
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic C–H silylation of heteroarenes by using trialkylhydrosilanes†
Shihui
Liu
a,
Peng
Pan
a,
Huaqiang
Fan
a,
Hao
Li
 a,
Wei
Wang
*ab and
Yongqiang
Zhang
*a
a,
Wei
Wang
*ab and
Yongqiang
Zhang
*a
aState Key Laboratory of Bioengineering Reactor, Shanghai Key Laboratory of New Drug Design and School of Pharmacy, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: yongqiangzhang@ecust.edu.cn; wwang@pharmacy.arizona.edu
bDepartment of Pharmacology and Toxicology, BIO5 Institute, University of Arizona, Tucson, Arizona 85721-0207, USA
First published on 18th February 2019
Abstract
The efficient and selective C–H silylation of heteroarenes, especially the pharmaceutically relevant electron-deficient heteroarenes, represents a great challenge in organic synthesis. Herein we wish to report a distinctive visible light-promoted photocatalytic C–H silylation approach that enables the direct coupling of trialkylhydrosilanes with both electron-deficient and -rich heteroarenes as well as with cyano-substituted arenes in moderate to high yields and with good regioselectivity. The protocol features operational simplicity, mild reaction conditions, and the use of safe and readily available Na2S2O8, bis(trimethylsilyl) peroxide (BTMSPO) or iPr3SiSH as the radical initiators. Notably, the challenging bulky and inert trialkylhydrosilanes, such as (t-butyldimethyl)silane (tBuMe2SiH) and (triisopropyl)silane (iPr3SiH), work smoothly with the protocol. Moreover, despite the higher stability of tBuMe2Si silylation products, our studies revealed their great reactivity and versatility in diverse C–Si-based chemical transformations, providing an operationally simple, low-cost, and environmentally benign synthetic technology for molecule construction and elaboration.
Introduction
Heteroaryltrialkylsilanes, especially the electron-deficient heteroaryltrialkylsilanes, represent a class of promising therapeutic agents with interesting biological properties (see the representative examples shown in Scheme 1).1–4 Moreover, these organosilicon functionalities serve as versatile heteroaryl handles for complex molecule synthesis owing to their high air and moisture stability, low toxicity, and ease of manipulation.5–9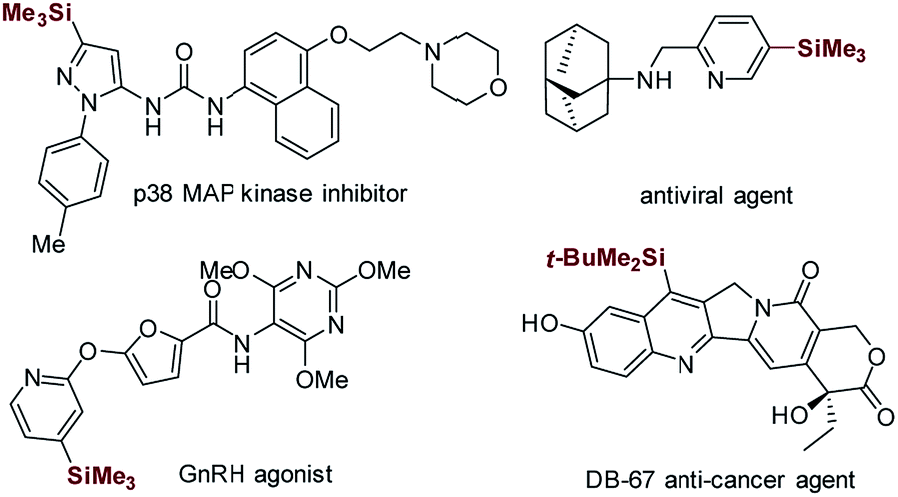 | ||
| Scheme 1 Selected bioactive compounds that contain heteroaryltrialkylsilane functionality. | ||
The direct coupling of readily available and unfunctionalized heteroarenes with simple trialkylhydrosilanes offers a step-efficient and atom-economical synthetic tool to access this class of valuable targets. The quest for such synthetic methodologies underscores significant efforts leading to elegant and powerful catalytic C–H silylation approaches, including the transition-metal-catalyzed approach,10–14 Friedel–Crafts-type reaction,15–18 and a rather unorthodox method promoted by KOtBu.19–21 However, these methods often rely on the use of precious iridium or ruthenium species at high temperature (80–135 °C) or the use of extremely strong Lewis acids or bases under strict water- and oxygen-free reaction conditions. Moreover, these methods are only compatible with electron-rich heteroarenes and small trialkylhydrosilanes, such as triethylhydrosilane (Et3SiH) and diethylmethylhydrosilane (Et2MeSiH). These drawbacks have significantly restricted the synthetic application of these methods, especially in the pharmaceutical sector.
Minisci-type C–H silylation offers a better solution (Scheme 2).22–26 The copper-catalyzed approach enables the introduction of trialkylsilyl groups to both electron-rich and -deficient heteroarenes in synthetically useful yields (30–76%) (Scheme 2a).24,25 It is noted that the electron-deficient heteroarenes display inferior reaction efficiency (32–40% yields) and only two pyridine examples were demonstrated. Furthermore, this process is carried out at a high reaction temperature (130 °C) in the presence of explosive peroxide (di-tert-butyl peroxide, DTBP), which causes significant safety concerns. Recently, inspired by the pioneering work of the Curran group,4 Maruoka and co-workers disclosed an interesting thiol-catalyzed approach for the C–H silylation of electron-deficient heteroarenes, such as pyridines, quinolones, pyrazines and quinoxalines, which could give the mono-silylation products in low yields (4–53%) accompanied with the formation of a significant amount of bis-silylation products (9–42% yield) (Scheme 2b).26 Notably, the in situ generated silyl radicals are highly reactive and non-clustered,27,28 thus enabling the construction of sterically demanding C–Si bonds and the synthesis of heteroaryltrialkylsilanes with bulky trialkylsilyl groups, such as t-butyldimethylsilyl and triisopropylsilyl groups. Nevertheless, the process is also performed at high temperature (110 °C) using a large excess of DTBP (7.0 equiv.), which causes significant safety concerns, as well as the difficulty controlling regioselectivity. Therefore, in this field, there is an unmet synthetic challenge for the development of a mild and truly efficient approach for the selective C–H silylation of heteroarenes, especially the pharmaceutically relevant electron-deficient heteroarenes. Such a method enabling the incorporation of the challenging bulky trialkylsilyl functionalities into heteroarenes will streamline the synthesis of synthetically and medicinally valuable heteroaryltrialkylsilanes, such as the anticancer drug candidate camptothecin derivative DB-67 (Scheme 1).4
 | ||
| Scheme 2 Minisci-type approaches for C–H silylation of heteroarenes. | ||
Towards this end, we wish to report a distinctive visible light-promoted photocatalytic Minisci-type approach that enables the efficient and selective C–H silylation of both electron-deficient and -rich heteroarenes in moderate to high yields and with good regioselectivity by using trialkylhydrosilanes (Scheme 2c). The protocol features operational simplicity, mild reaction conditions, and the use of a significantly reduced amount of Na2S2O8 (2.0 equiv.), bis(trimethylsilyl)-peroxide (BTMSPO) (2.0 equiv.) or an alternative safe iPr3SiSH (1.2 equiv.) as the radical initiators. This study, therefore, provides a toolbox of powerful solutions for the synthesis of structurally diverse heteroaryltrialkylsilanes. While Na2S2O8 and iPr3SiSH serve as efficient promoters for the C–H silylation of electron-deficient heteroarenes, BTMSPO was proved effective for both electron-deficient and electron-rich heteroarenes as a more promising radical initiator. It is of note that the thiol-mediated sila-Minisci-type reaction is carried out under peroxide-free conditions with superior regioselectivity. The cyano-substituted arenes were also amenable to this method. Notably, the challenging bulky and inert trialkylhydrosilanes, such as (t-butyldimethyl)silane (tBuMe2SiH) and (triisopropyl)silane (iPr3SiH), work smoothly with the protocol. Moreover, despite the higher stability of tBuMe2Si silylation products, our studies revealed their great reactivity and versatility in diverse C–Si-based chemical transformations, providing an operationally simple, low-cost, and environmentally benign synthetic technology for molecule construction and elaboration.
Results and discussion
Photocatalytic Minisci-type reactions for the construction of heteroaryl C–C bonds from two different and inert C–H bonds have been well developed.29–31 We questioned whether this strategy could be extended for the formation of heteroaryl C–Si bonds. This process is generally carried out under mild reaction conditions, which may provide an opportunity to address the issue of regioselectivity and minimize the production of undesired bis-silylation products in the present Minisci-type C–H silylation methods.24–26Optimization of reaction conditions
The initial investigation of the proposed photocatalytic C–H silylation reaction started by subjecting 6-methylisoquinoline 1a and t-butyldimethylsilane (tBuMe2SiH) 2a to the benzaldehyde-mediated photoredox reaction conditions established in our previous study using Na2S2O8 as the radical initiator.31 To our delight, the desired product was obtained in a tractable yield (20%). This outcome inspired us to systematically investigate the interesting reaction. Extensive reaction condition screening, including the screening of photocatalysts (Table S1†), the screening of oxidants (Table S2†), the screening of solvents (Table S3†), and the screening of the amount of 2a (Table S4†), identified the optimal conditions as follows: in the presence of 23 W CFL, 1 mol% of [Ir(ppy)2(dtbbpy)]PF6, and 2 equiv. of Na2S2O8, the reaction of 6-methylisoquinoline 1a with 5 equiv. of tBuMe2SiH 2a is conducted in a solvent mixture of DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCE = 1:1 (0.1 M) for 24 h at 30 °C (Table 1, entry 1). Under the optimal reaction conditions, the mono-silylation product 3a was produced in high yield (77%, entry 1, Table 1). Nevertheless, a very small amount of the bis-silylation product 3a′ was also observed (3a:3a′ = 15:1). The decrease of the amount of tBuMe2SiH 2a (3 equiv.) was detrimental to the reaction efficiency, while the regioselectivity remained unaffected (69% yield, 3a:3a′ = 14:1, entry 2, Table 1). Employing trifluoroacetic acid (TFA)32,33 as an additive or a stronger 34 W LED as an alternative visible light source resulted in slightly decreased yields and regioselectivity (70–74% yields, 3a:3a′ = 9:1 to 12:1, entries 3–4, Table 1). The use of thiol as an additive in this process, which was shown an efficient promotor in sila-Minisci-type reactions,4,26 had little effect on the reaction outcome (77% yield, 3a:3a′ = 13:1, entry 5, Table 1). Interestingly, the removing of Na2S2O8 in the presence of thiol was also able to produce 3a in a synthetically useful yield with better regioselectivity (27% yield, 3a only, entry 6, Table 1). This study offers the possibility of developing a peroxide-free photocatalytic Minisci-type C–H silylation reaction with better site selectivity. Oxygen is not essential for this process (76–77% yields, entries 7–8, Table 1). Control experiments show that visible light, photocatalyst, and Na2S2O8 are essential for this process, again suggesting the visible-light-driven photochemical nature of this reaction (entries 9–11, Table 1).
:DCE = 1:1 (0.1 M) for 24 h at 30 °C (Table 1, entry 1). Under the optimal reaction conditions, the mono-silylation product 3a was produced in high yield (77%, entry 1, Table 1). Nevertheless, a very small amount of the bis-silylation product 3a′ was also observed (3a:3a′ = 15:1). The decrease of the amount of tBuMe2SiH 2a (3 equiv.) was detrimental to the reaction efficiency, while the regioselectivity remained unaffected (69% yield, 3a:3a′ = 14:1, entry 2, Table 1). Employing trifluoroacetic acid (TFA)32,33 as an additive or a stronger 34 W LED as an alternative visible light source resulted in slightly decreased yields and regioselectivity (70–74% yields, 3a:3a′ = 9:1 to 12:1, entries 3–4, Table 1). The use of thiol as an additive in this process, which was shown an efficient promotor in sila-Minisci-type reactions,4,26 had little effect on the reaction outcome (77% yield, 3a:3a′ = 13:1, entry 5, Table 1). Interestingly, the removing of Na2S2O8 in the presence of thiol was also able to produce 3a in a synthetically useful yield with better regioselectivity (27% yield, 3a only, entry 6, Table 1). This study offers the possibility of developing a peroxide-free photocatalytic Minisci-type C–H silylation reaction with better site selectivity. Oxygen is not essential for this process (76–77% yields, entries 7–8, Table 1). Control experiments show that visible light, photocatalyst, and Na2S2O8 are essential for this process, again suggesting the visible-light-driven photochemical nature of this reaction (entries 9–11, Table 1).
|
|
|||
|---|---|---|---|
| Entry | Reaction conditions | Yield (3a)b | Ratio (3a:3a′)c |
|
a Standard reaction conditions: 23 W CFL, 1a (0.5 mmol), 2a (2.5 mmol), photocatalyst (1 mol%), Na2S2O8 (1.0 mmol), solvent mixture (5.0 mL, DMSO:DCE = 1:1), air, 30 °C, 24 h, unless otherwise noted.
b Isolated yields were reported.
c Regiomeric ratio (r.r.) determined using 1H NMR spectroscopy.
d TFA, trifluoroacetic acid.
e Performed in the absence of Na2S2O8.
f The reaction mixture was degassed via freeze–pump–thaw (three times) and refilled with N2.
g Not detected.
|
|||
| 1 | Standard conditions | 77% | 15:1 |
| 2 | 2a (3 equiv.) | 69% | 14:1 |
| 3 | TFAd (1.0 equiv.) | 70% | 9:1 |
| 4 | 34 W blue LED | 74% | 12:1 |
| 5 | t BuSH (20 mol%) | 77% | 13:1 |
| 6 | t BuSH (20 mol%)e | 27% | 3a only |
| 7 | Under O2 (1 atm) | 76% | 13:1 |
| 8 | Under N2 (1 atm)f | 77% | 15:1 |
| 9 | In darkness | n.d.g | — |
| 10 | No photocat. | 16% | — |
| 11 | No Na2S2O8 | n.d.g | — |

Reaction scope
:3b13 = 6:1). We developed a new alternative thiol-mediated photocatalytic C–H silylation reaction with better regioselectivity to solve this issue (see Scheme 5). Interestingly, blocking the C-1 position of isoquinoline mainly results in the formation of C-6 silylation products, albeit with slightly decreased reaction efficiency (3b14–b15, 35–61% yields). Furthermore, 6-bromoisoquinoline, which is not tolerated with this protocol, was also demonstrated as a suitable substrate of the thiol-mediated reaction, providing the exclusive mono-silylation product in good yield (3b16, 64%).
 | ||
| Scheme 3 Direct C–H silylation of diverse electron-deficient heteroarenes via Na2S2O8-mediated photocatalytic Minisci-type reaction. aSee general procedure A for the experimental details unless otherwise noted; isolated yields are reported. bRegiomeric ratio (r.r.) determined using 1H NMR spectroscopy; 3b13′, mono-Si (C-1):bis-Si (C-1 and C-6) = 1:6 (r.r.); 3e′, mono-Si product (C-2):bis-Si product (C-2 and C-4) = 1:3 (r.r.); 3p, mono-Si product (C-2):bis-Si product (C-2 and C-5) = 1.6:1 (r.r.); 3q′, mono-Si product (C-2):bis-Si product (C-2 and C-6) = 1:13 (r.r.). cThe thiol-mediated C–H silylation approach was employed; isolated yields are reported; please see general procedure B for the experimental details. dC-4 product:C-3 product = 2:1. | ||
Having demonstrated the utility of this protocol in the installation of the bulky t-butyldimethylsilyl group onto isoquinoline structures, we next examined other pharmaceutically relevant electron-deficient heteroarenes. The quinolines with substituted groups at the 2 or 4 positions worked smoothly, giving the mono-silylation products in high yields (3c–3d, 70–73%). However, the quinoline without any substituted groups also displayed poor regioselectivity accompanied with the formation of a significant amount of bis-silylation product (C-2 and C-4 positions) (3e′, 75% yield, 3e′:3e = 3:1). Phenanthridine, β-carboline, as well as the pyridine structures fused with five-membered nitrogen-containing heteroarenes, such as imidazole and 1,2,4-triazole, also proved to be effective reactants (3f–3i, 52–82% yields), while a poor result was observed for 6-azaindole (pyridine structure fused with pyrrole). Nonetheless, the thiol-mediated approach is compatible with this substrate, albeit with slightly decreased yield (3j, 58%). Notably, the silyl group was installed at the pyridine ring with excellent site selectivity (C-1 position), which provides a complementary C–H silylation approach to that of Grubbs's, where a silyl group is incorporated on the pyrrole ring of 6-azaindole (C-5 position).19 Widely used ligand molecules, including 2,2′-bipyridine and 1,10-phenanthroline, were also identified as suitable substrates for this protocol. The mono-silylation product at the C-1 position was observed for 2,2′-bipyridine (3k, 54% yield), while 1,10-phenanthroline is highly reactive, providing the bis-silylation product (3l′, C-1 and C-1′ positions, 62% yield). The thiol-mediated approach cannot improve the regioselectivity (data not shown here). In combination with diverse C–Si-based transformations, this protocol holds great potential for ligand modification. Various pyridine substrates also worked well to give the mono-silylation products at the most electrophilic sites (C-2, C-4, or C-6 position) (3m–3p, 40–72% yields). However, the introduction of a phenyl group at the C-4 position of pyridine simultaneously activates both C-2 and C-6 positions, thus mainly giving the bis-silylation product (3q′, 65% yield, 3q:3q′ = 1:13). The thiol-mediated approach cannot further improve this site selectivity either (data not shown here). The pyrimidine and pyrazine substrates were also amenable but with low yields (3r–3s, 39–43%). However, the formation of only trace products was observed for quinazoline, quinoxaline, benzothiazole, benzimidazole and benzoxazole structures (data not shown here), which might be attributed to the instability of the products under the reaction conditions.
Various trialkylsilanes were then examined to further extend the substrate scope. Notably, the large silyl groups, including di-tert-butylsilyl (tBu2SiH–) and triisopropylsilyl (iPr3Si–) were readily introduced at the C-1 position of 6-methyl-isoquinoline 1a in moderate to high yields and with excellent site selectivity (3t–3u, 51–77%). A limitation is also realized for the protocol. Only trace products were observed for small trialkylsilanes, such as triethylsilane and isopropyldimethylsilane. The thiol-mediated approach was not compatible with these small triethylsilanes either. This result might also be attributed to the instability of the resulting products under the reaction conditions (strong acid and oxidant) (Table S5, ESI†).
The observed regioselective installation of a silyl group onto the benzene ring of isoquinolines (C-6 position, 3b14–b15, Scheme 3) inspired us to explore the potential of our method in the C–H silylation of non-heteroaromatic substrates. Various substituted benzenes were screened (data not shown here). However, only 1,4-dicyanobenzene was identified as a suitable substrate, giving the mono-silylation product in high yield (5a, 71%, Scheme 4). The use of the thiol-mediated approach cannot further improve the reaction (5a, 65% yield, Scheme 4) accompanied by the use of more tBuMe2SiH (10 equiv.). Polycyano substitution was proved essential for this transformation. Good to high yields were observed (5d–5f, 50–74% yields, Scheme 4). It should be noted that cyano-substituted benzenes represent a class of frequently used coupling partners in reported photoredox reactions, where the cyano group is directly converted to other functional groups via substitution.34,35 However, the substitution of the cyano group with a silyl group was not observed in this process.
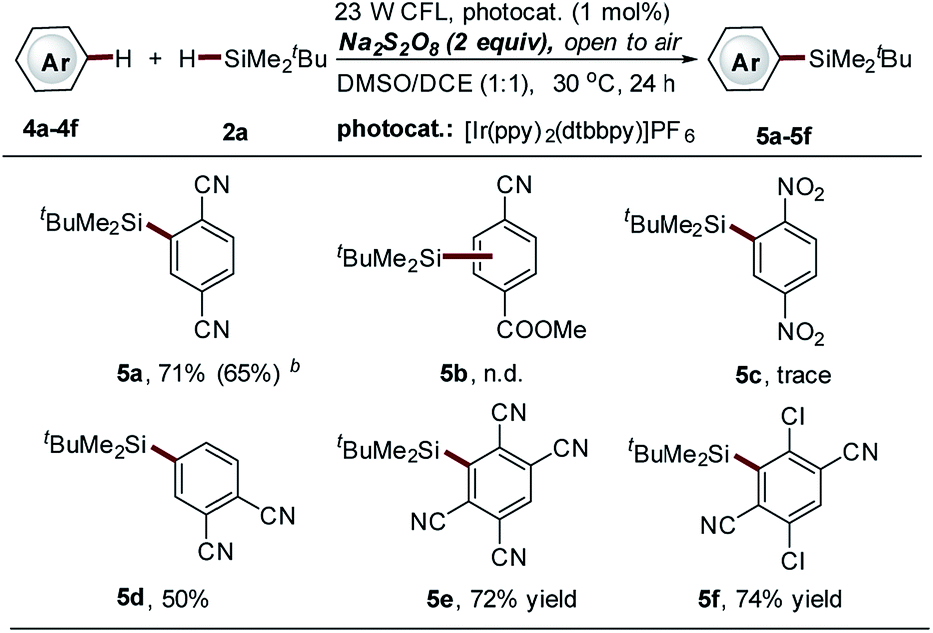 | ||
| Scheme 4 Direct C–H silylation of cyano-substituted benzenes via Na2S2O8-mediated photocatalytic Minisci-type reaction. aSee general procedure A for the experimental details, unless otherwise noted; isolated yields are reported. bThiol-mediated photocatalytic C–H silylation approach was employed; isolated yields are reported; see general procedure B for the experimental details. | ||
:3b13′ > 20:1, Scheme 5A). The same trend was also observed in the C–H silylation of various substituted isoquinoline substrates (Scheme S1†). Meanwhile, the excellent regioselectivity of this approach was further supported by the fact that the silyl group was mainly installed at the C-2 position of quinoline with moderate yield (3e, 66%, 3e:3e′ = 8:1, Scheme 5B). It should be noted that the poor site selectivity and the use of explosive peroxides represent two challenging issues of Minisci-type reactions.32,33 This thiol-mediated photocatalytic process provides a powerful alternative for the development of peroxide-free Minisci-type reactions with significantly improved regioselectivity.
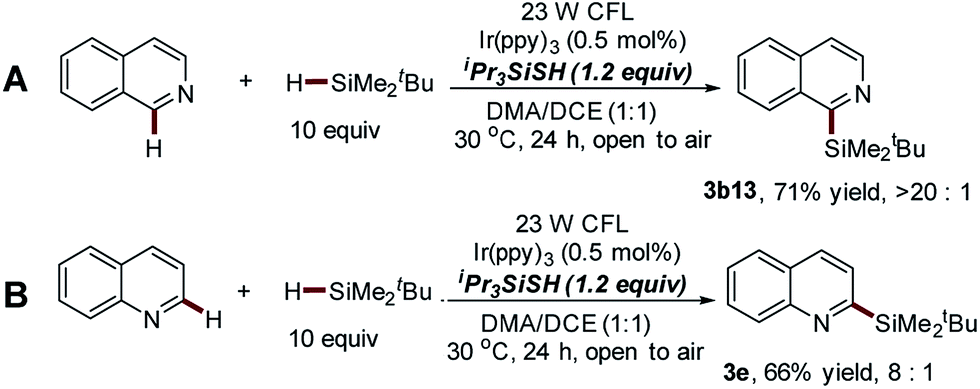 | ||
| Scheme 5 Direct C–H silylation of non-substituted isoquinoline and quinoline via thiol-mediated photocatalytic Minisci-type reaction. | ||
Mechanistic studies
To understand the mechanistic nature of the Na2S2O8-mediated photocatalytic Minisci-type C–H silylation reaction, we firstly performed a study to probe and verify the radical engaged process. The formation of silanol and siloxane structures was observed by GC-MS analysis (Fig. S1 and Scheme S2†). This fact and the radical trapping experiments (Scheme S3†) suggest the formation of a silyl radical in the process. Inferior reaction efficiency was observed in the control reactions employing bis(tert-butyldimethylsilyl)-peroxide (tBuMe2SiOOSiMe2tBu) as the radical initiator (43–45% yields, Scheme S4B and 4C†), which might be in situ generated in the process. Considering the fact that 2 equiv. of Na2S2O8 was typically required for effective transformation, it is reasonable to conclude that the sulfate radical anion (generated from the photocatalytic decomposition of Na2S2O8) might function as the major driving force to perform HAT of t-butyldimethylsilane owing to the polar effect.34 Based on these studies and the emission quenching experiment (Fig. S2†), as well as the established mechanism of photocatalytic Minisci-type reaction,29,31 a plausible reaction mechanism is proposed (Scheme 6A). The photocatalytic decomposition of Na2S2O8 produces a sulfate radical anion, which promotes the formation of a t-butyldimethylsilyl radical via HAT. The resulting silyl radical undergoes nucleophilic addition with the protonated 6-methylisoquinoline at the C-1 position, followed by deprotonation and oxidation to provide the desired product.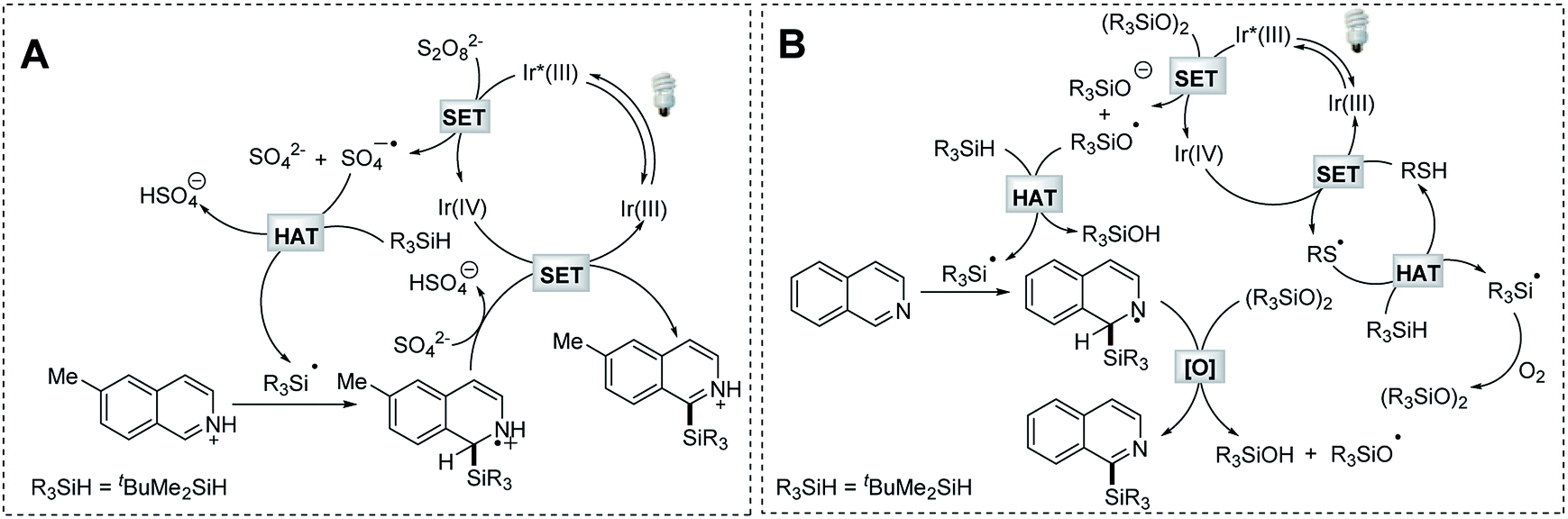 | ||
| Scheme 6 Proposed mechanisms. | ||
The formation of a silyl radical was also validated in the thiol-mediated photocatalytic Minisci-type C–H silylation reaction through radical trapping and GC-MS experiments (Fig. S3, Scheme S5†). Furthermore, both oxygen and t-butyldimethylsilane proved essential for this transformation (Scheme S6B and S6C†). The replacement of thiol with tBuMe2SiOOSiMe2tBu, which might be in situ generated in this process, provided the product in a comparable yield (62%, Scheme S6D†). Considering the fact that peroxides are typically required in Minisci-type reactions32,33 and oxygen-centered radical species are able to perform HAT more efficiently,36 as well as the emission quenching experiment (Fig. S4†), a mechanism employing this newly formed organic peroxide as the radical initiator was proposed (Scheme 6B). The sulfur radical generated from the photoredox process promotes the formation of the organic peroxide tBuMe2SiOOSiMe2tBu, which provides the siloxyl radical via photocatalytic decomposition. This oxygen-centered radical serves as an efficient mediator for HAT of t-butyldimethylsilane. The resulting silyl radical undergoes nucleophilic addition with isoquinoline at the C-1 position, followed by oxidation to give the desired product.
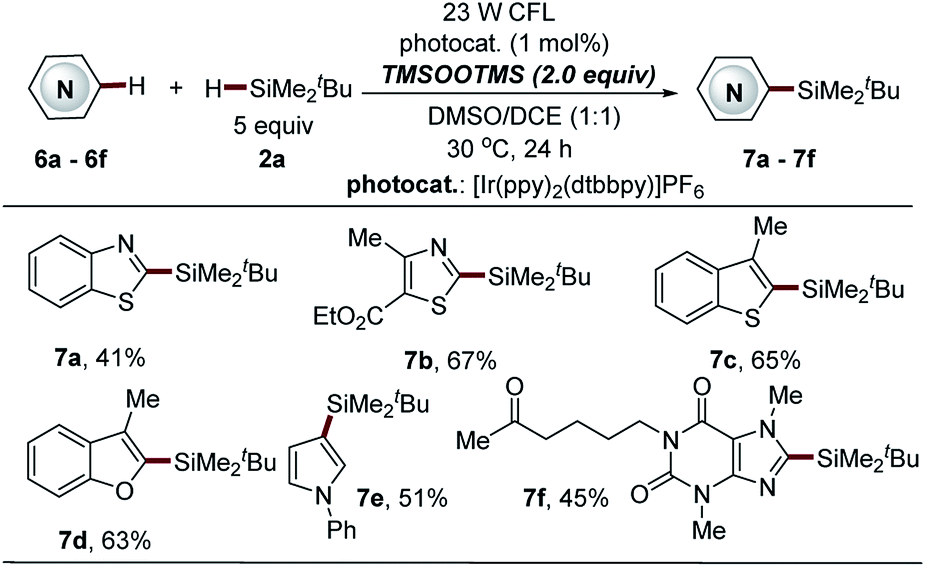 | ||
| Scheme 7 Direct C–H silylation of heteroarenes via BTMSPO-mediated photocatalytic Minisci-type reaction. aSee general procedure C for the experimental details in the ESI,† unless otherwise noted; isolated yields are reported. | ||
Bis(trimethylsilyl)-peroxide (BTMSPO), which is safe and highly soluble in most organic solvents, is widely used in various oxidation reactions in place of hydrogen peroxide (H2O2).37–40 The investigation of the reaction mechanism triggered us to further explore the feasibility of the use of BTMSPO as a radical initiator in photocatalytic sila-Minisci-type C–H silylation. Surprisingly, electron-deficient heteroarenes, such as benzothiazole, thiazole and a naturally occurred imidazole structure (pentoxifylline), as well as the electron-rich heteroarenes (benzofuran, benzothiophene and pyrrole structures) were demonstrated as suitable substrates (7a–7f, 41–67% yields, Scheme 7). It is of note that these heteroarenes are not amenable to both Na2S2O8- and thiol-mediated reactions. However, this method displayed inferior reaction efficiency in the C–H silylation of isoquinoline, quinoline and pyridine structures (data not shown here). The small trialkylsilanes were also not amenable to this protocol. Further investigation of this promising C–H silylation approach is still ongoing in our laboratory.
Synthetic applications
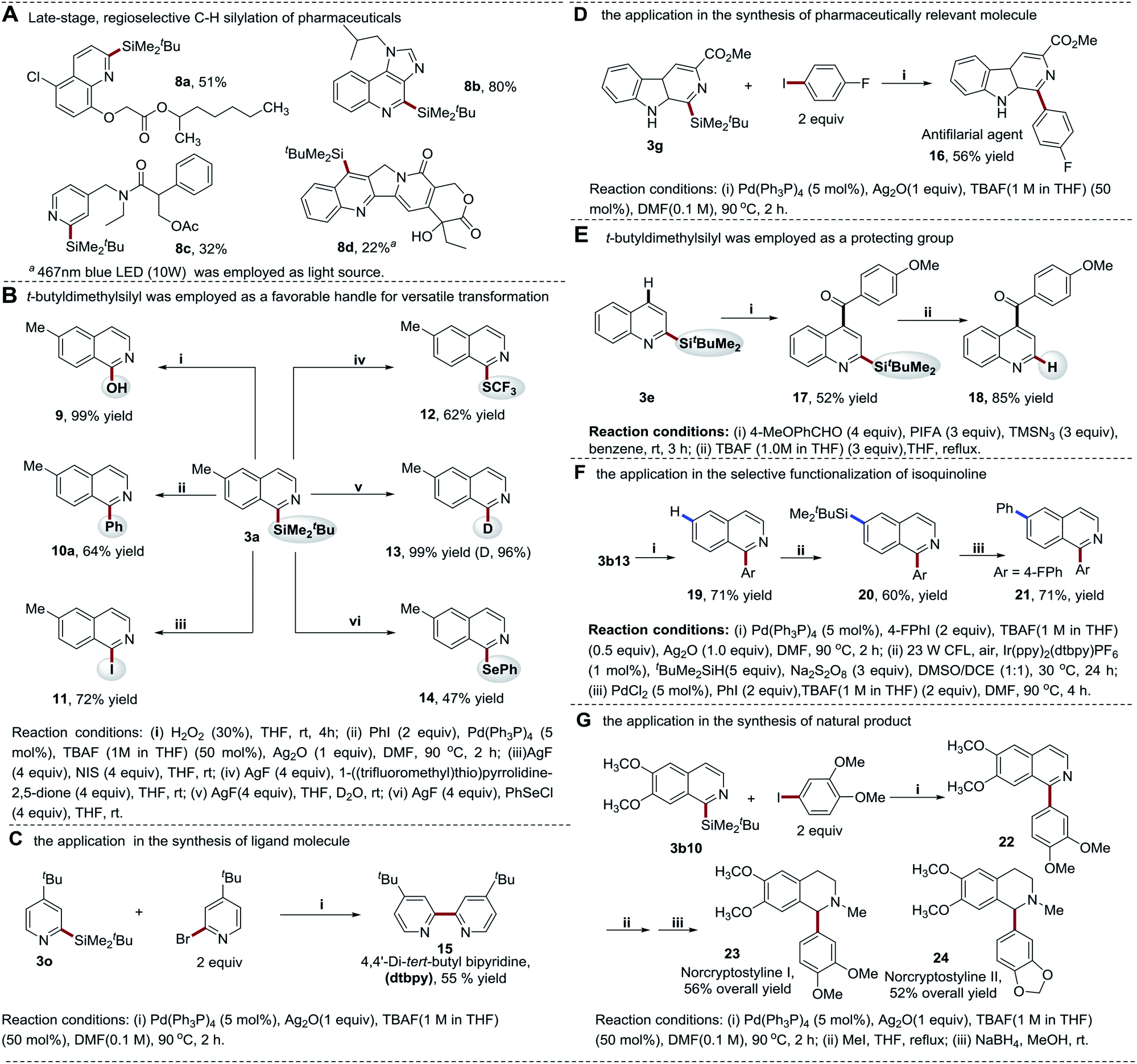 | ||
| Scheme 8 Synthetic applications. | ||
In addition to the cross-coupling reactions, the silyl group can also be converted to other functionalities. With silver fluoride (AgF) as an activating reagent, the silyl moiety was smoothly converted to iodine (I), thiotrifluoromethyl (SCF3), phenylselanyl (SePh), as well as deuterium (D) at the C-1 position of isoquinoline in 47–99% yields via nucleophilic substitutions (Scheme 8B). This provides a powerful tool to facilely access the heteroarenes with diverse substituted groups, enabling the quick study of the structure–activity relationship (SAR) of bioactive molecules. Furthermore, the excellent site selectivity of thiol-mediated photocatalytic C–H silylation approach enables the selective manipulation of different C–H bonds of heteroarenes in combination with the new coupling technology (Scheme 8E and F). It is of note that fewer reaction steps and higher yields enable the quick diversification of quinoline and isoquinoline structures. To further demonstrate the utility of our strategy, a new synthetic route towards the synthesis of benzylisoquinoline alkaloids norcryptostyline I–II was developed (Scheme 8G). These two naturally occurring target compounds were readily assembled via a modular synthetic approach in 52–56% yields (overall yields of three steps), which provides an alternative efficient method for the synthesis of this class of natural products.46,47
Conclusions
In conclusion, motivated by the lack of efficient methods for the direct coupling of the bulky and inert trialkylhydrosilanes with unfunctionalized heteroarenes, especially the medicinally valuable electron-deficient heteroarenes, we have developed a novel photocatalytic Minisci-type C–H silylation approach employing Na2S2O8, BTMSPO, or an alternative safe iPr3SiSH as the radical initiators. While Na2S2O8 and iPr3SiSH serve as efficient promoters for the C–H silylation of electron-deficient heteroarenes, BTMSPO was proved effective for both electron-deficient and electron-rich heteroarenes as a more promising radical initiator. This method employs visible light as the energy source under mild reaction conditions and features broad substrate scope and operational simplicity, allowing the synthesis of structurally diverse heteroaryltrialkylsilanes in moderate to high yields and with good regioselectivity. It is of note that the thio-mediated reaction displays superior regioselectivity and cyano-substituted arenes were also amenable to this method. Furthermore, the silylation products with a large tBuMe2Si group show great synthetic versatility in diverse C–Si-based chemical transformations. To the best of our knowledge, the employment of this silyl functionality in cross-coupling reactions has not been reported before. Moreover, the applications of our method are further demonstrated in the synthesis of natural products and other complex molecules, as well as the late-stage functionalization. Further studies of this powerful radical-based chemistry for new C–Si bond formation are being pursued in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this research from the program of the National Natural Science Foundation of China (21738002 and 21572055, W. W. and 21602060 and 21871086, Y.-Q. Z.) is gratefully acknowledged. Acknowledgement is also made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this research (PRF#: 57164-ND1).Notes and references
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779 CrossRef CAS PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- Y.-M. Hu, Y.-X. Wang, F. Li, C.-L. Ma and J. Wang, Eur. J. Med. Chem., 2017, 135, 70 CrossRef CAS PubMed.
- W. Du, B. Kaskar, P. Blumbergs, P. K. Subramanian and D. P. Curran, Bioorg. Med. Chem., 2003, 11, 451 CrossRef CAS PubMed.
- Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC.
- H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC.
- T. Komiyama, Y. Minami and T. Hiyama, Angew. Chem., Int. Ed., 2016, 55, 15787 CrossRef CAS PubMed.
- N. V. Wolff, J. Char, X. Frogneux and T. Cantat, Angew. Chem., Int. Ed., 2017, 56, 5616 CrossRef PubMed.
- X. Frogneux, N. V. Wolff, P. Thuery, G. Lefevre and T. Cantat, Chem.–Eur. J., 2016, 22, 2930 CrossRef CAS PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 Search PubMed.
- C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592 CrossRef CAS PubMed.
- K. Devaraj, C. Sollert, C. Juds, P. J. Gates and L. T. Pilarski, Chem. Commun., 2016, 52, 5868 RSC.
- B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508 CrossRef CAS PubMed.
- H. Fang, L. Guo, Y. Zhang, W. Yao and Z. Huang, Org. Lett., 2016, 18, 5624 CrossRef CAS PubMed.
- Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868 CrossRef CAS PubMed.
- H. F. Klare, M. Oestreich, J. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312 CrossRef CAS PubMed.
- S. Wubbolt and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 15876 CrossRef PubMed.
- S. Bahr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52 CrossRef PubMed.
- A. A. Toutov, W. B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80 CrossRef CAS PubMed.
- S. Banerjee, Y.-F. Yang, I. D. Jenkins, Y. Liang, A. A. Toutov, W.-B. Liu, D. P. Schuman, R. H. Grubbs, B. M. Stoltz, E. H. Krenske, K. N. Houk and R. N. Zare, J. Am. Chem. Soc., 2017, 139, 6880 CrossRef CAS PubMed.
- A. A. Toutov, W. B. Liu, K. N. Betz, B. M. Stoltz and R. H. Grubbs, Nat. Protoc., 2015, 10, 1897 CrossRef CAS PubMed.
- The silyl anion engaged C-H silylations were also reported: A. Postigo and R. A. Rossi, Org. Lett., 2001, 3, 1197 CrossRef CAS PubMed and also see ref. 23.
- Y.-T. Gu, Y.-Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127 CrossRef CAS PubMed.
- Z. Xu, L. Chai and Z.-Q. Liu, Org. Lett., 2017, 19, 5573 CrossRef CAS PubMed.
- J. Gu and C. Cai, Chem. Commun., 2016, 52, 10779 RSC.
- R. Sakamoto, B.-N. Nguyen and K. Maruoka, Asian J. Org. Chem., 2018, 7, 1085 CrossRef CAS.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed.
- C. P. Jasperse, D. P. Curran and T. L. Fevig, Chem. Rev., 1991, 91, 1237 CrossRef CAS.
- J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed.
- Y.-Q. Zhang, K. B. Teuscher and H. Ji, Chem. Sci., 2016, 7, 2111 RSC.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135 RSC.
- J. Tauber, D. Imbri and T. Opatz, Molecules, 2014, 19, 16190 CrossRef PubMed.
- Z.-W. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef CAS PubMed.
- A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114 CrossRef CAS PubMed.
- P. B. Roberts, Chem. Soc. Rev., 1999, 28, 25 RSC.
- C. W. Jefford, A. Jaber and J. Boukouvalas, Synthesis, 1988, 5, 391 CrossRef.
- G. A. Olah and T. D. Ernst, J. Org. Chem., 1989, 54, 1204 CrossRef CAS.
- A. Casarinia, P. Dembech, G. Reginato, A. Ricci and G. Seconi, Tetrahedron Lett., 1991, 32, 2169 CrossRef.
- R. Sakamoto, S. Sakurai and K. Maruoka, Chem. Commun., 2017, 53, 6484 RSC.
- P. Pierrat, P. Gros and Y. Fort, Org. Lett., 2005, 7, 697 CrossRef CAS PubMed.
- S. Napier, S. M. Marcuccio, H. Tye and M. Whittaker, Tetrahedron Lett., 2008, 49, 6314 CrossRef CAS.
- F. Louerat, H. Tye, S. Napier, M. Garrigou, M. Whittaker and P. C. Gros, Org. Biomol. Chem., 2011, 9, 1768 RSC.
- G. R. Jones and Y. Landais, Tetrahedron, 1996, 52, 7599 CrossRef CAS.
- S. K. Srivastava, A. Agarwal, P. M. S. Chauhan, S. K. Agarwal, A. P. Bhaduri, S. N. Singh, N. Fatima and R. K. Chatterjee, J. Med. Chem., 1999, 42, 1667 CrossRef CAS PubMed.
- F. Crestey, A. A. Jensen, M. Borch, J. T. Andreasen, J. Andersen, T. Balle and J. L. Kristensen, J. Med. Chem., 2013, 56, 9673 CrossRef CAS PubMed.
- R. Zhu, Z. Xu, W. Ding, S. Liu, X. Shi and X. Lu, Chin. J. Chem., 2014, 32, 1039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S6, Scheme S1–S8, Fig. S1–S4, experimental procedures and characterization for all of the new compounds. See DOI: 10.1039/c9sc00046a |
| This journal is © The Royal Society of Chemistry 2019 |