
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroxy group-enabled highly regio- and stereo-selective hydrocarboxylation of alkynes†
Chaofan
Huang
a,
Hui
Qian
a,
Wanli
Zhang
*a and
Shengming
Ma
 *ab
*ab
aResearch Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P. R. China. E-mail: dxdzwl@sina.com
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
First published on 17th April 2019
Abstract
Here we present an example of utilizing hydroxy groups for regioselectivity control in the addition reaction of alkynes—a highly efficient Pd-catalyzed syn-hydrocarboxylation of readily available 2-alkynylic alcohols with CO in the presence of alcohols with an unprecedented regioselectivity affording 3-hydroxy-2(E)-alkenoates. The role of the hydroxy group has been carefully studied. The synthetic potential of the products has also been demonstrated.
Introduction
As one of the most abundant and fundamental chemical feedstock, alkynes are widely applied in biochemistry, materials sciences, pharmacology, and medicine.1 Among many reactions, their addition reactions with another molecule, X–Y, perfectly suit the demand for green chemistry due to the 100% atom economy, thus leading to tremendous interest in this area due to the high importance of stereo-defined olefins.2 However, regioselectivity is the issue when it comes to non-symmetric alkynes (Scheme 1a). Electronic and steric effects help in solving this type of problem (Scheme 1b and c).3 Using a pre-installed directing group, such as carbonates, pyridyl groups, amides, alkenes, etc., is another feasible way to control regioselectivity through coordination with metal catalysts (Scheme 1d).4,5 As we know hydrogen bonding interactions have been widely used in organocatalysis,6 and recent publications also demonstrate their capacity in regioselective addition reactions.7 Herein, we report our recent observation on hydroxy group-enabled regioselectivity control in highly stereoselective hydrocarboxylation of readily available 2-alkynylic alcohols affording highly functionalized 3-hydroxy-2(E)-alkenoates (Scheme 1e).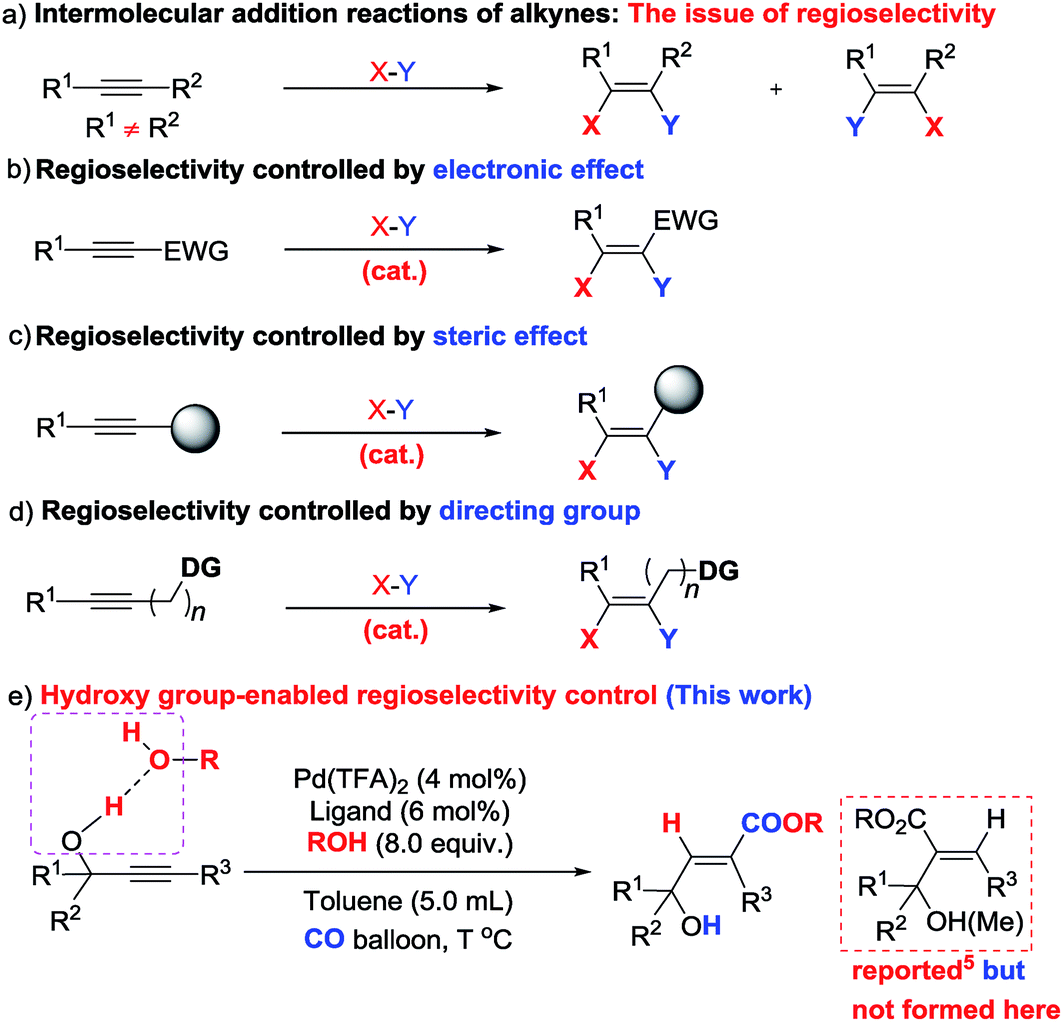 | ||
| Scheme 1 Addition reactions of alkynes—approaches for regioselectivity control (only syn-additions are shown for clarity). | ||
Results and discussion
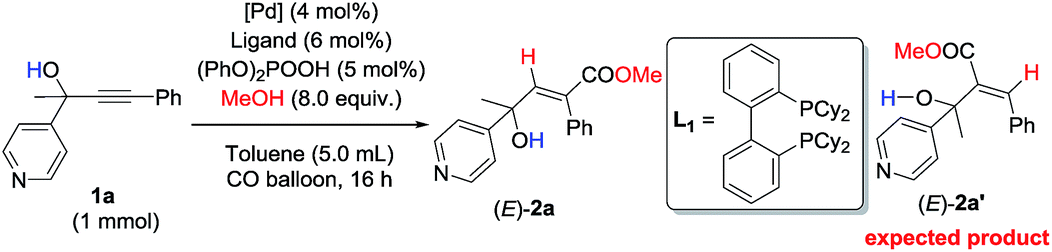
Initially, 1-phenyl-2-(pyridin-4-yl)but-3-yn-2-ol (1a) was treated with 8 equiv. of MeOH in the presence of 2 mol% [PdCl(π-allyl)]2, 6 mol% DPEphos, and 5 mol% (PhO)2POOH with a CO balloon. Surprisingly, the expected syn-hydrocarboxylation product (E)-2a′5 was not detected, while 66% of its regioisomeric product (E)-2a was exclusively formed unexpectedly together with 25% recovery of 1a (Table 1, entry 1). The regio- and stereo-selectivity were further established by single-crystal X-ray diffraction analysis of (E)-2a (Fig. 1). Various palladium catalysts were then screened with no obvious improvement except for Pd(TFA)2, which afforded 82% yield of (E)-2a (Table 1, entries 2–4). Pd(0) pre-catalysts were also examined, affording (E)-2a with 57–82% yields (Table 1, entries 5–7). Among all the ligands examined, DPEphos was still the best (Table 1, entries 8–10). When the reaction was conducted at 60 °C, the yield of (E)-2a was improved to 90% (Table 1, entry 11). Besides, the reaction could also occur efficiently without the help of (PhO)2POOH (Table 1, entry 13).8|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | T/°C | (E)-2aa (%) | Recoverya (%) |
| a Yield and recovery were determined by 1H-NMR analysis using CH2Br2 as the internal standard. b The reaction was carried out without (PhO)2POOH. | |||||
| 1 | [PdCl(π-allyl)]2 | DPEphos | 80 | 66 | 25 |
| 2 | Pd(PPh3)2Cl2 | DPEphos | 80 | 0 | 100 |
| 3 | Pd(TFA)2 | DPEphos | 80 | 82 | 6 |
| 4 | Pd(OAc)2 | DPEphos | 80 | 70 | 4 |
| 5 | Pd2(dba)3 | DPEphos | 80 | 57 | 34 |
| 6 | Pd(t-Bu2-PPh)2 | DPEphos | 80 | 78 | 12 |
| 7 | Pd(PPh3)4 | DPEphos | 80 | 82 | 15 |
| 8 | Pd(TFA)2 | BINAP | 80 | 26 | 67 |
| 9 | Pd(TFA)2 | DPPB | 80 | 33 | 43 |
| 10 | Pd(TFA)2 | L1 | 80 | 32 | 69 |
| 11 | Pd(TFA)2 | DPEphos | 60 | 90 | 0 |
| 12 | Pd(TFA)2 | DPEphos | 50 | 45 | 53 |
| 13b | Pd(TFA)2 | DPEphos | 60 | 89 | 0 |
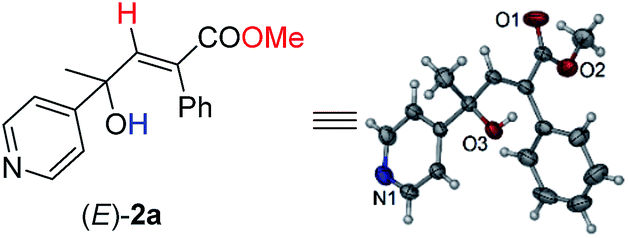 | ||
| Fig. 1 ORTEP representation of (E)-2a. | ||
With the optimized conditions in hand and the importance of such 2-alkenoates, we set out to explore the scope of this reaction (Table 2). To our delight, this highly regioselective syn-hydrocarboxylation reaction delivered 3-hydroxy-2(E)-alkenoates as the sole product for various 2-alkynylic alcohols. Substitution of the pyridine ring at different positions made no difference (Table 2, entries 1–3). Quinolinyl-containing substrates were also compatible, efficiently furnishing (E)-2d in 71% yield (Table 2, entry 4). An electron-rich 3-aryl-substituted 2-alkynylic alcohol gave a higher yield (Table 2, entry 5). Compared with aromatic groups, 3-alkyl-substituted substrates 1f and 1g were less reactive and required a higher catalyst loading and temperature (Table 2, entries 6 and 7). Notably, the reaction could also be executed with more sterically hindered 2-alkynylic alcohols, delivering (E)-2h and (E)-2i in good yields (Table 2, entries 8 and 9). Interestingly, reaction with a 2-alkynylic alcohol with a p-nitrophenyl group instead of a pyridyl group also proceeded smoothly to give (E)-2j in 58% yield (Table 2, entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | T/°C | (E)-2 yielda/% |
| a Isolated yield. b With 6 mol% Pd(TFA)2 and 9 mol% DPEphos. c With 6 mol% Pd(TFA)2, 9 mol% DPEphos, and 6.0 mmol of MeOH for 24 h. d With 4 mol% Pd(TFA)2, 8 mol% DPEphos, and 4.0 mmol of MeOH. | |||||
| 1 | Me | 4-pyridyl | Ph (1a) | 60 | 82 (2a) |
| 2 | Me | 3-Pyridyl | Ph (1b) | 60 | 80 (2b) |
| 3 | Me | 2-Pyridyl | Ph (1c) | 60 | 62 (2c) |
| 4 | Me | 3-Quinolinyl | Ph (1d) | 60 | 71 (2d) |
| 5 | Me | 4-Pyridyl | 4-MeC6H4 (1e) | 60 | 89 (2e) |
| 6b | Me | 4-Pyridyl | n-Bu (1f) | 70 | 86 (2f) |
| 7b | Me | 4-Pyridyl | n-C8H17 (1g) | 70 | 81 (2g) |
| 8 | Et | 4-Pyridyl | Ph (1h) | 60 | 88 (2h) |
| 9c | Ph | 4-Pyridyl | n-Bu (1i) | 75 | 81 (2i) |
| 10d | Me | 4-O2NC6H4 | n-C6H13 (1j) | 70 | 58 (2j) |
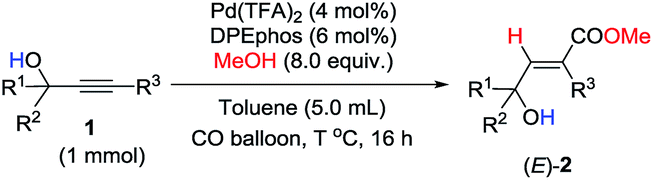
Unfortunately, this set of reaction conditions did not work very efficiently for 2-alkynylic alcohols with R1 and R2 both being alkyl groups—the reaction of 1k resulted in the formation of the desired (E)-2k in only 35% yield (Table 3, entry 1). Lowering the temperature increased the yield up to 49% (Table 3, entry 2). Then, the ligand effect was re-investigated to address this issue. As shown in Table 3, mono-phosphine ligands were not efficient for the hydrocarboxylation (Table 3, entries 3 and 4). It is also worth noting that the efficiency strongly depends on the electronic properties and the backbone structure of the bisphosphine ligands. Compared to 2,2′-bis(dicyclohexyl-phosphino)-1,1′-biphenyl (L1), more rigid or more flexible backbone structures both made the reaction slower (Table 3, entries 5–7). Furthermore, a relatively electron-deficient ligand, BIPHEP, gave only 5% yield of the product with 95% recovery of 1k (Table 3, entry 8). Finally, when the reaction was carried out with 4 equiv. of MeOH at 60 °C, (E)-2k could be obtained with the highest yield (Table 3, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T/°C | (E)-2ka (%) | Recoverya (%) |
| a Yield and recovery were determined by 1H-NMR analysis using CH2Br2 as the internal standard, and the isolated yield is shown in parentheses. b The reaction was carried out on a 1 mmol scale with 5 mL of toluene. c With 12 mol% mono-phosphine ligands. d With 4.0 equiv. of MeOH for 24 h. | |||||
| 1b | DPEphos | Toluene | 60 | 35 | 0 |
| 2b | DPEphos | Toluene | 50 | 49 | 0 |
| 3c | Zheda-phos | Toluene | 50 | 1 | 93 |
| 4c | Sphos | Toluene | 50 | 11 | 88 |
| 5 | BINAP | Toluene | 50 | 40 | 44 |
| 6 | DPPB | Toluene | 50 | 17 | 66 |
| 7 | L1 | Toluene | 50 | 58 | 42 |
| 8 | BIPHEP | Toluene | 50 | 5 | 95 |
| 9b,d | L1 | Toluene | 60 | 99 (95) | 0 |
| 10b,d | L1 | THF | 60 | 34 | 66 |
| 11b,d | L1 | 1,2-DCE | 60 | 30 | 70 |
| 12b,d | L1 | CH3CN | 60 | 8 | 92 |
| 13b,d | L1 | DMF | 60 | 5 | 90 |
| 14b,d | L1 | DMSO | 60 | — | 98 |

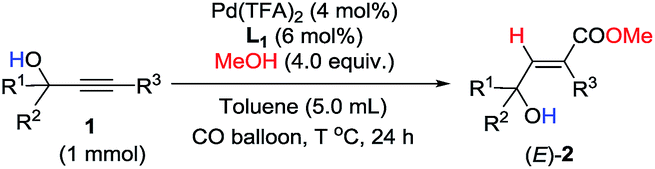
Under this set of new optimal reaction conditions, more examples of 2-alkynylic alcohols were examined. As shown in Table 4, R2 and R3 are both compatible with an alkyl or aryl group (Table 4, entries 1–9). The structure of (E)-2q was further confirmed by its single crystal X-ray diffraction analysis (Fig. 2). In addition, a 2-alkynylic alcohol with a four-membered cyclo-butyl ring also survived affording (E)-2s in 87% yield (Table 4, entry 10). Reaction with more sterically hindered 1,1-diphenylhept-2-yn-1-ol proceeded smoothly to give (E)-2t in 86% yield and 9% recovery of 1t (Table 4, entry 11). It is noteworthy that secondary 2-alkynylic alcohols also afforded the target products in good to excellent yields with the same regio- and stereo-selectivity (Table 4, entries 12–15). Interestingly, even the C–Br bond could survive in this reaction (Table 4, entries 2, 3, and 15). The reaction could be easily executed on a gram scale, delivering (E)-2l in 89% yield (Table 4, entry 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | T/°C | (E)-2 yielda/% |
| a Isolated yield. b The reaction was carried out on a 4 mmol scale. c With 5 mol% Pd(TFA)2, 10 mol% L1, and 5.0 mmol of MeOH. d Reaction time 48 h; 19% recovery of 1m was detected. e With 6 mol% Pd(TFA)2, 12 mol% L1, and 5.0 mmol of MeOH. f Reaction time 48 h; 27% recovery of 1n was detected. g 36% recovery of 1o was detected. h Reaction time 48 h; 34% recovery of 1p was detected. i Reaction time 48 h; 21% recovery of 1r was detected. j Reaction time 32 h; 9% recovery of 1t was detected. | |||||
| 1 | Me | n-C5H11 | Ph (1k) | 60 | 95 (2k) |
| 2 | Me | n-C5H11 | 4-BrC6H4 (1l) | 60 | 92 (2l) |
| 3b | Me | n-C5H11 | 4-BrC6H4 (1l) | 60 | 89 (2l) |
| 4c,d | Me | n-C5H11 | n-Bu (1m) | 80 | 66 (2m) |
| 5e,f | Me | n-Pr | n-C8H17 (1n) | 75 | 71 (2n) |
| 6c,g | Me | (CH2)2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
n-Pr (1o) | 75 | 41 (2o) |
| 7e,h | Me | n-Pr | (CH2)4Cl (1p) | 75 | 60 (2p) |
| 8 | Me | Ph | Ph (1q) | 70 | 93 (2q) |
| 9c,i | Me | Ph | n-Bu (1r) | 75 | 63 (2r) |
| 10 | –(CH2)3– | Ph (1s) | 25 | 87 (2s) | |
| 11c,j | Ph | Ph | n-Bu (1t) | 80 | 86 (2t) |
| 12 | H | n-C11H23 | Ph (1u) | 60 | 93 (2u) |
| 13c | H | Ph | Ph (1v) | 70 | 69 (2v) |
| 14c | H | Ph | 4-MeOC6H4 (1w) | 70 | 69 (2w) |
| 15 | H | n-C11H23 | 4-BrC6H4 (1x) | 60 | 87 (2x) |
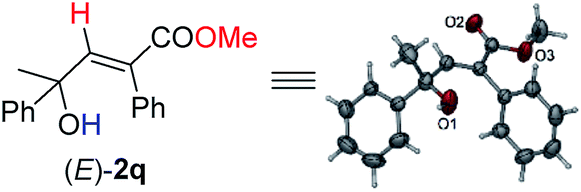 | ||
| Fig. 2 X-ray crystal structure of (E)-2q. | ||
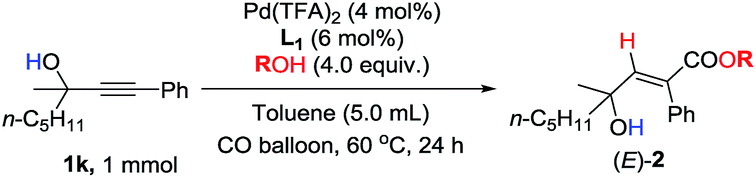
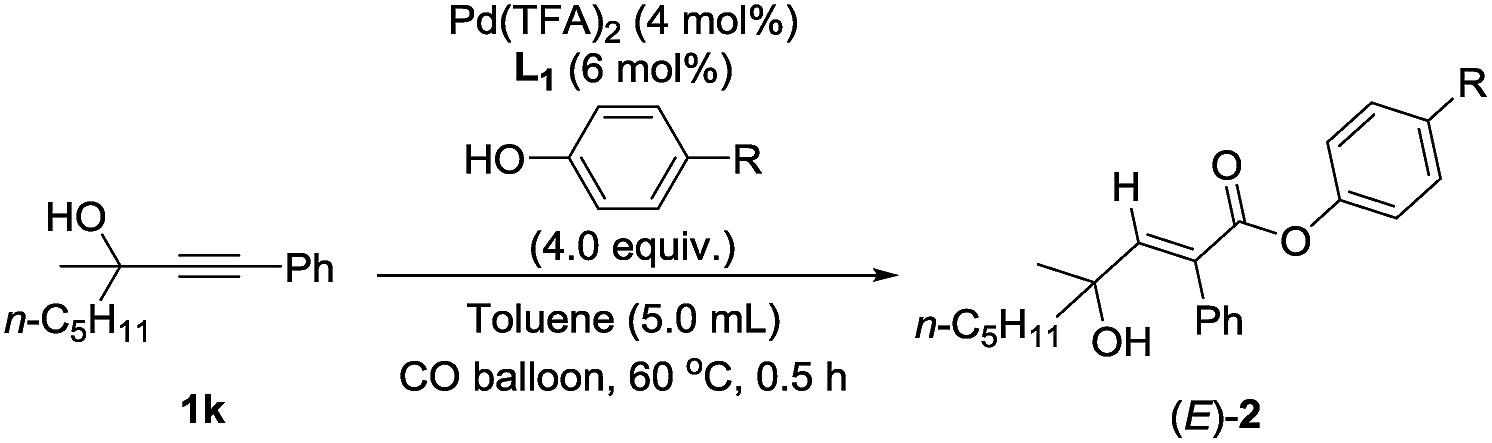
In addition to methanol, some other alcohols were also examined. Ethanol and TMSCH2OH work well to obtain the target products in 95–97% yield (Table 5, entries 1 and 2). Sterically hindered i-PrOH is also tolerated with 76% yield (Table 5, entry 3). Phenol behaves worse, and only 30% yield was detected (Table 5, entry 4).9
|
|
||
|---|---|---|
| Entry | ROH | Yield of (E)-2a/% |
| a Isolated yield. b 24% recovery of 1k was determined by 1H-NMR analysis using CH2Br2 as the internal standard. c 70% recovery of 1k was determined by 1H-NMR analysis using CH2Br2 as the internal standard. | ||
| 1 | EtOH | 95 (2ka) |
| 2 | TMSCH2OH | 97 (2kb) |
| 3b | i-PrOH | 76 (2kc) |
| 4c | PhOH | 30 (2kd) |
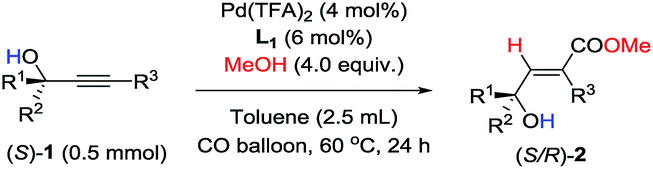
Furthermore, as shown in Table 6, racemization of the chiral center in substrates (S)-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 was not observed—the reaction of optically active propargylic alcohols afforded optically active 3-hydroxy-2(E)-alkenoates with excellent ee values and high yields.
10 was not observed—the reaction of optically active propargylic alcohols afforded optically active 3-hydroxy-2(E)-alkenoates with excellent ee values and high yields.
|
|
|||||
|---|---|---|---|---|---|
| Entry | (S)-1 | 2 | |||
| R1 | R2 | R3 | Yield/% | ee/% | |
| a Isolated yield; ee values were determined by chiral HPLC analysis. b The reaction was carried out at 70 °C. c With 4 mol% Pd(TFA)2 and 8 mol% L1. d With 5 mol% Pd(TFA)2, 10 mol% L1, and 5.0 equiv. of MeOH. | |||||
| 1b,c | H | n-C11H23 | Ph (1u, 98) | 90 (2u) | 99 |
| 2 | H | n-C11H23 | 4-BrPh (1x, >99) | 86 (2x) | 99 |
| 3b,d | H | Ph | Ph (1v, >99) | 68 (2v) | 99 |
| 4b,d | H | Ph | 4-MeOPh (1w, >99) | 70 (2w) | 99 |
| 5b | Me | n-C5H11 | Ph (1k, 97) | 89 (2k) | 96 |
As we know that 2-alkenoates are important intermediates in organic synthesis, their synthetic potential has been further demonstrated for the synthesis of different stereo-defined functionalized olefins. Owing to the presence of the C–Br bond in (E)-2l, Suzuki coupling reactions could easily afford (E)-7 in 80% yield.11 The ester unit could be hydrolyzed with KOH at 50 °C for 2 hours to afford the corresponding acid (E)-8 in 80% yield,12 or reduced with DIBAL-H at −78 °C delivering the corresponding 1,4-diol (E)-9 in 80% yield.13 Fluorination of the hydroxyl group could also be easily conducted with DAST to furnish (E)-10 in 94% yield14 (Scheme 2).
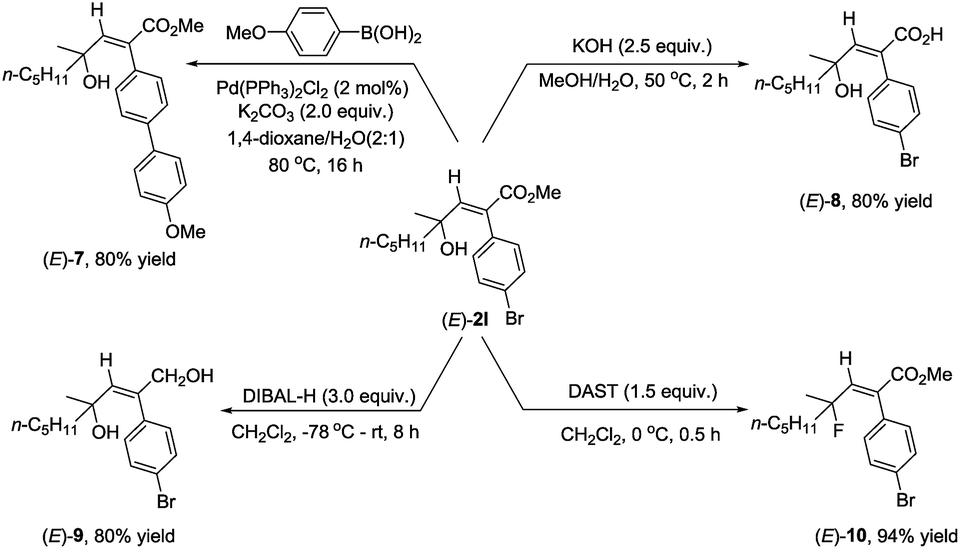 | ||
| Scheme 2 Synthetic applications of (E)-2l. | ||
To gain insight into the reaction mechanism and the effect of the hydroxyl group, a couple of control experiments were conducted (Scheme 3). No desired hydrocarboxylation products were obtained when propargylic methyl ether 3, acetate 4, or internal alkyne 5 was utilized (Scheme 3a and b), indicating that the hydrogen bonding originating from the free hydroxyl groups in propargylic alcohol and methanol might have played a critical role in this transformation. Isotopic labeling studies reinforce the notion that methanol was the hydrogen donor (Scheme 3c). We reasoned that the low D incorporation was caused by adventitious water in the reaction mixture. Furthermore, the 1H NMR signals of 1-phenyl-3-methyloctyn-3-ol 1k were measured with respect to different amounts of MeOH and 1k: an obvious shift of the hydroxy signal in 1k and MeOH was observed, indicating hydrogen bonding between the two hydroxyl groups (Fig. 3).
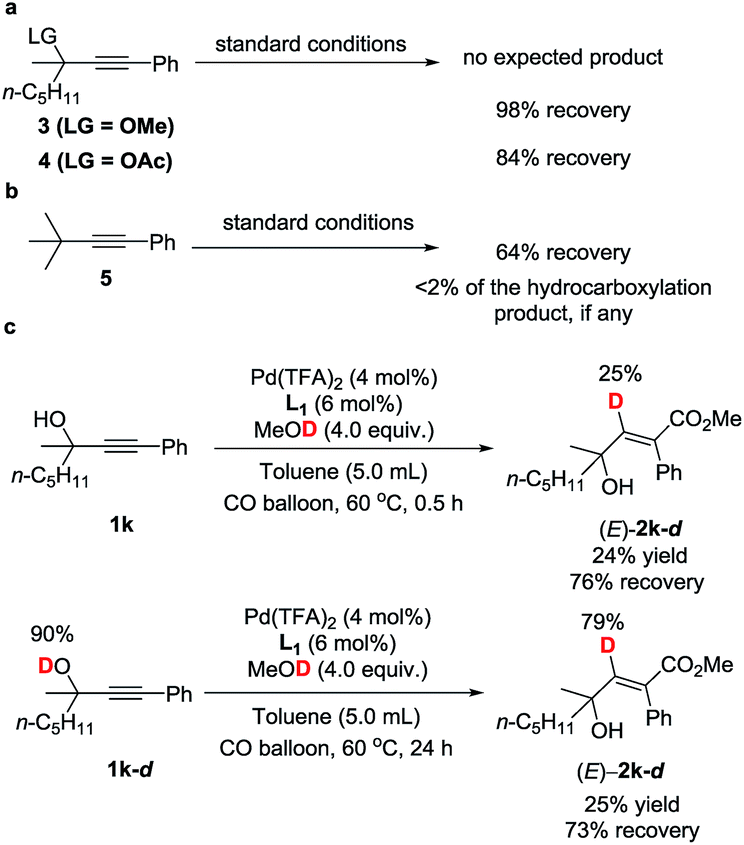 | ||
| Scheme 3 Mechanistic studies. | ||
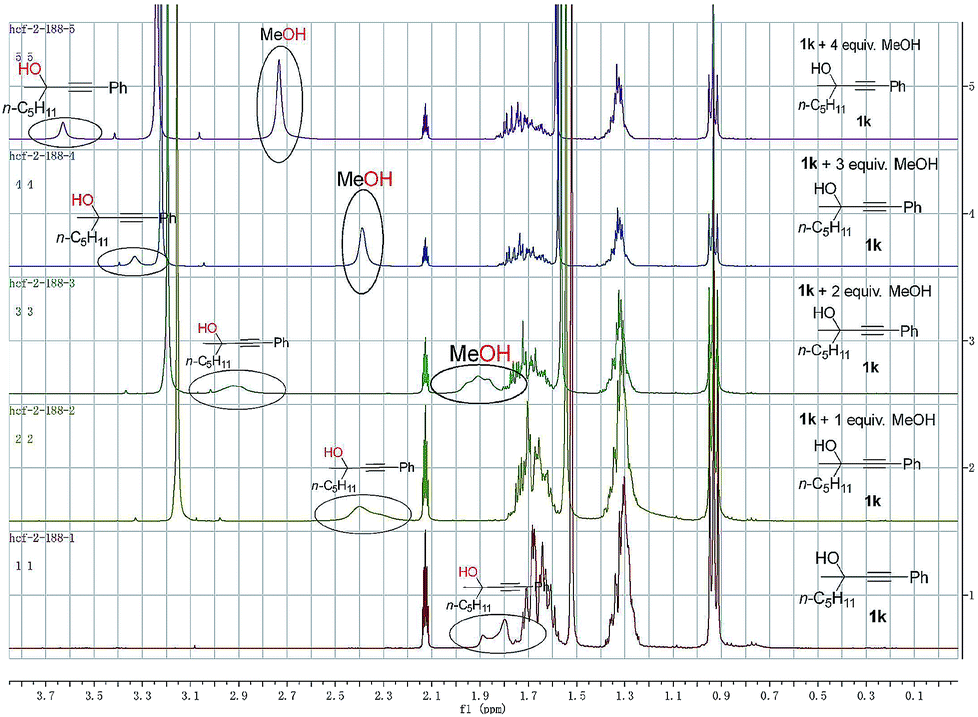 | ||
| Fig. 3 NMR investigation on hydrogen bonding. | ||
In addition, kinetic studies were also carried out. Linear relationships were obtained for ln{c0/(c0 − [(E)-2k])} vs. reaction time (co is the initial concentration of 1k), even with 10-fold excess of MeOH to ensure pseudo zero order in MeOH, indicating first-order dependence of the reaction rate with respect to propargylic alcohol (Fig. 4b). An experiment was also carried out to measure the rate of H/D-scrambling. By adding MeOD into the solution of 1k in CDCl3 and then subjecting the mixture to 1H NMR analysis immediately, the H/D-exchange process was found to reach an equilibrium state within 3 minutes (for details, see the ESI†), which is much faster than the rate of this hydrocarboxylation reaction (Fig. 4a).
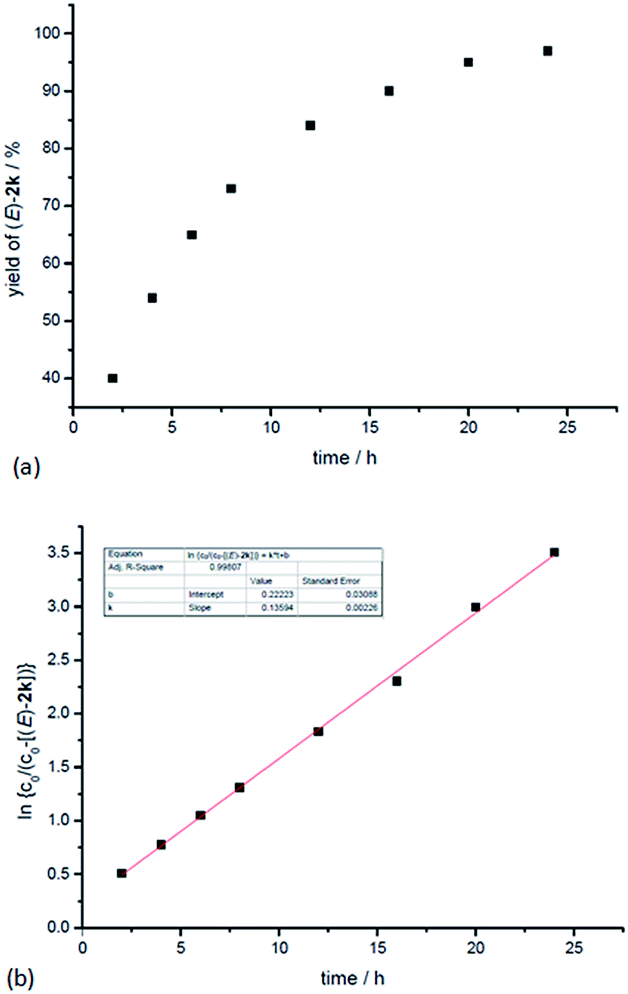 | ||
| Fig. 4 Determination of the reaction order of propargylic alcohol. (a) Yield of (E)-2kvs. time. (b) ln{c0/(c0 − [(E)-2k])} vs. time (R-squared is the coefficient of determination). | ||
Based on this, parallel reactions of 1k and 1k-d in separate reaction vessels monitored by 1H NMR analysis of the reaction profile could help determine the value of kH and kD, and then the KIE was calculated to be kH/kD = 16 (Fig. 5), indicating the primary isotope effect of H/D.
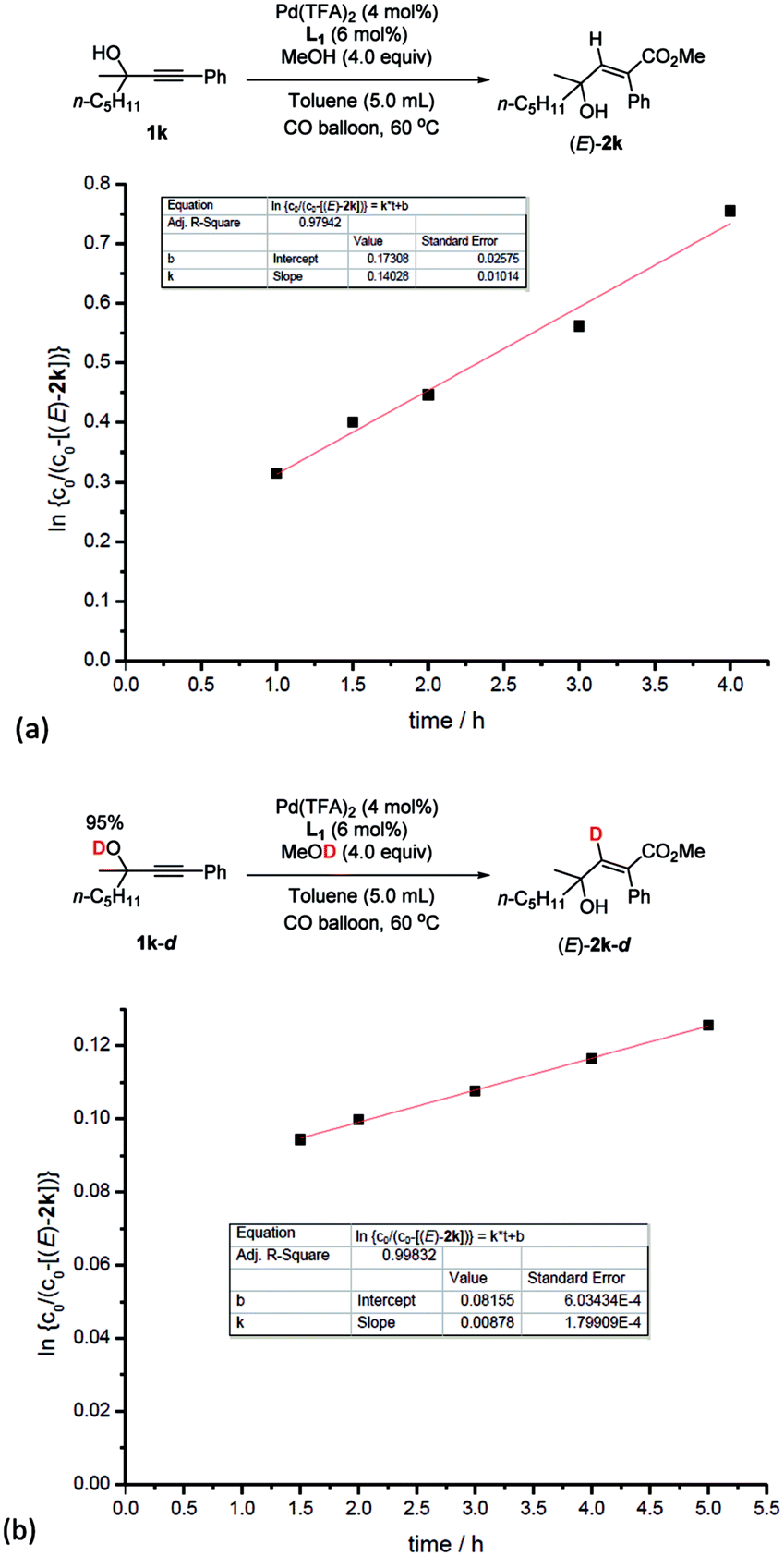 | ||
| Fig. 5 Kinetic isotope effect experiments. (a) Linear function fit for the reaction rate of 1k to obtain kH. (b) Linear function fit for the reaction rate of 1k-d to obtain kD. kH/kD = 16. | ||
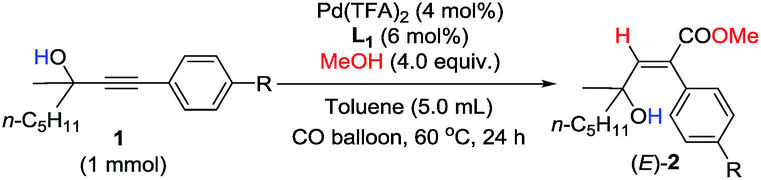
In order to further identify the rate-determining step, the electronic effect of substrates on the Pd–H insertion step was investigated (Table 7). Then, kinetic studies of the substrates with different substituents on the para-position of the phenyl ring such as Br, CO2Me, Me, and OMe were carried out. Linear relationships were obtained for ln{c0/(c0 − [(E)-2k])} vs. reaction time, and show significantly different reaction rates, that is, the more electron-rich the substituent is, the faster the reaction rate is (Fig. 6). These results also indicate that Pd–H insertion has a large effect on the reaction rate. However, we are still not able to exclude the oxidative addition of O–H with Pd as the rate-determining step.
 | ||
| Fig. 6 ln{c0/(c0 − [(E)-2k-R])} vs. time (R-squared is the coefficient of determination). | ||
In order to further unveil the mechanism, solvents without hydrogen bonding7b were also screened—lower yields were detected in comparison with the data for toluene. The stronger the polarity of the solvent is, the lower the yield would be, and nothing but a large amount of substrate recovery was observed when using DMSO, further supporting the irreplaceable role of hydrogen bonding in this transformation (Table 3, entries 10–14).
Other than this, a Hammett study with phenols bearing various substituents has also been carried out (Table 8). The negative value for ρ points out that the rate-determining step favors phenols with electron-donating groups (Fig. 7).15 This seems reasonable to us because phenols with electron-donating groups would result in a higher electron density on the oxygen atom, thus leading to stronger hydrogen bonding with the hydrogen atom in the hydroxyl group and/or nucleophilicity (see step 3 in Scheme 4).
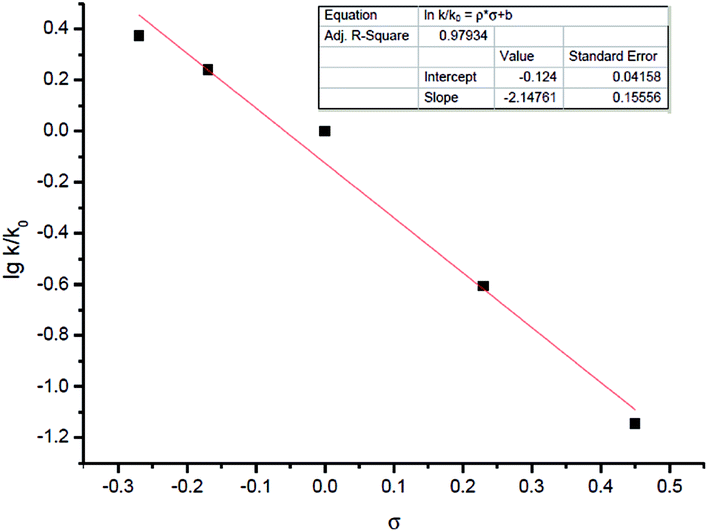 | ||
| Fig. 7 Hammett equation of phenols with varying acidities. | ||
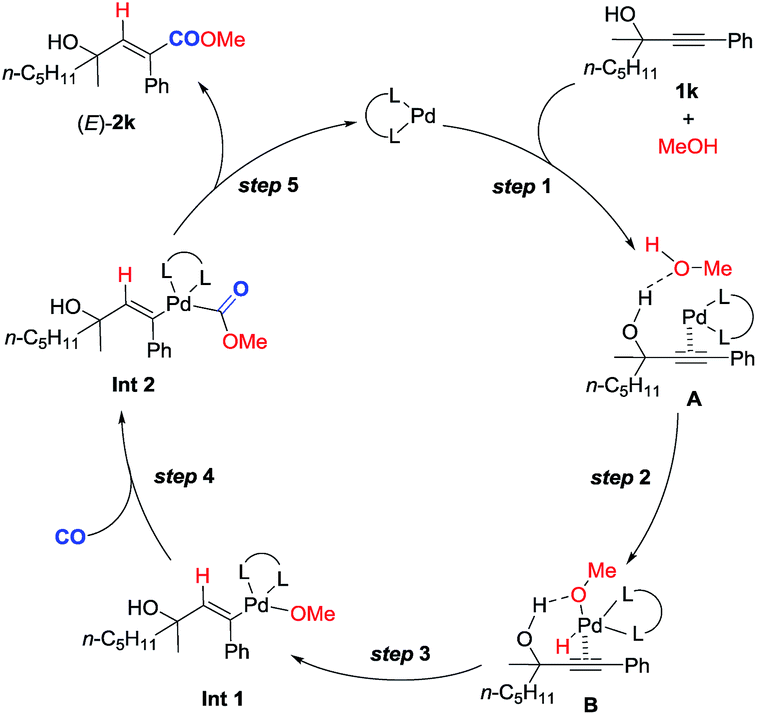 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Based on these studies, a plausible mechanism is proposed (Scheme 4). Hydrogen bonding between the hydroxyl group of methanol and that of 2-alkynol combined with the coordination of the C–C triple bond to the Pd0 species would form complex A. Subsequent oxidative addition of the O–H bond in methanol with Pd0 in A affords complex B. Subsequent regioselective syn-hydropalladation of the C–C triple bond delivers the H atom to the sp carbon atom closer to the hydroxy group in 1k, and then nucleophilic attack of CO by the methoxy anion generates Int 2. Reductive elimination would then furnish (E)-2k and regenerate the Pd0 species to finish the catalytic cycle. Of course, further studies are needed to fully verify this mechanism.
Conclusions
In summary, we have developed hydroxy group-enabled highly regio- and stereo-selective hydrocarboxylation of 2-alkynylic alcohols, exploiting a previously unrecognized regioselectivity control strategy. The remarkable substrate scope, atom economy, and good to excellent yields make this reaction a facile synthetic method to produce highly functionalized 3-hydroxy-2(E)-alkenoates and the observed regioselectivity may arise from hydrogen bonding, which needs further investigation. Due to the versatility of the functionality in the products, the importance of the stereo-selective construction of CC bonds, and the nature of regio-selectivity control, this method will be of high interest to organic and medicinal chemists. Further studies in this area are currently ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (Grant No. 21690063) is greatly appreciated. We also thank Mr Qi Liu in this group for reproducing the results for (E)-2j in Table 2, (E)-2m in Table 4, and (S, E)-2x in Table 6.Notes and references
- For selected books and reviews, see: (a) Modern Alkyne Chemistry, ed. B. M. Trost and C.-J. Li, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed; (b) H. Schobert, Chem. Rev., 2014, 114, 1743 CrossRef CAS PubMed; (c) I. T. Trotuş, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761 CrossRef PubMed; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed; (e) R. Salvio, M. Moliterno and M. Bella, Asian J. Org. Chem., 2014, 3, 340 CrossRef CAS.
- (a) E.-I. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef CAS PubMed; (b) G. Boije af Gennas, V. Talman, J. Yli-Kauhaluoma, R. K. Tuominen and E. Ekokoski, Curr. Top. Med. Chem., 2011, 11, 1370 CrossRef CAS PubMed; (c) J. Duchek, D. R. Adams and T. Hudlicky, Chem. Rev., 2011, 111, 4223 CrossRef CAS PubMed; (d) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2017, 50, 988 CrossRef CAS PubMed; (e) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564 CrossRef CAS PubMed; (f) J.-J. Shie, J.-M. Fang and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 5788 CrossRef CAS PubMed.
- For selected books and reviews, see: (a) L. Hintermann, in Topics in Organometallic Chemistry, Springer-Verlag, Heidelberg, 2010, vol. 31, pp. 123–155 Search PubMed; (b) W. Li and J. Zhang, in Comprehensive Organic Synthesis, Elsevier, Amsterdam, 2nd edn, 2014, vol. 4, pp. 342–391 Search PubMed; (c) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed; (d) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed; (e) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086 CrossRef CAS PubMed; (f) V. P. Boyarskiy, D. S. Ryabukhin, N. A. Bokach and A. V. Vasilyev, Chem. Rev., 2016, 116, 5894 CrossRef CAS PubMed; (g) J. J. Feng and J. Zhang, ACS Catal., 2016, 6, 6651 CrossRef CAS; (h) M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240 CrossRef CAS PubMed; (i) M. B. Ansell, O. Navarro and J. Spencer, Coord. Chem. Rev., 2017, 336, 54 CrossRef CAS; (j) Y. Zheng and W. Zi, Tetrahedron Lett., 2018, 59, 2205 CrossRef CAS.
- For selected examples of using directing groups, see: (a) S. Wu, X. Huang, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2015, 6, 7946 CrossRef PubMed; (b) S. Wu, X. Huang, C. Fu and S. Ma, Org. Chem. Front., 2017, 4, 2002 RSC; (c) M. Lautens and M. Yoshida, Org. Lett., 2002, 4, 123 CrossRef CAS PubMed; (d) N. Kim, K. S. Kim, A. K. Gupta and C. H. Oh, Chem. Commun., 2004, 618 RSC; (e) B. Gourdet, D. L. Smith and H. W. Lam, Tetrahedron, 2010, 66, 6026 CrossRef CAS; (f) S. Li, W. Yuan and S. Ma, Angew. Chem., Int. Ed., 2011, 50, 2578 CrossRef CAS PubMed; (g) Y. Kawasaki, Y. Ishikawa, K. Igawa and K. Tomooka, J. Am. Chem. Soc., 2011, 133, 20712 CrossRef CAS PubMed; (h) A. L. Moure, P. Mauleón, R. G. Arrayás and J. C. Carretero, Org. Lett., 2013, 15, 2054 CrossRef CAS PubMed; (i) K. H. Huang and M. Isobe, Eur. J. Org. Chem., 2014, 4733 CrossRef CAS; (j) Z. Liu, J. Derosa and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 13076 CrossRef CAS PubMed.
- (a) T. Nogi and J. Tsuji, Tetrahedron, 1969, 25, 4099 CrossRef CAS; (b) Y. Tsuji, T. Kondo and Y. Watanabe, J. Mol. Catal., 1987, 40, 295 CrossRef CAS; For Pd-catalyzed dehydration-hydrocarboxylation of terminal propargylic alcohols affording alkadienoic acids, see: (c) K.-T. Huh, A. Orita and H. Alper, J. Org. Chem., 1993, 58, 6956 CrossRef CAS; For Ni(CO)4-mediated carboxylation of propargyl alcohol in the presence of acetic acid and MeOH affording a regioisomeric mixture of carboxylic acids, see: (d) R. W. Rosenthal, L. H. Schwartzman, N. P. Greco and R. Proper, J. Org. Chem., 1963, 28, 2835 CrossRef CAS.
- For selected reviews, see: (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed; (b) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed; (c) C. Min and D. Seidel, Chem. Soc. Rev., 2017, 46, 5889 RSC.
- For utilizing hydrogen bonding to explain selectivities in Pd-catalyzed allylic alkylation reactions, see: (a) B. M. Trost, R. C. Bunt, R. C. Lemoine and T. L. Calkins, J. Am. Chem. Soc., 2000, 122, 5968 CrossRef CAS; (b) G. R. Cook, H. Yu, S. Sankaranarayanan and P. S. Shanker, J. Am. Chem. Soc., 2003, 125, 5115 CrossRef CAS PubMed; For utilizing hydrogen bonding with a catalyst to control regioselectivities in Rh-catalyzed hydroformylations, see: (c) T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 311 CrossRef PubMed; For utilizing hydrogen bonding to explain selectivities in photocycloaddition reactions, see (d) H. Maeda, K. Chiyonobu and K. Mizuno, Photochem. Photobiol. Sci., 2011, 10, 1445 RSC; For utilizing hydrogen bonding with a chloride ligand to explain selectivities in Ru-catalyzed trans-hydrometalations, see: (e) S. M. Rummelt, K. Radkowski, D.-A. Roşca and A. Fürstner, J. Am. Chem. Soc., 2015, 137, 5506 CrossRef CAS PubMed.
- W. Zhang, C. Huang, Y. Yuan and S. Ma, Chem. Commun., 2017, 53, 12430 RSC.
- C. Godard, B. K. Muñoz, A. Ruiz and C. Claver, Dalton Trans., 2008, 853 RSC.
- (a) D. Xu, Z. Li and S. Ma, Tetrahedron Lett., 2003, 44, 6343 CrossRef CAS; (b) W. Zhang and S. Ma, Chem. Commun., 2018, 54, 6064 RSC.
- (a) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866 RSC; (b) N. Eleya, A. Mahal, M. Hein, A. Villinger and P. Langer, Adv. Synth. Catal., 2011, 353, 2761 CrossRef CAS.
- (a) J. Kenyon, Org. Synth., 1926, 6, 68 CrossRef; (b) X. Tang, X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, Y. Tang, J. Zhang, Q. Yu, C. Fu and S. Ma, Org. Chem. Front., 2015, 2, 688 RSC.
- (a) A. E. G. Miller, J. W. Biss and L. H. Schwartzman, J. Org. Chem., 1959, 24, 627 CrossRef CAS; (b) S. Rajaram, U. Ramulu, S. Aravind and K. S. Babu, Helv. Chim. Acta, 2015, 98, 650 CrossRef CAS.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS.
- M. B. Smith and J. March, Advanced Organic Chemistry, Wiley, New Jersey, 6th edn, 2007, ch. 9, pp. 401–412 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1571775 and 1845002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05743e |
| This journal is © The Royal Society of Chemistry 2019 |