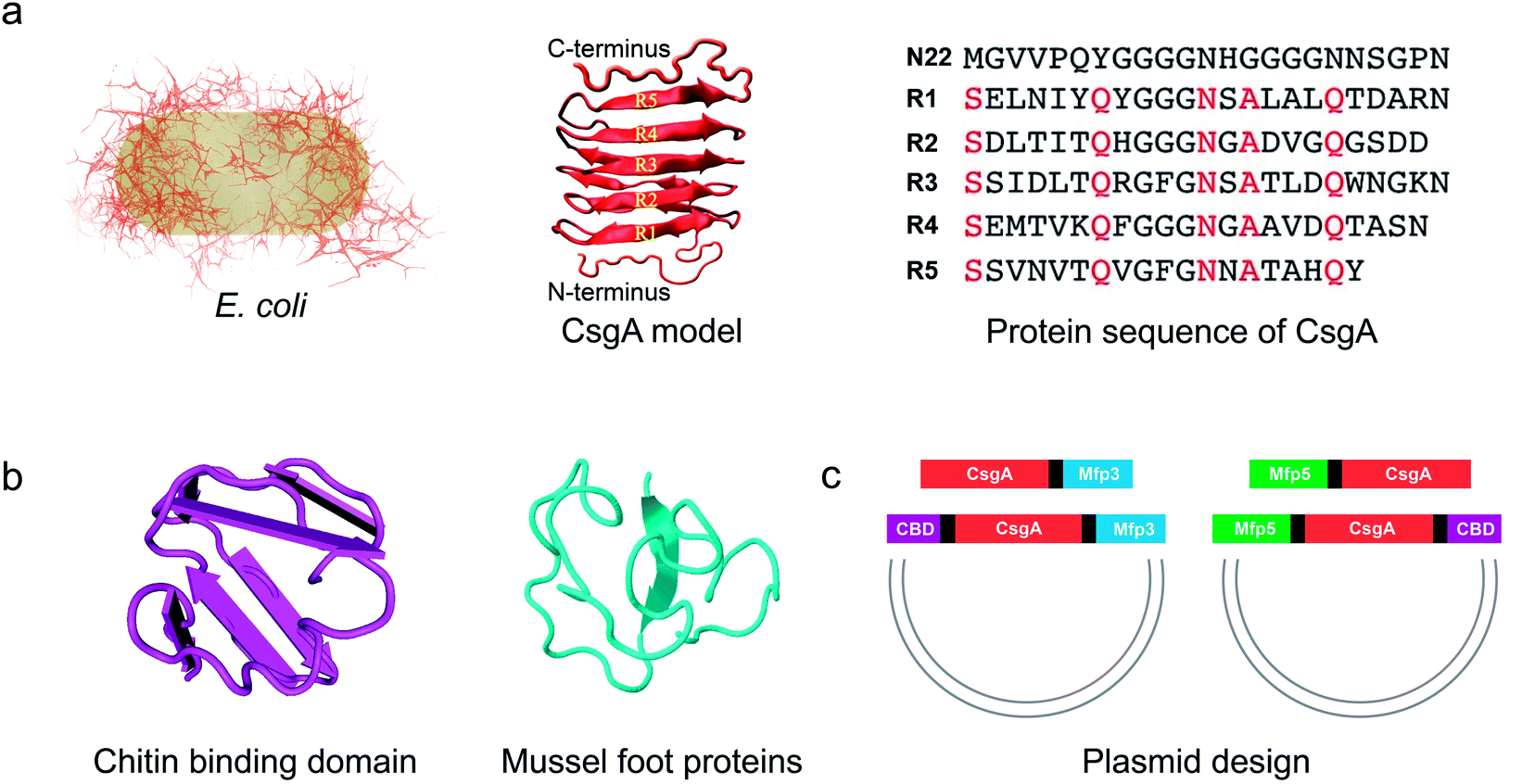 | ||
| Fig. 1 Construction of tunable functional amyloids with variable domain structures through a modular genetic design using E. coli curli as a model amyloid system. (a) Schematic illustration of E. coli curli fibrils, β-helix model of CsgA and amino-acid sequences for CsgA. Curli, adhesive amyloid fibrils of E. coli biofilms, are constituted by the major subunits, CsgA. CsgA contains N-terminal 22 residues that act as a secretion signal through the outer membrane, and five repeating strand-loop-strand motifs mediated by conserved residues (marked in red color), that form the amyloid core.25–27 The glutamine and asparagine residues in each repeating unit are predicted to form a hydrogen bond network that contributes to the extreme stability of the fibrils. (b) Structural illustration of chitin binding domains (CBD), which are C-terminal domains of Bacillus circulans chitinase that can specifically bind to insoluble chitin (PDB code: 1ED7),21 and mussel foot proteins (Mfps), which are interfacial adhesion proteins of mussels.28,29 The CBD is a rigid and compact twisted β-sandwich structure that contains two antiparallel β-sheets,21 and Mfp is an intrinsically disordered protein in solution.30 (c) Two groups of gene constructs (CsgA-Mfp3, CBD-CsgA-Mfp3 and Mfp5-CsgA, CBD-Mfp5-CsgA) were created using one-pot isothermal Gibson assembly and tagged with poly-histidine residues to facilitate purification, respectively. Note: N-terminal or C-terminal fusion of a flanking domain (R) onto CsgA proteins was denoted as R-CsgA or CsgA-R, respectively. In a typical construct (CBD-CsgA-Mfp3), CBD, Mfp3 domain was extended from CsgA's N-terminal and C-terminal, respectively. | ||
We first genetically fused CsgA with either one or two functional domains at the C and/or N terminus, respectively. Four genetic fusion constructs, namely, CsgA-Mfp3, Mfp5-CsgA, CBD-CsgA-Mfp3, and Mfp5-CsgA-CBD were created using isothermal one-step Gibson DNA assembly (Fig. 1c and ESI Fig. 1–6†). The purified proteins were confirmed to be the ones as designed using various biological assays (ESI Fig. 7†).
Atomic force microscopy (AFM) confirmed that all proteins formed self-assembled nanofibrils after aging for 18 hours, yet the morphologies of the fibrils varied among samples (Fig. 2a). Specifically, the fibril diameters slightly increased with increasing number of fusion domains (Fig. 2b). The diameters of the resultant fibrils comprising triple domains were larger than those of their counterparts containing two domains. Thus, it was reasonable to hypothesize that the fusion domains might be external to CsgA amyloid cores in the CsgA-fusion proteins, and would consequently enlarge fibril diameters to a certain extent. Furthermore, the fibril lengths among samples differed significantly (Fig. 2c). While CsgA protein tended to form the longest fibrils with an average fibril length of 3 to 4 μm (3197.2 ± 1169.4 nm), proteins containing two domains tended to form fibrils with intermediate lengths (1281.1 ± 446.1 and 719.9 ± 222.3 nm for CsgA-Mfp3 and Mfp5-CsgA, respectively). In contrast, nanofibrils assembled by proteins containing three different domains had the shortest lengths, with 576.3 ± 266.3 nm and 260.3 ± 125.9 nm for CBD-CsgA-Mfp3 and Mfp5-CsgA-CBD, respectively (Fig. 2b and 4c). These observations thus implied that a steric hindrance effect increased for fibril assembly as the number of domains increased in the fusion proteins. Conceivably, the random contact and interaction of the side domains with the β-sheet cores would disturb or even decrease the hydrogen bonds (thus lower the average hydrogen bonding density) within the amyloid nanofibril core.32 In addition, fibrils containing Mfp3 domains (46 amino acids) were longer than their counterparts containing Mfp5 domains (76 amino acids), both for the two-domain or triple-domain structures, suggesting that larger domains more significantly interfered with fibril assembly. Interestingly, the heights of irregular knots/dots, usually surpassing the average height of nanofibrils, were frequently visualized at fibril ends in both two-domain and triple-domain nanofibrils rather than CsgA fibrils (Fig. 2a, dark arrows). It was likely that such dots formed as a result of “incorrect” assembly among monomers, for example, via non-specific inter/intramolecular interactions between fused domains, thus interfering with CsgA–CsgA amyloid core interactions and thereby delaying or even blocking further growth of the nanofibrils. Notably, for all the CsgA-fusion proteins that we studied, we did not observe a twisted morphology of the resultant fibrils, which was often found associated with certain functional amyloids containing fusion domains, such as SH3-cytochrome b562 fusion proteins18 and a chimeric fusion protein, Rd-HET-s (218–289), containing a prion domain and a redox protein.15
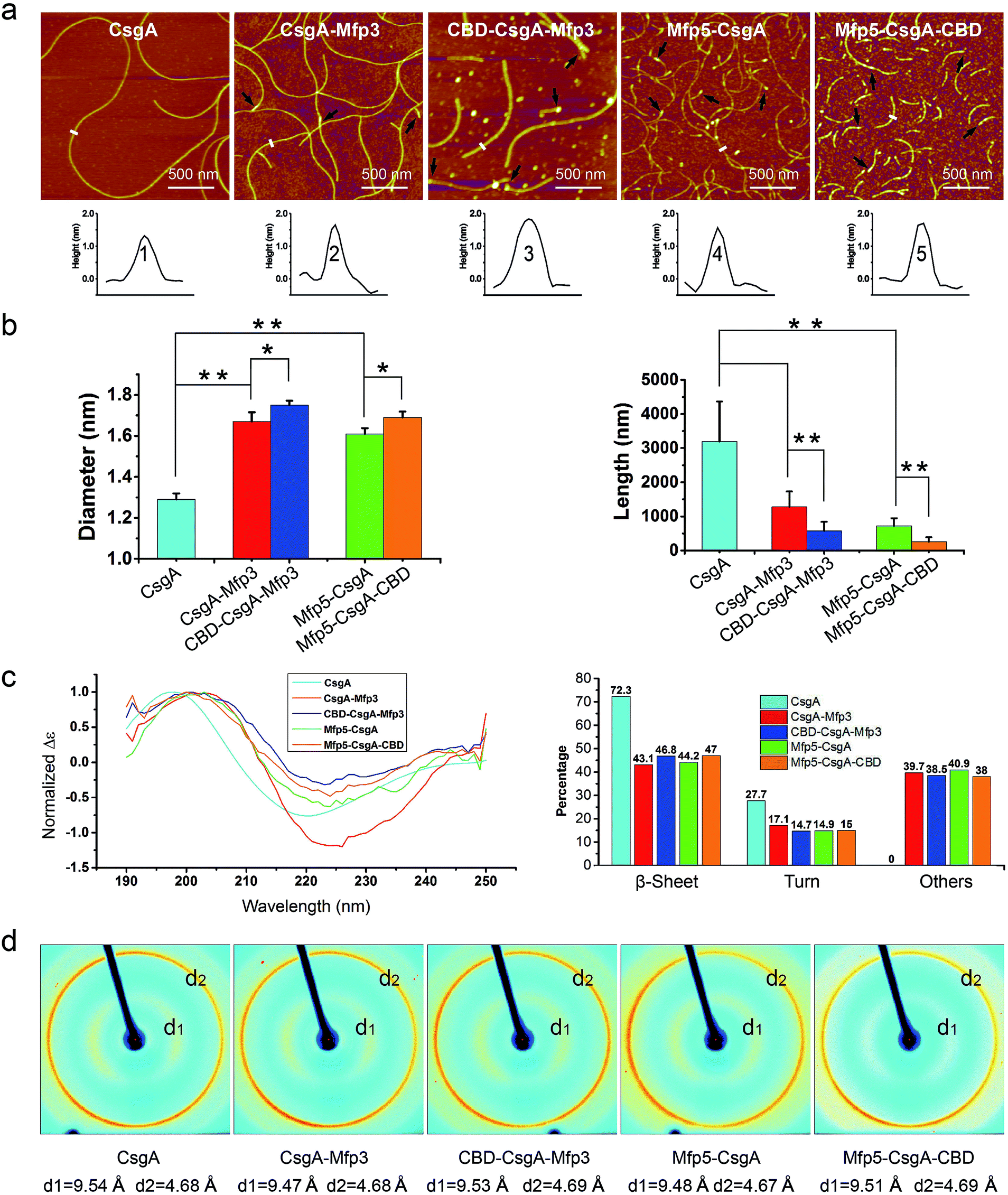 | ||
| Fig. 2 Morphological and structural comparison of CsgA and CsgA-fusion proteins comprising one or two additional domains. (a) AFM height images (top) and corresponding cross-sectional height profiles (bottom) of protein fibrils (after 18 h solution incubation) with increasing number of functional domains. Scale bars are 500 nm. The black arrows referred to the irregular knots/dots frequently forming at the ends of fibrils. The white short lines and numbers marked in the height images correspond to the specific sites of nanofibrils where the cross-sectional height profiles of different nanofibrils were measured (shown below); (b) the fibril diameter (left) and fibril length (right) histograms of functionalized proteins statistically summarized according to the AFM height images in (a). The diameters of the fibrils are: 1.39 ± 0.48 nm for CsgA; 1.66 ± 0.50 nm and 1.61 ± 0.45 nm for CsgA-Mfp3 and Mfp5-CsgA respectively, and 1.75 ± 0.46 nm and 1.67 ± 0.43 nm for CBD-CsgA-Mfp3 and Mfp5-CsgA-CBD respectively (the lengths are summarized in Fig. 4c). The statistical analysis for each sample was based upon 40 single fibrils. The error bars represent standard deviations (s.d.), *P < 0.05, **P < 0.01, student's t-test. (c) Circular Dichroism (CD) spectra of protein solutions containing different fibrils (left) and portions of secondary structures for each of the fibril solutions calculated by a previous method (right).31 (d) X-ray fibril diffraction of fibrils. The figure exhibits typical diffraction pattern of amyloid fibrils in which the meridional reflection (d2) is at ∼4.68 Å and the equatorial reflection (d1) is ∼9.47–9.54 Å. | ||
To study whether the size of a third domain would influence the morphology of triple-domain proteins, we constructed Spytag-CsgA-CBD by replacing the Mfp5 domain with a much smaller peptide, Spytag (13 amino acids) at the N-terminus of CsgA-CBD (ESI Fig. 10†). The fibril length, diameter, and thioflavin T (ThT) fibril formation kinetic curves of Spytag-CsgA-CBD and Mfp5-CsgA-CBD were identical, suggesting that the size of a third domain had less influence on fibril assembly compared with the number of fusion domains.
Next, we conducted far-UV Circular Dichroism (CD) measurements for all the protein solutions containing varied amyloid fibrils (Fig. 2c). The appearance of a minimum at ∼220 nm and a maximum at ∼200 nm in the spectra indicated that all samples were rich in β-sheet structures. Also, the slight difference in the minimum of CD signal (at ∼220 nm for CsgA, and at ∼224 for others) among these samples suggested that they differed somewhat structurally because of the introduction of different functional domains. Specifically, of all the secondary structures formed in CsgA solution, β-sheet and β-turn structures constituted approximately 70% and 30%, respectively. The two-domain proteins contained 60% of β-sheet/β-turn structures and 40% of random coils, owing to the introduction of random coil Mfps. Compared with their two-domain counterparts, the three-domain fibrils possess more β-sheet structures, as the newly added CBD were reported to be rich in antiparallel β-sheet structures.21
To further probe the architecture of fibrils formed by different fusion proteins, we carried out X-ray fibril diffraction experiments (Fig. 2d). All of the protein samples displayed typical cross-beta diffraction patterns, a pattern that is the characteristic of amyloid fibrils.33,34 No change was observed in meridional reflection (denoted as d2 in the diffraction patterns shown in Fig. 2d) at 4.8 Å corresponding to the spacing between β-strands within each layer of β-sheets in the fibril. However, the intersheet packing distances reflected by equatorial reflections (denoted as d1 in the diffraction patterns shown in Fig. 2d) for the two-domain and three-domain fibrils were slightly smaller than that of CsgA alone. This slight distortion in intersheet packing distance could be attributed to the stress exerted on the amyloid backbone by the fused domains.6,18,35 Collectively, these observations thus suggest that fusion of one or two functional domains at varied positions slows down fibril assembly and affect fibril morphology, but does not disrupt amyloid assembly or the typical cross-beta fibril structures of CsgA.
Amyloid proteins typically assemble into mature fibrils following a dynamic process from original monomeric protein to the oligomer and finally to the mature fibril.9,36 To test whether this observation would hold true for the CsgA-fusion proteins constructed here (Fig. 3a), we deposited newly purified protein solutions on mica and probed the morphological evolution of the proteins as incubation proceeded (Fig. 3b). All proteins seemed to follow the similar assembly pathway of typical amyloid fibrils, as revealed by a morphological transition from dotted particles (2 h or 6 h) to short fibrils (6 h), and finally to long fibrils (≥18 h) (Fig. 3b). However, their evolution processes were still roughly distinguishable from each other. In particular, both three-domain proteins showed delayed fibril assembly, as the only structures detected were spherical particles for 6 hour samples, and short protofibrils along with dotted particles for 18 hour samples, which were morphologically different from the other samples (Fig. 3b). In fact, neither CBD-CsgA-Mfp3 nor Mfp5-CsgA-CBD proteins formed very long fibrils even after aging for more than 48 hours (ESI Fig. 9†). These observations thus suggested that fibril assembly rates decrease as the number of fusion domains added onto the CsgA backbone increases. A similar phenomenon was reported previously associated with Aβ42 variant amyloid protein, in which N-terminus extensions were found to retard Aβ42 fibril formation.11
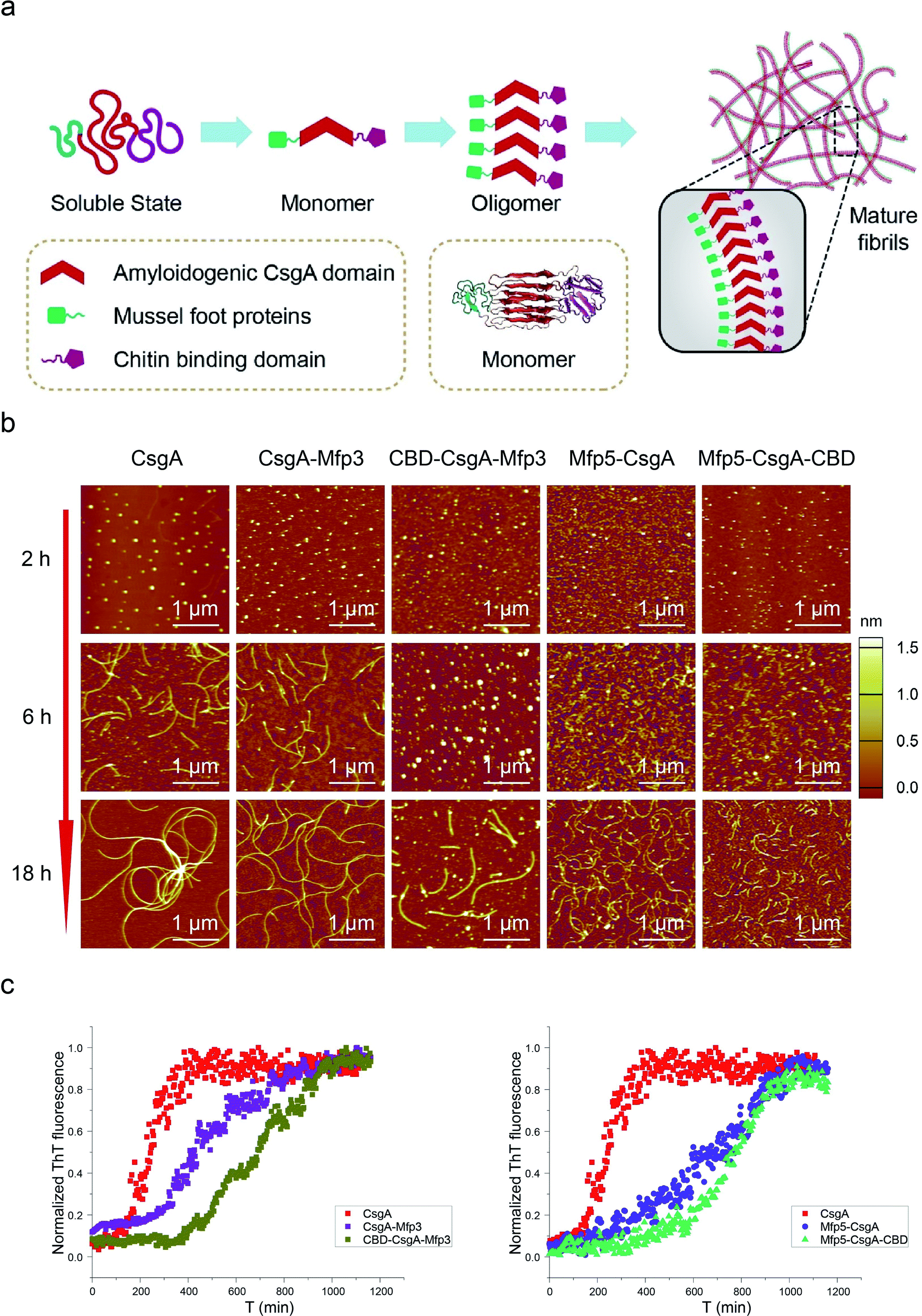 | ||
| Fig. 3 Self-assembly kinetics and morphological comparison for CsgA and CsgA-fusion proteins containing two or three domains. (a) Schematic illustration of the self-assembly pathway for CsgA-fusion proteins, which follow a typical amyloid assembly pathway from monomer to oligomer and finally to mature fibrils. (b) AFM morphological evolution for the differently functionalized CsgA-fusion proteins after aqueous incubation on mica for 2 h, 6 h and 18 h, respectively. (c) Thioflavin T (ThT) assay revealing fibril formation kinetics for CsgA, CsgA-Mfp3, CBD-CsgA-Mfp3 (left) and CsgA, Mfp5-CsgA and Mfp5-CsgA-CBD (right), respectively. | ||
Transitions of soluble proteins to insoluble amyloid aggregates for CsgA-fusion proteins can also be monitored using ThT, an amyloid-specific dye commonly used to assay amyloid formation.37 The ThT fluorescence of all samples followed a sigmoidal curve with distinguishable lag, growth, and stationary phases (Fig. 3c). However, the polymerization lag phases and time to reach stationary phases for CsgA-fusion proteins were typically longer than those for CsgA (Fig. 3c), suggesting that fusion of the Mfp and/or CBD domains to CsgA indeed retard both of the nucleation and growth processes. Such trends appear more pronounced for three-domain proteins than for two-domain proteins. Interestingly, a similar trend was also found associated with the comparison group for CsgA, CsgA-CBD, and Mfp5-CsgA-CBD (ESI Fig. 11†).
The mechanism of amyloid assembly involves a series of microscopic events, including primary nucleation, fibril elongation, and secondary nucleation processes.38,39 Conceivably, the flanking domains posed steric hindrance effects to the molecular recognition and aggregation of CsgA amyloids cores in the CsgA-fusion proteins, thus resulting in delayed formation of amyloid oligomers (longer lag time) and lower nucleation rates for the primary nucleation processes. As a result, more nuclei would form in the solutions of CsgA-fusion proteins compared to CsgA under similar protein concentrations. In addition, the flanking domains external to the amyloid cores may also cause interference, for example, by disturbing the addition or incorporation of amyloids monomers into the growing fibrils through random contact and/or interactions with fibril ends, therefore resulting in lower fibril elongation rate of CsgA fusion proteins. Consequently, the increased number of nuclei in the solutions and lower elongation rates would therefore produce shorter nanofibrils for CsgA-fusion proteins compared to CsgA, as revealed by the AFM morphological observations discussed above (Fig. 2a and 3b).
Based on these observations, we concluded that CsgA-fusion proteins all formed self-assembled fibrils through typical amyloid aggregation pathway from monomers, through oligomers to mature fibrils. The additional domains did not participate in the construction of the CsgA amyloid cores, but exerted some constrains on the aggregation of monomer proteins and therefore directly affected the fibril assembly kinetics and the morphology of the resultant fibrils. Moreover, the structures and assembly kinetics of amyloid structures were more heavily affected as the number of flanking domains increase in the CsgA system, and such general trend seems to be independent of the specific flanking domains to be fused in the system.
Mechanical properties are critical to our understanding of how domains contribute to a synthetic material system, and determining whether a given structure satisfies requirements for specific applications.40,41 The mechanical properties of fibril variants were determined through DMT modulus channel based upon PF-QNM measurement (Fig. 4a).36,42 All these values fall into the normal range of the Young's modulus typically for amyloid fibrils in gigapascal scale.40,41,43,44 In particular, the Young's modulus of fibrils increased when incorporated with β-sheet structure domain but decreased when fused with random coil domains, with an average value of 6.99 ± 0.72, 5.04 ± 0.48 and 7.84 ± 1.01 GPa for CsgA, CsgA-Mfp3, and CBD-CsgA-Mfp3, respectively (Fig. 4b and c). A similar descending trend is also found for CsgA, Mfp5-CsgA, and Mfp5-CsgA-CBD fibrils (ESI Fig. 12b†).
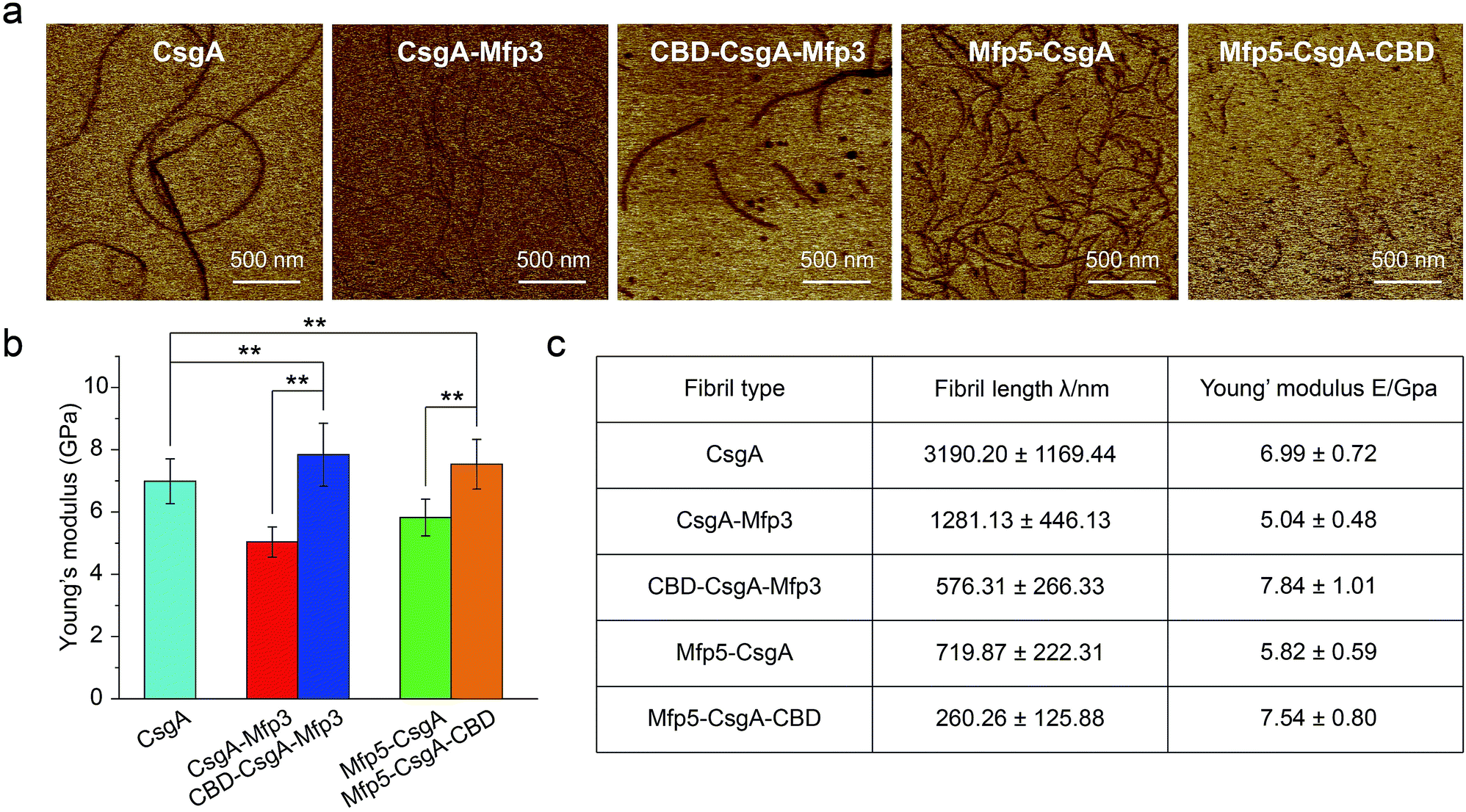 | ||
| Fig. 4 Comparison of the Young's modulus for the functionalized CsgA-fusion proteins. (a) Modulus mapping images of fibril variants performed by AFM peak force quantitative nanomechanics (PF-QNM) following a Derjaguin, Muller, Toropov (DMT) model. Scale bars, 500 nm. (b) Comparison of the Young's modulus of CsgA and CsgA-fusion fibrils extracted based upon DMT modulus images shown in (a). (c) Statistical comparison of fibril features of fibril variants, including fibril length and the Young's modulus. Fibril length was obtained by statistics from 40 randomly picked fibril spots. The Young's modulus was obtained by statistics from 25 randomly picked fibril spots. The error bars represent standard deviations (s.d.), **P < 0.01, student's t-test. | ||
The orientation of β-sheets – regular structures made of β-strands connected laterally by several backbone hydrogen bonds – and the inter-sheet packing of amyloid structures are believed to effectively contribute to mechanical properties of peptide fibrils.40,45 The CsgA-fusion fibrils comprised CsgA amyloidogenic cores and side fusion domains, which were thought to be external to the amyloid cores and exposed on the fibril surfaces.46 Therefore, the amyloidogenic cores mostly contributed to the elastic modulus of entire structures. Functional domains did not participate in the formation of amyloid cores and also did not affect the fibril structures based upon X-ray fibril diffraction data (Fig. 2d). However, fusion of flanking domains indeed influenced the Young's modulus to a certain extent based upon PF-QNM measurement (Fig. 4b and c). Noticeably, the two-domain CsgA-fusion proteins contain a random-coil Mfp domain, while the three-domain fusion proteins comprise a third rigid (β-sheet structure) CBD domain either at C or N-terminus other than the Mfp domain. Among the three most common types of secondary structures – i.e. α-helices, β-strands and random coil – random coil, in principle, is the one possessing least number of intermolecular hydrogen bonds, followed by α-helices and β-sheets.45,47 Previous studies suggested that a high amount of ordered β-sheets increased in the rigidity of the fibrils, whereas a high amount of ordered α-helices and random coils seemed to weaken the structure by softening the self-assembled fibrils.36,45 The content of β-sheets in the fusion proteins decreased when fused with mfp3 or mfp5, and increased when fused with CBD, thus explaining that the stiffness of CsgA-fusion fibrils in its entirety was indeed dependent on the nature of the incorporated functional domains.
To further assess how the flanking CBD domain affects the function of CsgA-fusion proteins, we measured the functional activities of CsgA-fusion proteins containing a CBD domain and compared with their counterparts without CBD domain. Specifically, we tested the binding behaviors of amyloid fibrils towards chitin substrates based upon quartz crystal microbalance with dissipation (QCM-D) monitoring (Fig. 5a). QCM-D monitors the absorption of fibrils towards chitin-coated Au surfaces under dynamic flow conditions, thereby providing useful information regarding how differently the CsgA-fusion fibrils bind towards chitin substrates when the absorption reaches dynamic equilibrium. The adsorption of protein fibrils was registered as a decrease in the frequency (ΔF). The marked change in ΔF at around 0.5 h indicated rapid absorption of the proteins on the surface. The change in ΔF was much slower afterwards, suggesting that the absorption capacity of substrate was saturated. After removing the non-specific binding proteins using distilled water, ΔF reached a steadily numerical value, which represented the actual adsorption of protein fibrils onto chitin surfaces. The results indicated that CsgA-fusion proteins with either specific CBD domain or adhesive domains Mfp3 or Mfp5 all showed higher affinity than CsgA (Fig. 5b). Mfp5-CsgA-CBD had the highest mass absorption towards chitin surfaces, while Mfp5-CsgA showed lower absorption. The absorption capacities of CsgA-Mfp3 and CBD-CsgA-Mfp3 were identical to each other and lay between those of the other two aforementioned proteins.
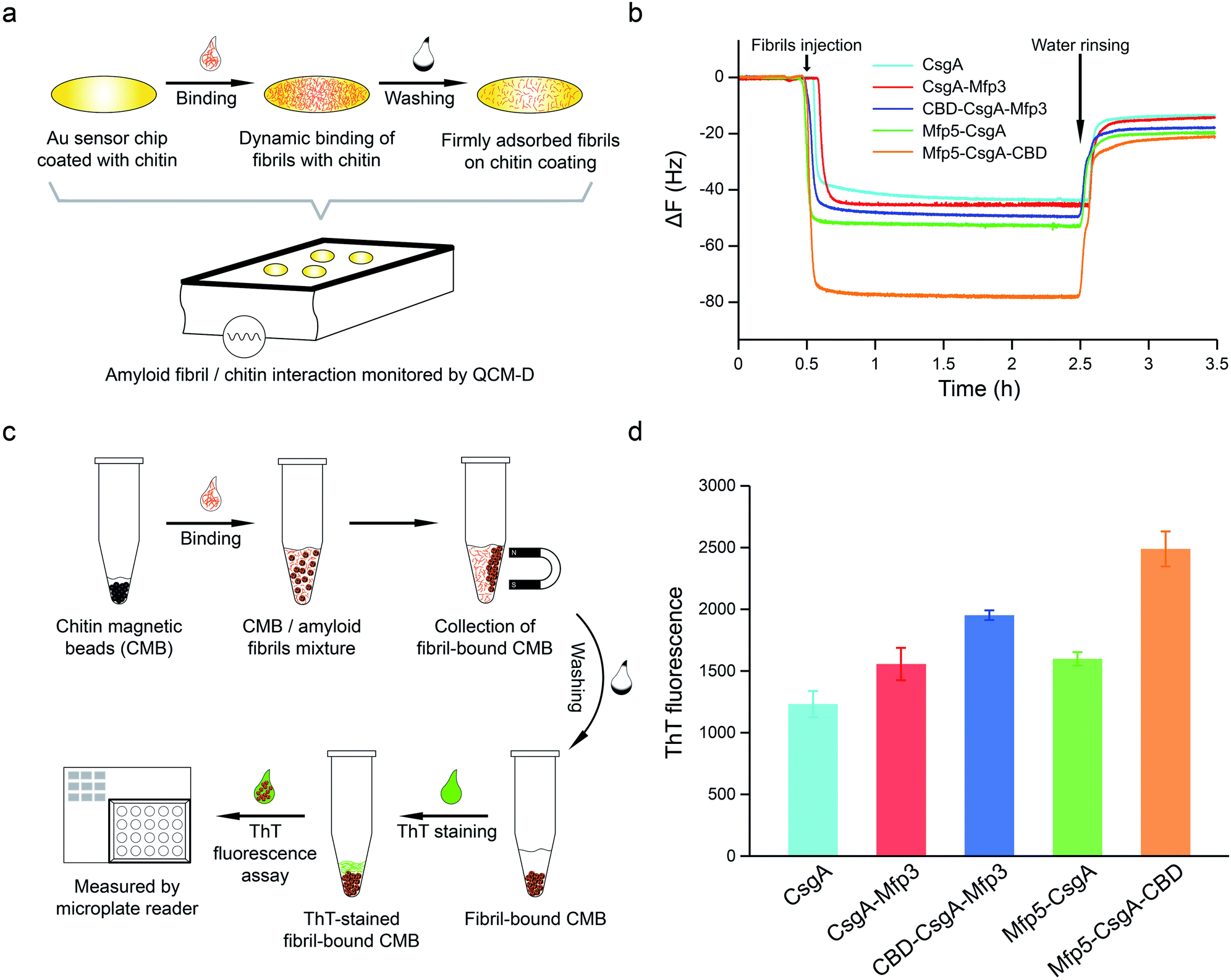 | ||
| Fig. 5 Functional assessment of CsgA and CsgA-fusion proteins based on molecular interactions between proteins and chitin substrates with QCM-D experiments (a and b) and thioflavin T (ThT) quantitative fluorescence assay (c and d). (a) Schematic of QCM-D experiment designed to analyze the absorption behaviors of different amyloid fibrils towards chitin-coated gold chips. The frequency signal change over time was recorded by QCM-D. (b) The frequency (ΔF/n for overtone n = 3) time curves showing the different adsorption capacities of CsgA and CsgA-fusion amyloid nanofibrils to chitin-coated surface. (c) Schematic of ThT fluorescence assay to assess the specific interaction between fibrils and chitin magnetic beads. Amyloid fibrils (1.0 mM) were incubated with beads, which were then mixed with 50 mM ThT solution and tested with Microplate Reader. (d) The ThT fluorescence strength comparison of CBD-containing fibrils Mfp5-CsgA-CBD and CBD-CsgA-Mfp3, with CsgA, Mfp5-CsgA and CsgA-Mfp3 fibrils as controls. Fluorescence strength in each sample was correlated to the amount of amyloid fibrils absorbed onto the chitin beads. The statistical analysis for each sample was average of 5 independent tests with s.d. error shown. | ||
To further support our results, we performed a ThT fluorescence assay, in which ThT can be specifically recognized and absorbed by amyloid fibrils and the fluorescence strength correlates of amyloid fibrils that have bound onto the chitin beads. This assay therefore allows the indirect assessment of how efficiently amyloid fibrils bind towards chitin magnetic beads (Fig. 5c). Interestingly, in consistence with the QCM-D results, the ThT assay indicated that Mfp5-CsgA-CBD displayed significantly higher fluorescence compared to the other proteins, while the fluorescence strength of the other two-domain proteins was slightly lower than that of CBD-CsgA-Mfp3 (Fig. 5d). Collectively, these results strongly suggested that the binding affinity for chitin surfaces of the two three-domain amyloid proteins containing the CBD domain was higher than that of their two-domain counterparts without the CBD domain, yet CBD-CsgA-Mfp3 did not show much increased affinity compared with CsgA-mfp3. This latter observation could possibly be explained as a result of the unfolded secondary and tertiary structure of CBD domain in the CBD-CsgA-Mfp3 protein, as revealed by molecular dynamic simulations presented below. These interesting results also indicated that the flanking domains, more or less, could endow the assembled CsgA-fusion amyloids with functional activity, implying the promise of a modular genetic design for molecular materials design.
We next carried out molecular dynamics simulations to study an approximation of how individual domains behave, both in the form of monomers or fibrils, with simulation time of 1 μs and 100 ns respectively (Fig. 6). Simulations of both monomers showed that the CsgA core retained its structure during 1 μs of simulation time, suggesting that the CsgA stacked beta-sheets retained their structure in the presence of the CBD and Mfp3/5 domains. Since there do not exist atomic-level resolution crystal or NMR structures of the CsgA domain, we used a previously published structure of the CsgA core domain5 that was derived from a homology model with an amyloid–beta fibril core structure. Its structure has subsequently been found to be in good agreement with models derived from pure molecular dynamics simulations.48 In the Mfp5-CsgA-CBD fibril simulations, the CBD domains retained secondary and tertiary structure similar to the initial model (PDB ID 1ED7), despite self-associating through motion of the flexible linkers connecting them to the CsgA fibril core (Fig. 6). Mfp5 domains also self-associated, much like the Mfp5-CsgA and CsgA-Mfp3 fibril models, but did not interfere with the CBD domains or their structure over the course of the 100 ns of simulation time. In contrast, the Mfp3 domains did not self-associate in the CBD-CsgA-Mfp3 fibrils and remained fairly extended, as did the CBD domains, which unfolded over the course of the simulation. Interestingly, the results seem to be supported by CD spectra, in which the overall percentage of β-sheet structures of CBD-CsgA-Mfp3 is indeed slightly lower than that of Mfp5-CsgA-CBD fibrils (Fig. 2c).
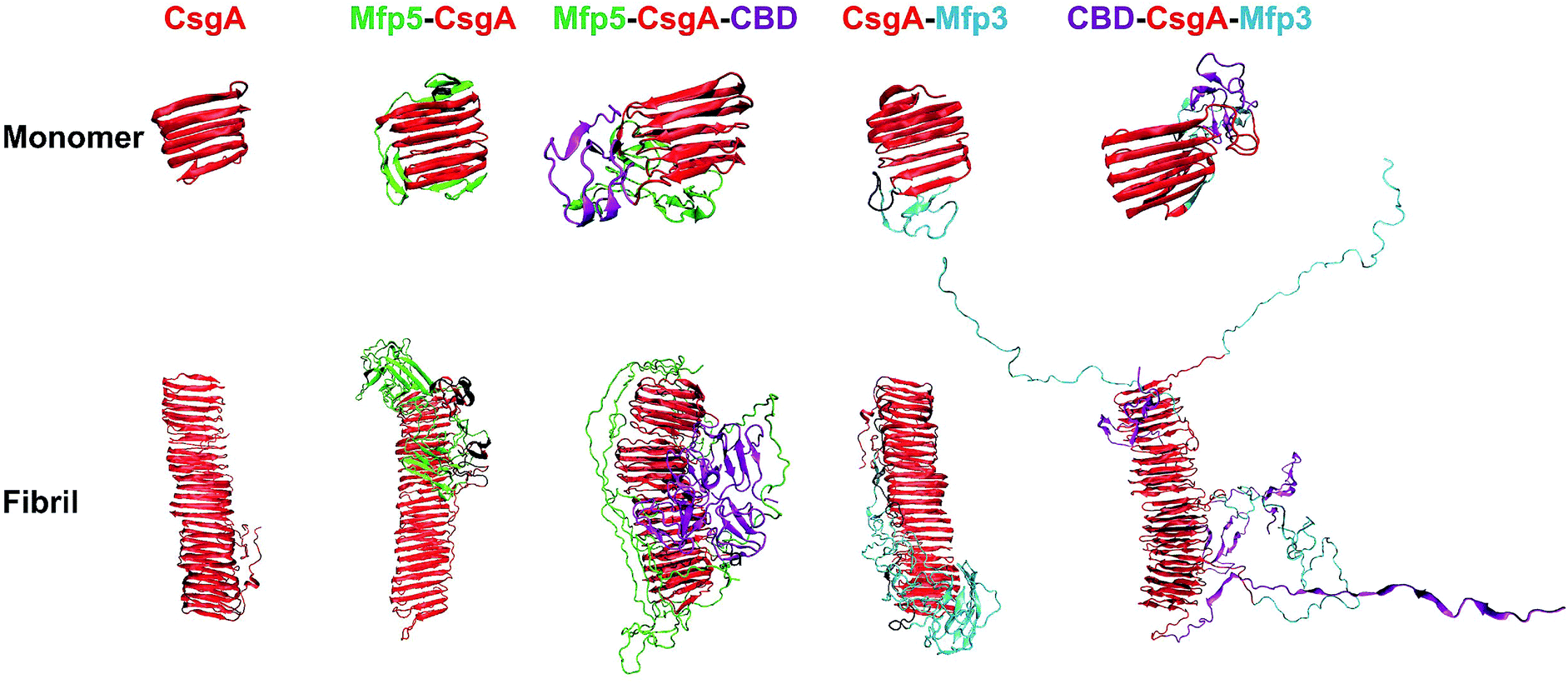 | ||
| Fig. 6 Comparison of protein monomers and individual fibril structures based upon molecular dynamics simulations. The structures obtained from simulation for the one, two-, and three-component CsgA-fusion protein monomers (top) and fibrils (bottom). Note: for both monomeric and fibrillar states of the constructs, the structures are similar: the CsgA domain always dominate the well-ordered amyloid cores, while disordered Mfp5 or Mfp3 domains as well as CBD are external to the amyloid cores. In particular, the CBD domain in Mfp5-CsgA-CBD retained secondary and tertiary structure similar to the initial folding, while the secondary and tertiary structure of CBD-CsgA-Mfp3 unfolded over the course of simulation. The simulation times for the monomeric and the fibrillar structures were 1 μs and 100 ns, respectively. | ||
These results based upon molecular dynamics simulations are thus in good agreement with the functional activity of the CBD domain associated with the three-domain amyloid fibrils, in which CBD-CsgA-Mfp3 displays significantly lower binding affinity towards chitin surfaces than Mfp5-CsgA-CBD. In addition, the predicted structures based on molecular dynamics simulations also implied that fusion domains might impose steric hindrance and thus interfere amyloid monomer/monomer interaction by having random contacts and/or interacting with the core β-sheet structures, therefore more or less affecting the nanofibril assembly kinetics and fibril morphology as observed in our experiments.
In summary, we have demonstrated that all CsgA-fusion proteins are able to self-assemble into typical amyloid fibrils, but the introduction of additional flanking domains retards fibril formation and leads to shortened fibrils. Moreover, the incorporation of side domains also affected the stiffness of nanofibrils, which are directly correlated to the structural features of the flanking domains. In addition, fibrils containing CBD domains indeed showed higher chitin-binding activity compared with their CBD-free counterparts. In particular, the three-domain fibril Mfp5-CsgA-CBD displays stronger specific chitin-binding activity compared with CBD-CsgA-Mfp3 owing to better retention of secondary and tertiary structures, in agreement with results based on molecular dynamic modeling. Collectively, these observations suggest that the structures, assembly kinetics and functions of amyloid structures are more heavily affected as the number of flanking domains increase. In addition, the domain sequences and arrangement are both critical to the functional performance of the final assembled structures even though morphologically they look identically. Our studies therefore underscore that careful designs and measurements are needed to engineer multi-domain functional amyloids and to implement such structures for new applications relevant to nanotechnology and materials science.
The identification of functional amyloids in diverse organisms has recently prompted a new research paradigm of creating functional amyloid-based molecular materials through a modular genetic strategy.17 This paradigm represents an unparalleled potential to create multifunctional materials with both predictable functionalities and precise tunability, properties that are lacking in amyloid structures either derived from denatured proteins or synthetic peptides. Given the diverse library of functional amyloids in organisms that are ripe for incorporation into new amyloid-based molecular materials, we believe that the insights provided by this study will provide new insights the future design of multi-domain functional amyloids. Last but not least, the molecular assembly principles of rationally designed functional amyloids via a modular genetic design described here may also serve as the basis for developing a new generation of living functional materials based on engineered bacterial biofilms with emerging functions.24,49–53
Author contributions
C. Z. directed the research, M. K. C., Q. Q. and C. Z. designed and participated in all the experiments; M. K. C. and Q. Q. carried out AFM peak-force QNM test, AFM imaging and participate in most of the experiments. T. G. constructed the initial models and conducted the molecular dynamics simulations, T. X. Z. performed in protein expression, purification, X. R. G. helped to collect the data of X-ray fibril diffraction; C. L. assisted in analyzing the result of X-ray fibril diffraction, B. A. collected all the CD spectra with the help from X. R. G., A. C. constructed the plasmids for the two-domain amyloid fusion proteins. C. Z., M. K. C. and Q. Q. performed in analyzing all of the data; and C. Z., M. K. C., Q. Q. and T. G. wrote the paper with help from all authors. All authors contributed to revising the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Xinyu Wang for providing plasmid CBD-CsgA-Mfp3 and Susu Ren for providing plasmid SpyTag-CsgA-CBD. Tapping-mode and PK-QNM mode AFM was performed on a Dimension Icon AFM (Bruker Co.) in Analytical Instrumentation Center, School of Physical Science and Technology, ShanghaiTech University, and National Center for Protein Science (Shanghai). X-ray fibril diffraction was performed with a Rigaku Micromax-007 X-ray generator in Shanghai Institute Organic Chemistry, Chinese Academy of Sciences. Special thanks for access to Synchrotron X-ray fibril diffraction on beamline 14B on Shanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, China. This work was primarily supported by the Joint Funds of the National Natural Science Foundation of China (Seed Grant No. U1532127). This work was also supported in part by National Natural Science Foundation of China (Grant No. 31570972). C. Z. acknowledges start-up funding support from ShanghaiTech University and 1000 Youth Talents Program, granted by the Chinese Central Government.References
- R. O. Hynes, Science, 2009, 326, 1216–1219 CrossRef CAS PubMed
.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed
- F. M. Watt and W. T. S. Huck, Nat. Rev. Mol. Cell Biol., 2013, 14, 467 CrossRef CAS PubMed
- R. L. DiMarco and S. C. Heilshorn, Adv. Mater., 2012, 24, 3923–3940 CrossRef CAS PubMed
- C. Zhong, T. Gurry, A. A. Cheng, J. Downey, Z. Deng, C. M. Stultz and T. K. Lu, Nat. Nanotechnol., 2014, 9, 858 CrossRef CAS PubMed
- J.-M. Malho, S. Arola, P. Laaksonen, G. R. Szilvay, O. Ikkala and M. B. Linder, Angew. Chem., Int. Ed., 2015, 54, 12025–12028 CrossRef CAS PubMed
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103 CrossRef CAS
- C. M. Dobson, Nature, 2003, 426, 884 CrossRef CAS PubMed
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed
- L. Chaoxu, B. Sreenath and M. Raffaele, Adv. Mater., 2013, 25, 3694–3700 CrossRef
- O. Szczepankiewicz, B. Linse, G. Meisl, E. Thulin, B. Frohm, C. Sala Frigerio, M. T. Colvin, A. C. Jacavone, R. G. Griffin, T. Knowles, D. M. Walsh and S. Linse, J. Am. Chem. Soc., 2015, 137, 14673–14685 CrossRef CAS PubMed
- C. Li, J. Adamcik and R. Mezzenga, Nat. Nanotechnol., 2012, 7, 421–427 CrossRef CAS PubMed
- X. Wang, Y. Li and C. Zhong, J. Mater. Chem. B, 2015, 3, 4953–4958 RSC
- T. P. Knowles and R. Mezzenga, Adv. Mater., 2016, 5, 961–977 Search PubMed
- L. Altamura, C. Horvath, S. Rengaraj, A. Rongier, K. Elouarzaki, C. Gondran, A. L. B. Maçon, C. Vendrely, V. Bouchiat, M. Fontecave, D. Mariolle, P. Rannou, A. Le Goff, N. Duraffourg, M. Holzinger and V. Forge, Nat. Chem., 2016, 9, 157 CrossRef PubMed
- B. An, X. Wang, M. Cui, X. Gui, X. Mao, Y. Liu, K. Li, C. Chu, J. Pu, S. Ren, Y. Wang, G. Zhong, T. K. Lu, C. Liu and C. Zhong, ACS Nano, 2017, 11, 6985–6995 CrossRef CAS PubMed
- Y. Wang, J. Pu, B. An, T. K. Lu and C. Zhong, J. Mol. Biol., 2018, 430, 3720–3734 CrossRef CAS PubMed
- C. J. Forman, A. A. Nickson, S. J. Anthony-Cahill, A. J. Baldwin, G. Kaggwa, U. Feber, K. Sheikh, S. P. Jarvis and P. D. Barker, ACS Nano, 2012, 6, 1332–1346 CrossRef CAS PubMed
- H. Zhao and J. H. Waite, J. Biol. Chem., 2006, 281, 26150–26158 CrossRef CAS PubMed
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed
- M. S. e. a. Takahisa Ikegami, J. Biol. Chem., 2000, 275, 13654–13661 CrossRef
- X. Wang and M. R. Chapman, J. Mol. Biol., 2008, 380, 570–580 CrossRef CAS PubMed
- A. Y. Chen, Z. Deng, A. N. Billings, U. O. Seker, M. Y. Lu, R. J. Citorik, B. Zakeri and T. K. Lu, Nat. Mater., 2014, 13, 515–523 CrossRef CAS PubMed
- P. Q. Nguyen, Z. Botyanszki, P. K. R. Tay and N. S. J. N. c. Joshi, Nat. Commun., 2014, 5, 4945 CrossRef CAS PubMed
- S. K. Collinson, J. M. R. Parker, R. S. Hodges and W. W. Kay, J. Mol. Biol., 1999, 290, 741–756 CrossRef CAS PubMed
- M. R. Chapman, L. S. Robinson, J. S. Pinkner, R. Roth, J. Heuser, M. Hammar, S. Normark and S. J. Hultgren, Science, 2002, 295, 851–855 CrossRef CAS PubMed
- M. M. Barnhart and M. R. Chapman, Annu. Rev. Microbiol., 2006, 60, 131–147 CrossRef CAS PubMed
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed
- L. Petrone, A. Kumar, C. N. Sutanto, N. J. Patil, S. Kannan, A. Palaniappan, S. Amini, B. Zappone, C. Verma and A. Miserez, Nat. Commun., 2015, 6, 8737–8748 CrossRef CAS
- D. S. Hwang and J. H. J. P. S. Waite, Protein Sci., 2012, 21, 1689–1695 CrossRef CAS PubMed
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed
- R. Nassar, E. Wong, J. Gsponer and G. Lamour, J. Am. Chem. Soc., 2019, 141, 58–61 CrossRef CAS PubMed
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef CAS PubMed
- J. Greenwald and R. Riek, Structure, 2010, 18, 1244–1260 CrossRef CAS PubMed
- X. M. Zhou, A. Entwistle, H. Zhang, A. P. Jackson, T. O. Mason, U. Shimanovich, T. P. Knowles, A. T. Smith, E. B. Sawyer and S. Perrett, ChemCatChem, 2014, 6, 1961–1968 CrossRef CAS PubMed
- F. S. Ruggeri, J. Adamcik, J. S. Jeong, H. A. Lashuel, R. Mezzenga and G. Dietler, Angew. Chem., Int. Ed. Engl., 2015, 54, 2462–2466 CrossRef CAS PubMed
- L. P. Blanco, M. L. Evans, D. R. Smith, M. P. Badtke and M. R. Chapman, Trends Microbiol., 2012, 20, 66–73 CrossRef CAS PubMed
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. J. Knowles and S. Linse, Chem. Sci., 2015, 6, 4215 RSC
- G. Meisl, X. Yang, C. M. Dobson, S. Linse and K. Tpj, Chem. Sci., 2017, 8, 4352–4362 RSC
- T. P. Knowles and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS
- T. P. Knowles and M. J. Buehler, Nat. Nanotechnol., 2011, 6, 469–479 CrossRef CAS
- J. Adamcik, C. Lara, I. Usov, J. S. Jeong, F. S. Ruggeri, G. Dietler, H. A. Lashuel, I. W. Hamley and R. Mezzenga, Nanoscale, 2012, 4, 4426–4429 RSC
- J. F. Smith, T. P. Knowles, C. M. Dobson, C. E. Macphee and M. E. Welland, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15806–15811 CrossRef CAS
- J. Adamcik, A. Berquand and R. Mezzenga, Appl. Phys. Lett., 2011, 98, 193701 CrossRef
- C. Bortolini, N. C. Jones, S. V. Hoffmann, C. Wang, F. Besenbacher and M. Dong, Nanoscale, 2015, 7, 7745–7752 RSC
- S. Sambashivan, Y. Liu, M. R. Sawaya, M. Gingery and D. Eisenberg, Nature, 2005, 437, 266–269 CrossRef CAS PubMed
- S. Xiao, S. Xiao and F. Grater, Phys. Chem. Chem. Phys., 2013, 15, 8765–8771 RSC
- E. P. Debenedictis, D. Ma and S. Keten, RSC Adv., 2017, 7, 48102–48112 RSC
- J. Huang, S. Liu, C. Zhang, X. Wang, J. Pu, F. Ba, S. Xue, H. Ye, T. Zhao, K. Li, Y. Wang, J. Zhang, L. Wang, C. Fan, T. K. Lu and C. Zhong, Nat. Chem. Biol., 2019, 15, 34–41 CrossRef CAS PubMed
- P. Q. Nguyen, N. M. D. Courchesne, A. Duraj-Thatte, P. Praveschotinunt and N. S. J. A. M. Joshi, Adv. Mater., 2018, 30, 1704847 CrossRef
- A. Y. Chen, C. Zhong and T. K. Lu, ACS Synth. Biol., 2015, 8–11 CrossRef CAS
- C. Gilbert and T. Ellis, ACS Synth. Biol., 2019, 8, 1–15 CrossRef CAS PubMed
- C. Zhang, J. Huang, J. Zhang, S. Liu, M. Cui, B. An, X. Wang, J. Pu, T. Zhao, C. Fan, T. K. Lu and C. Zhong, Mater. Today, 2019 DOI:10.1016/j.mattod.2018.12.039
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00208a |
| ‡ M. K. C., Q. Q. and T. G. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |