
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWidely applicable background depletion step enables transaminase evolution through solid-phase screening†
Matteo
Planchestainer
 a,
Eimear
Hegarty
a,
Christian M.
Heckmann
a,
Louise J.
Gourlay
b and
Francesca
Paradisi
*ac
a,
Eimear
Hegarty
a,
Christian M.
Heckmann
a,
Louise J.
Gourlay
b and
Francesca
Paradisi
*ac
aUniversity of Nottingham, School of Chemistry, University Park, Nottingham NG7 2RD, United Kingdom
bStructural Biology Unit, Dep. Biosciences, Università degli Studi di Milano, Via Celoria 26, 20133 Milano, Italy
cUniversity of Bern, Department of Chemistry and Biochemistry, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: francesca.paradisi@dcb.unibe.ch
First published on 9th May 2019
Abstract
Directed evolution of transaminases is a widespread technique in the development of highly sought-after biocatalysts for industrial applications. This process, however, is challenged by the limited availability of effective high-throughput protocols to evaluate mutant libraries. Here we report a rapid, reliable, and widely applicable background depletion method for solid-phase screening of transaminase variants, which was successfully applied to a transaminase from Halomonas elongata (HEWT), evolved through rounds of random mutagenesis towards a series of diverse prochiral ketones. This approach enabled the identification of transaminase variants in viable cells with significantly improved activity towards para-substituted acetophenones (up to 60-fold), as well as tetrahydrothiophen-3-one and related substrates. Rationalisation of the mutants was assisted by determination of the high-resolution wild-type HEWT crystal structure presented herein.
Introduction
Transaminases have attracted considerable attention for the production of compounds containing chiral amino groups, which are widespread in the pharmaceutical, agrochemical and fine chemical industry.1,2 Of particular interest, in terms of synthetic applications, are the amine transaminases (ATAs), (R)- or (S)-selective enzymes that are capable of aminating carbonyls (both aldehydes and ketones) in the absence of a neighbouring carboxylic group.3 Despite the enormous potential of ATAs, the widespread application of these biocatalysts in their wild type form is hindered by a number of challenges including limited substrate specificities, substrate and product inhibition, and unfavourable reaction equilibria.4Protein engineering by rational design5–7 and directed evolution8,9 have played a major role in the ability to address these issues. Evolutionary strategies, in particular, are ideally suited to the optimisation of the physical properties of enzymes for large-scale manufacturing processes, as well as enabling fine-tuning of substrate and stereochemical preferences.10,11 The evolution of ATAs by Codexis and Merck12,13 pioneered subsequent engineering efforts of many ATAs, resulting in the production of an array of highly specialised ATAs for chiral amine synthesis.8,9,14
Even though a fully randomised approach to mutagenesis, targeting the whole length of the protein, has become less common, directed evolution clearly requires high-throughput screening methods to evaluate variable size libraries of engineered variants. The generation of smaller, smart libraries designed by rational targeted approaches such as CASTing,15 or saturation mutagenesis of specific residues identified in silico, still yield a sizable number of clones to screen (saturation at three positions affords 8000 variants). The identification of hits within live cells is advantageous for library screening because, if it is efficient, it prevents culturing of large number of clones followed by assays with cell extracts, which, in the absence of robotics, is very time-consuming.16 Recently, microfluidic screening,17,18 in addition to the application of desorption electrospray ionisation mass spectrometry19 on whole colonies, have emerged as powerful tools for screening large libraries, however, the equipment required in both cases is still highly specialised and expensive, therefore they are not yet suitable for widespread implementation. Solid-phase screening (SPS), on the other hand, is a simpler approach that facilitates the screening of a medium-large number of mutant variants based on colorimetric changes that are easily observed by the naked eye.20 SPS was successfully applied in the evolution of monoamine oxidase (MAO-N) enzymes, using two libraries that were fully saturated at two positions each (400 variants).21 SPS methods applicable to ATA mutant libraries, however, are currently limited to a few strategies to screen for amino donors. Wu and co-workers, for example, reported the use of SPS to screen libraries of approximately 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ATA variants for reduced inhibition towards methoxyisopropylamine.22 Later, Bornscheuer and colleagues applied SPS to evolve an ATA towards suitable amino donors (screening ca. 4600 colonies), however this required co-expression of a second enzyme as a coupled reporting system, increasing the complexity of the system.23,24 Similarly, Turner and co-workers evolved SPS for ATA library screening, specifically to identify either (R)- or (S)-selective ATAs (screening ca. 1000 colonies), which also required multiple reporting enzymes, as well as flash-freezing in liquid nitrogen, to increase permeability.25
000 ATA variants for reduced inhibition towards methoxyisopropylamine.22 Later, Bornscheuer and colleagues applied SPS to evolve an ATA towards suitable amino donors (screening ca. 4600 colonies), however this required co-expression of a second enzyme as a coupled reporting system, increasing the complexity of the system.23,24 Similarly, Turner and co-workers evolved SPS for ATA library screening, specifically to identify either (R)- or (S)-selective ATAs (screening ca. 1000 colonies), which also required multiple reporting enzymes, as well as flash-freezing in liquid nitrogen, to increase permeability.25
Screening strategies for varying carbonyl acceptors, which are key substrates for ATAs to produce chiral amine products, appear to be much more challenging. ortho-Xylylenediamine26 and 4-nitrophenethylamine27 have been recently reported as amine donors in proof of concept studies to rapidly generate coloured by-products upon transamination. Both these strategies suffer from strong background interference in live cells due to endogenous pyruvate which is nearly always an excellent substrate for ATAs; in a mutant library screening, the number of false positives is certain to be significant without an efficient system to deplete background. Pyruvate is a primary metabolite, essential for cell growth, therefore knock-out strategies are not feasible, making it inherently difficult to minimise the background noise in SPS.
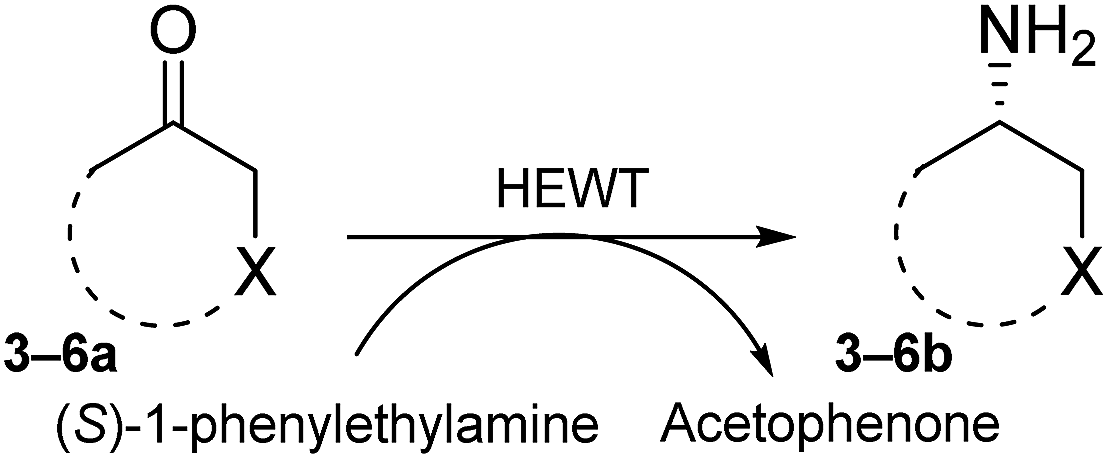
We here report the development of a simple and reliable SPS method that encapsulates (S)-1-phenylethylamine as a straightforward background depletion agent to minimize high levels of pyruvate activity. To highlight the power of this methodology, we successfully applied it to the screening of a fully randomised mutant library of an (S)-selective ATA from Halomonas elongata (HEWT)28 against a series of prochiral aromatic and cyclic ketones. In addition, the crystal structure of the wild-type HEWT is reported, which facilitated rationalisation of the mutations introduced.
Results and discussion
Depletion of background activity and test screening
Reliable methodology to quench background activity, which leads to a high number of false positives, was designed to specifically deplete endogenous pyruvate. Unlike previously discussed methods, our alternative approach is based on pre-reacting cellular pyruvate with (S)-1-phenylethylamine. The reaction is catalysed by the expressed transaminase to produce alanine and acetophenone with an extremely favourable equilibrium, effectively removing pyruvate as a non-specific acceptor from the environment. The screening was initially optimised with E. coli BL21(DE3) cells expressing wild-type HEWT, which were spread onto a nitrocellulose membrane deposited on a LB-agar plate. This permitted easy transfer of the colonies through the different assay steps (Fig. S1†). Following expression and dialysis (Experimental in the ESI†), the membrane was left for 30 minutes on filter paper soaked with (S)-1-phenylethylamine and then washed with 50 mM phosphate buffer, pH 8.Test screening of pre-treated colonies to establish timelines and reaction efficiency, were performed with both pyruvate and acetophenone and compared with a control plate not supplemented with any amino acceptor (Fig. S2†). ortho-Xylylenediamine was used in the first instance as an amino donor. The control plate did not give any colorimetric change up to two hours, after which time a slightly brown tinge could be observed likely due to the cell healthy metabolic system producing sufficient amounts of pyruvate detectable through the reaction. In the absence of the background depletion step, the cells almost immediately respond to the amino donor preventing any significant discrimination between exogenous amino acceptor and the endogenous pyruvate (Fig. 1), as was also observed by O'Reilly and co-workers.26 Likewise, the addition of pyruvate to the membrane as the amino acceptor, causes visible darkening of the colonies within 5 minutes of incubation. Acetophenone, which in liquid-phase assays had shown no detectable activity over 3 h with wild-tpe HEWT, performs similarly to the control plate, with no observable colour formation during total incubation (Fig. S2†). The mild treatment, preserved cell viability, which is essential for the direct growth and plasmid extraction, bypassing the need for a replica plate. To further evaluate the effectiveness of this strategy, 4-nitrophenethylamine was used as amino donor in the assay.27 Remarkably, a significant reduction in background noise was observed once more, highlighting the transferability of this methodology (should o-xylylenediamine not be accepted by the investigated TA). Indeed, the background was even further suppressed when compared to the ortho-xylylenediamine screen, with barely detectable darkening of the colonies after 24 h (Fig. S3†). Liquid nitrogen- and chloroform-induced permeabilization steps were attempted also,24,25 but in our case neither method was successful.
 | ||
| Fig. 1 ortho-Xylylenediamine solid screening control. E. coli BL21(DE3) cells expressing wild-type HEWT after 1 h of incubation with ortho-xylylenediamine at RT, following background depletion step (A), and in the absence of such pre-treatment (B). | ||
The use of amines other than (S)-1-phenylethylamine, such as isopropylamine and cadaverine, for the background depletion step was also investigated. Cadaverine, previously described as a “smart” amine donor,29 can be used in ATA reactions ex vivo and may effectively convert endogenous pyruvate in whole cells. Isopropylamine is a very inexpensive amine donor, commonly used in industrial processes;12 the generated acetone is produced in minimal quantities and expected to be harmless to colonies. While both amines also resulted in a significant reduction in background (Fig. S4†), (S)-1-phenylethylamine appeared to be most effective, possibly due to better diffusion of this substrate. However, this demonstrates that other amines can also be used to quench endogenous pyruvate, for example with aminotransferases that do not accept 1-phenylethylamine as a substrate.
Evolution towards substituted acetophenones
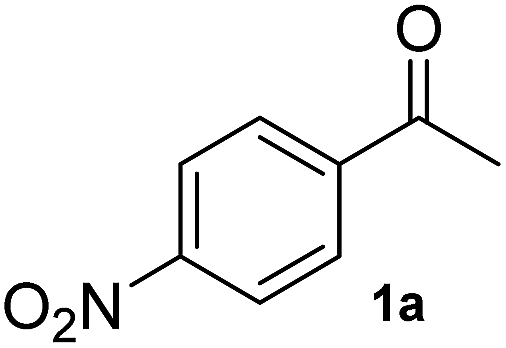
To test the methodology, HEWT was subjected to directed evolution using error-prone PCR to generate a completely randomised mutant library with a high number of mutations. The enzyme has minimal activity towards the aromatic substrate para-nitroacetophenone (1a) and this was selected as the amino acceptor for the initial screening, using ortho-xylylenediamine as the amino donor. DMSO (10% (v/v)) was required for substrate solubility and enhanced cell-permeability. The discrimination between wild-type and an improved variant relies on the rapidity with which the enzyme converts the substrate in vivo. Screening of ca. 15000 colonies provided two variants with enhanced catalytic activity towards para-nitroacetophenone. The two new variants (referred to as A1 and B1) were expressed and purified for further characterisation. They exhibited a 2-fold increase in turnover frequency with respect to the wild-type, achieved higher conversion after 2 hours, and maintained excellent enantioselectivity (Table 1). Both variants were subjected to a second round of error-prone PCR and the libraries screened as before with 1a. Interestingly, in this case the positive colonies were isolated after a significantly shorter incubation period (2–15 minutes compared to 25 minutes for the parental colony). While the A1 template did not provide any significant hits, an improved variant was isolated from the B1 library. The new mutant, identified as B2, afforded comparable molar conversion after 2 hours and a further 1.5-fold increase in turn-over frequency (Table 1). The e.e. measured at 24 h was in all cases >99% (though the overall yields were low due to experimental conditions needed to assess the kinetic behaviour of the mutants). A third round of mutagenesis, using the B2 variant as a template, initially failed to identify better variants, as the colour change on solid screening was too rapid and did not allow discrimination between parental activity and mutants. Reducing the concentration of para-nitroacetophenone and ortho-xylylenediamine from 10 mM to 1 mM, with just 5% (v/v) DMSO prolonged the screening window to ca. 1 h. An additional variant, B3, was isolated from the screening and while, upon purification, it displayed comparable reaction velocity and 2 h conversions to B2, it had 2-fold higher expression levels which justifies a more rapid colour development with respect to the parental variant. Each variant was also fully characterised in terms of activity and stability at different temperatures and pHs. In all the cases, the mutants generated did not show significant alterations when compared to the WT enzyme (Fig. S5–S8†).
|
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate | WT | A1 | B1 | B2 | B3 | ||||||||||
| TOF (10−3 s−1) | m.c. (%) | e.e. (%) | TOF (10−3 s−1) | m.c. (%) | e.e. (%) | TOF (10−3 s−1) | m.c. (%) | e.e. (%) | TOF (10−3 s−1) | m.c. (%) | e.e. (%) | TOF (10−3 s−1) | m.c. (%) | e.e. (%) | |
| a Biotransformation reactions were performed with 10 mM ketones, 500 mM L-alanine, 0.1 mM PLP, 0.1 mg mL−1 (1.9 µM) enzyme in 50 mM phosphate buffer pH 8 and 10% (v/v) DMSO at 37 °C (see Experimental in the ESI, Fig. S9). All experiments were conducted in triplicate and the standard error is reported accordingly. Mutations; A1: W56C, V435A; B1: W56C, L211V, L306M; B2: W56C, L211V, L306M, V361A, Q388R, P453L; B3: W56C, L211V, A254V, L306M, V361A, Q388R, P453L. | |||||||||||||||
|
36 ± 6 | 4 | >99 (S) | 66 ± 9 | 6 | >99 (S) | 65 ± 26 | 6 | >99 (S) | 92 ± 5 | 7 | >99 (S) | 84 ± 14 | 7 | >99 (S) |
|
4 ± 1 | 9 | 87 (S) | 50 ± 7 | 11 | 94 (S) | 127 ± 5 | 12 | >99 (S) | 229 ± 7 | 11 | >99 (S) | 246 ± 7 | 12 | >99 (S) |
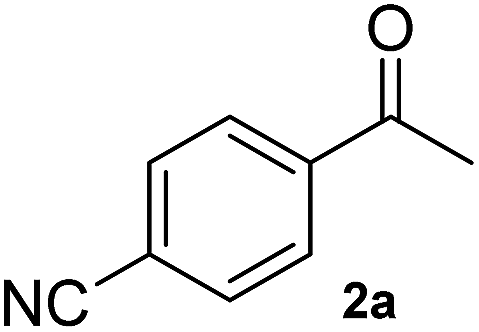
A second substrate, para-cyanoacetophenone (2a), was also investigated. HEWT wild-type has negligible activity towards this molecule, even lower than the nitro substitution, with a turnover frequency of 4 × 10−3 s−1. The B2 variant showed an impressive 60-fold increase in turn-over frequency (229 × 10−3 s−1), and enhanced stereoselectivity, affording >99% of the (S)-enantiomer (Table 1). Again, B3 showed virtually identical catalytic properties to B2.
Evolution towards small cyclic ketones
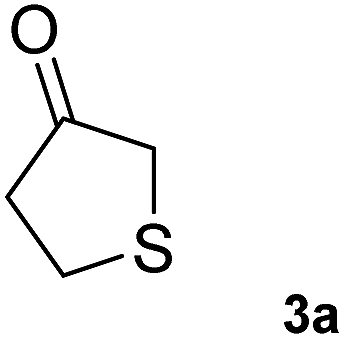
To further expand the catalytic scope of HEWT, the original mutant library was screened towards a structurally different substrate, tetrahydrothiophen-3-one (3a), a small cyclic ketone which if successfully aminated yields a chiral amine (3b) that has potential use as a building block for active pharmaceutical ingredients.30In this case, the wild-type enzyme already possesses some activity towards the target substrate (3a), with a turnover frequency of 206 × 10−3 s−1 and complete molar conversion (>99%) after 24 h using equimolar amounts of (S)-1-phenylethylamine as donor and higher catalyst concentration (1 mg mL−1 – 19 µM). The optimised solid phase screening with ortho-xylylenediamine yielded a new variant (C1) which, upon expression and purification, displayed a 2-fold increase in turnover frequency (Table 2). On this occasion, the mutant showed a colour change after 18 minutes, compared to 30 minutes for the wild-type control. Notably, the variant matched the excellent conversion displayed by the wild-type enzyme, affording the (S)-enantiomer of the corresponding amine with >99% conversion and 66% e.e.
|
|
||||||
|---|---|---|---|---|---|---|
| Substrate | WT | C1 | ||||
| TOF (10−3 s−1) | m.c. (%) | e.e. (%) | TOF (10−3 s−1) | m.c. (%) | e.e. (%) | |
| a Biotransformation reactions were performed with 10 mM ketones, 10 mM (S)-1-phenylethylamine, 0.1 mM PLP, 1 mg mL−1 (19 µM) enzyme in 50 mM phosphate buffer pH 8 and 10% (v/v) DMSO at 37 °C (see Experimental in the ESI, Fig. S10). All experiments were conducted in triplicate and the standard error is reported accordingly. Mutation in C1: D5E. | ||||||
|
206 ± 5 | >99 | 66 (S) | 460 ± 6 | >99 | 66 (S) |
|
430 ± 1 | >99 | 70 (S) | 580 ± 3 | >99 | 70 (S) |
|
18 ± 0.2 | 94 | 90 (S) | 11.6 ± 0.3 | 94 | 90 (S) |
|
10 ± 0.8 | 98 | 90 (S) | 19 ± 0.8 | 98 | 90 (S) |
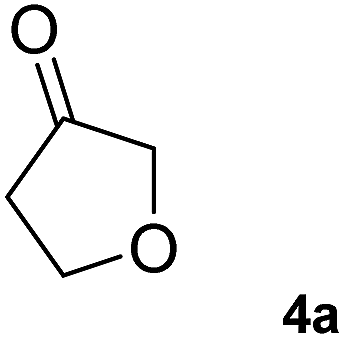
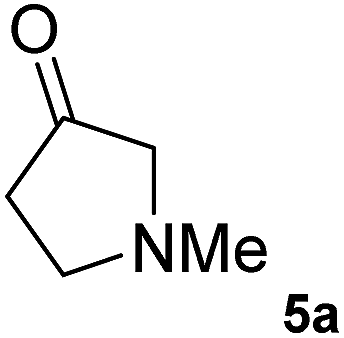
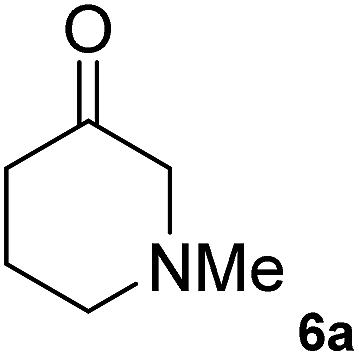
The C1 variant was subsequently screened against a panel of representative cyclic ketones (Table 2, 4a, 5a, 6a). The mutant had improved TOF with both tetrahydrofuan-3-one (4a) and 1-methylpiperidin-3-one (6a) (35% and 90% increase, respectively) but it was in fact slower with 1-methylpyrrolidin-3-one (5a) (see Table S1†). Similarly to what was observed with the library evolved towards para-nitroacetophenone, an additional round of mutagenesis did not permit the identification of better variants towards tetrahydrothiophen-3-one (3a) as the colour change on the plates was too rapid to discriminate further enhanced variants.
This clearly demonstrates that this solid phase screen is able to identify better variants, both in terms of TOFs and expression levels, from libraries that are of desirable size for semi-rational approaches to directed evolution.10 The application of this methodology was successful even with a fully randomised approach which can only afford limited improvements of the mutants, given the size of the library, due to the high probability of unsuitable mutations.31
3D structural analysis of wild type HEWT and mutant variants
In order to understand the effect of the various mutations on wild-type HEWT, the crystal structure was determined at a resolution of 2 Å, as described in the methods. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (www.rcsb.org) under PDB accession code 6GWI.Two identical HEWT monomers (chains A and B; RMSD 0.23 Å) were present in the asymmetric unit (Matthews coefficient = 2.14 Å3 Da−1), with an estimated solvent content of 42.5% (see Table S2†). This homodimer arrangement is biologically relevant as HEWT is a catalytically active dimer. Electron density was present for residues 2 to 451 (chain A) and 1 to 450 (chain B). The HEWT monomer comprises 3 sub-domains: (i) a principle PLP-dependent transferase-like domain (residues 82 to 318) that hosts a central mixed β-sheet, comprising 7 strands, (β9–β8–β10–β11–β12–β13–β7), with β-strand 13 being antiparallel to the rest, surrounded by 9 α-helices and two 310 helices (η1–2) (Fig. 2); (ii) domain 2 (residues 2–79; 320–451) that contains a small N-terminal sub-domain (residues 2 to 32; α1–β1–β2–α2–β3–5) which forms the main interactions with the opposing monomer. One molecule of pyridoxal-5′-phosphate (PLP), produced by the covalent attachment to the ε-amino group of the catalytic lysine (K284), via an aldimine linkage, is bound to each monomer. Dimerisation involves 100 residues contributed from each monomer, with an interface area of 5395 Å2. The main dimerisation interactions include 4 salt bridges and 47 hydrogen bonds, as calculated by PDBsum available from the European Bioinformatics Institute (www.ebi.ac.uk).
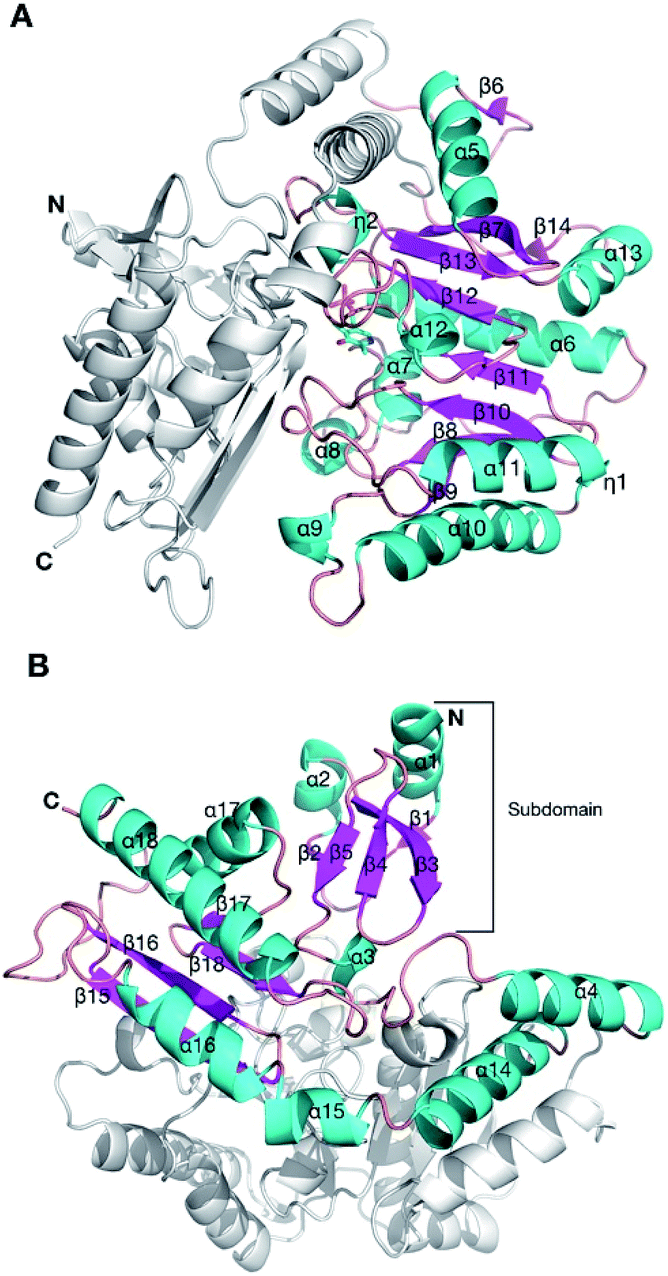 | ||
| Fig. 2 The crystal structure of the HEWT WT monomer. Cartoon secondary structure representation of the (A) PLP-dependent transferase-like domain (residues 82 to 318), with PLP indicated in sticks. For simplicity, the other domain is not coloured. The N-and C-termini are labelled. (B) The second domain of HEWT, comprising a subdomain (labelled) that mediates dimerisation with the opposing monomer. In this case, the PLP-dependent domain is not shown for clarity. This figure was generated using Chimera.36 | ||
Based on a secondary structure-based homology search, the DALI server identified the 4-aminobutyrate transaminase from Pseudomonas as the top hit (PDB entry 5KR632) with a RMSD of 1.3 Å over 445/460 aligned main chain residues.33 Sequence and structure conservation between HEWT and all other structures deposited in the PDB was assessed using ENDscript 2.0 (http://endscript.ibcp.fr).34 HEWT was compared with 124 structures with sequence conservation >30%. As expected, the highest conservation (sequence and structural) is located in the PLP-dependent transferase-like domain and active site region (Fig. 3A).
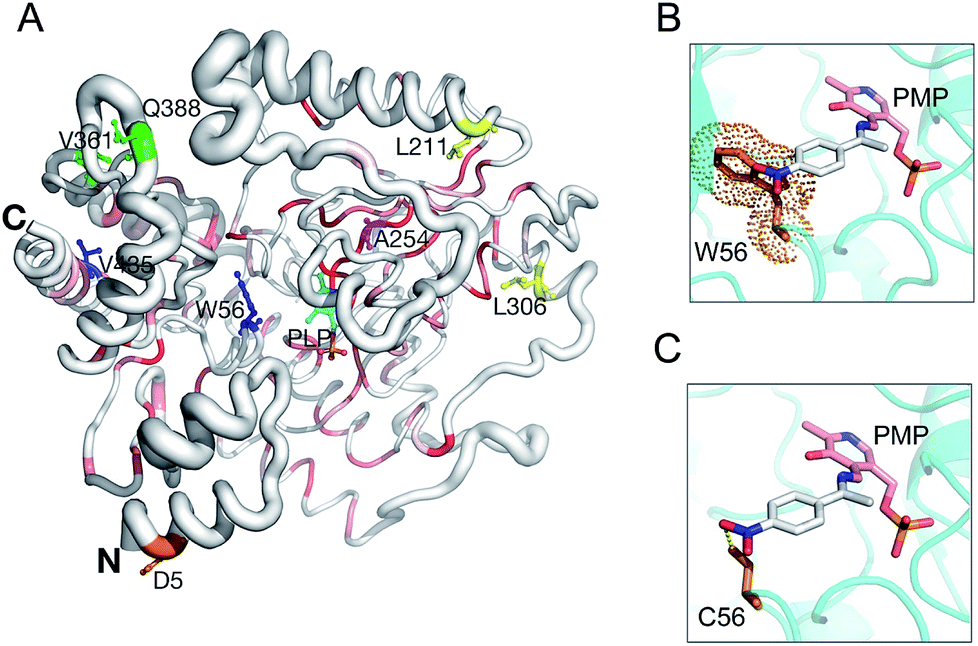 | ||
| Fig. 3 HEWT 3D structure conservation and mutant residue positions. Secondary structure of the HEWT monomer shown in sausage representation, as automatically generated by ENDscript 2.0 (http://endscript.ibcp.fr).34 Structure conservation between chain A of HEWT and 124 structure homologs present in the PDB is indicated by ribbon thickness, with regions of low conservation being thicker than highly conserved regions (thin regions). Sequence conservation is indicated by red shading; the redder the residue, the more conserved it is. Mutated residues in variants A1 (blue), B1 (yellow), B2 (green) and C1 (orange) are shown as sticks. The B3 mutant shares all B2 mutations and contains an extra A254V mutation (pink sticks). P453 from variants B2 and B3 is not present in the model. The N- and C-termini are indicated and PLP is shown in sticks. This figure was generated using Pymol 2.0.6. Modelling of the active site for the A1 and B1 mutants, where the bulky Trp (the steric encumbrance of the residue is shown as an orange cloud) (D) is substituted with a cysteine (E), allowing the aromatic ring substituent (para-nitroacetophenone in the aldimine intermediate complex with the pyridoxamine phosphate (PMP)) to be more easily accommodated. | ||
Sequencing of the selected mutants identified two amino acid changes in the A1 variant, (W56C, V435A), while the B1 variant displayed three amino acid substitutions (W56C, L211V, L306M) (notes in the ESI†). Interestingly, both mutants harbour a tryptophan to cysteine change in position 56, which is located in the active site. This residue has previously been identified as a hotspot in in silico rational design studies.7,35 In this case, removing the large indole ring and replacing it with the smaller cysteine side chain allows for easier substrate binding. Furthermore, the thiol can promote hydrogen bonding with the aromatic substituent and stabilise para-nitroacetophenone (Fig. 3B and C). Accordingly, W56 is located in a highly structurally conserved region (Fig. 3A), as is V435 (Fig. 3A). The L306M mutation is peripheral and not conserved with regards to sequence, and therefore less conserved than the A1 mutations with regards to structure, and therefore is somewhat more difficult to rationalise. The L306M mutation introduces a longer side chain that may form more stabilising hydrophobic interactions with surrounding hydrophobic residues (M126, V302 and F313). From the structure, this hydrophobic cluster may in-turn stabilise regions of the protein that mediate dimerisation (Fig. S11†). In agreement with this hypothesis, thermal stability analysis of the wild-type and mutant B2 indicates that the latter is more stable (Fig. S12†). The second round of evolution introduced three additional mutations (V361A, Q388R, P453L) in the B2 variant, which are also located outside of the active site and all are exposed to the solvent; their side-chains do not make any significant stabilising intramolecular interactions. B3 introduced mostly silent mutations, apart from a single A254V mutation neighbouring the D255 residue, forming a hydrogen bond with the pyridine-type nitrogen of the co-factor PLP.
On the other hand, structural analysis of the C1 variant revealed a single mutation (D5E) positioned at the N-terminus of the protein (Fig. 3A). Using the protein structure quasi-rigid domain decomposition (PiSQRD) web server (http://pisqrd.escience-lab.org),37 the HEWT crystal structure (chain A) was decomposed into four sub-domains that can behave as independent units during conformational fluctuations (Fig. S13†). D5 is located in domain 4 (residues 2–34 and 41–46) that adopts an α1-helix–loop–α2-helix structure. Based on these observations, an aspartate to glutamate mutation may alter the local protein dynamics that in-turn may cause alterations in the topology of the active site and hence substrate specificity and activity.
Conclusions
The introduction of an amine donor as a background depletion agent, which exploits the expressed ATA to quench endogenous pyruvate proved to be very effective in delivering an efficient screening platform for aminotransferase directed evolution. This high-throughput screening strategy was developed on the basis of the ortho-xylylenediamine assay described by O'Reilly and co-workers,26 which was a fundamental tool in the evolution of HEWT towards a diverse set of aromatic and cyclic ketones. After three rounds of directed evolution, a 2.5-fold increase in activity was obtained for para-nitroacetophenone, while with para-cyanoacetophenone the identified mutant exhibited a 60-fold higher turnover frequency coupled with a substantial improvement of the enantioselectivity with respect to the wild type. Furthermore, the evolution towards tetrahydrothiophen-3-one yielded a 2-fold increase in the overall catalytic efficiency. This enzyme also showed improved catalytic properties when screened against similarly challenging cyclic ketones. The background depletion step is straightforward and reliable, and can be applied to other screening methods where endogenous pyruvate interferes with detection making the evolution of these enzymes a much less challenging task. The use of 4-nitrophenethylamine as an alternative reporter, as well as alternative amine donors for the depletion step, proved successful. The limit of this screen is set by the power of discrimination between the mutant and the parent enzyme; if the substrate conversion is too fast, the colonies turn black rapidly, which leads to false positives. Tuning the screening conditions to slow the reaction can extend the screening window. In addition, through modulation of the parental activity, by regulating the expression levels (with the use of a promoter that allows for stricter control),38 this screening method could be a very powerful tool in further rounds of evolution towards the identification of more and more active mutants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council [grant number BB/P002536/1 and BB/M008770]. The authors thank the University of Nottingham through the Green Chemicals Beacon for support. Structural studies were supported by Università degli Studi di Milano Linea 2 funding.References
- I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 2015, 6965–6982 CrossRef CAS PubMed.
- F. Steffen-Munsberg, C. Vickers, H. Kohls, H. Land, H. Mallin, A. Nobili, L. Skalden, T. van den Bergh, H. J. Joosten, P. Berglund, M. Höhne and U. T. Bornscheuer, Biotechnol. Adv., 2015, 33, 566–604 CrossRef CAS PubMed.
- I. Slabu, J. L. Galman, C. Iglesias, N. J. Weise, R. C. Lloyd and N. J. Turner, Catal. Today, 2018, 306, 96–101 CrossRef CAS.
- K. S. Midelfort, R. Kumar, S. Han, M. J. Karmilowicz, K. McConnell, D. K. Gehlhaar, A. Mistry, J. S. Chang, M. Anderson, A. Villalobos, J. Minshull, S. Govindarajan and J. W. Wong, Protein Eng. Des. Sel., 2013, 26, 25–33 CrossRef CAS PubMed.
- L. Skalden, C. Peters, J. Dickerhoff, A. Nobili, H. J. Joosten, K. Weisz, M. Höhne and U. T. Bornscheuer, ChemBioChem, 2015, 16, 1041–1045 CrossRef CAS PubMed.
- K. E. Cassimjee, M. S. Humble, H. Land, V. Abedib and P. Berglund, Org. Biomol. Chem., 2012, 10, 5466–5470 RSC.
- H. Yun, B. Y. Hwang, J. H. Lee and B. G. Kim, Appl. Environ. Microbiol., 2005, 71, 4220–4224 CrossRef CAS PubMed.
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 1–7 Search PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- H. Renata, Z. J. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351–3367 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–310 CrossRef CAS PubMed.
- J. Limanto, E. R. Ashley, J. Yin, G. L. Beutner, B. T. Grau, A. M. Kassim, M. M. Kim, A. Klapars, Z. Liu, H. R. Strotman and M. D. Truppo, Org. Lett., 2014, 16, 2716–2719 CrossRef CAS PubMed.
- A. R. Martin, R. DiSanto, I. Plotnikov, S. Kamat, D. Shonnard and S. Pannuri, Biochem. Eng. J., 2007, 37, 246–255 CrossRef CAS.
- M. T. Reetz, J. D. Carballeira, J. Peyralans, H. Höbenreich, A. Maichele and A. Vogel, Chem. – Eur. J., 2006, 12, 6031–6038 CrossRef CAS PubMed.
- H. Xiao, Z. Bao and H. Zhao, Ind. Eng. Chem. Res., 2015, 54, 4011–4020 CrossRef CAS PubMed.
- R. Obexer, A. Godina, X. Garrabou, P. R. E. Mittl, D. Baker, A. D. Griffiths and D. Hilvert, Nat. Chem., 2017, 9, 50–56 CrossRef CAS PubMed.
- C. K. Longwell, L. Labanieh and J. R. Cochran, Curr. Opin. Biotechnol., 2017, 48, 196–202 CrossRef CAS PubMed.
- C. Yan, F. Parmeggiani, E. A. Jones, E. Claude, S. A. Hussain, N. J. Turner, S. L. Flitsch and P. E. Barran, J. Am. Chem. Soc., 2017, 139, 1408–1411 CrossRef CAS PubMed.
- F. H. Arnold and G. Georgiou, Directed Enzyme Evolution, Humana Press, New Jersey, 2003, vol. 230 Search PubMed.
- I. Rowles, K. J. Malone, L. L. Etchells, S. C. Willies and N. J. Turner, ChemCatChem, 2012, 4, 1259–1261 CrossRef CAS.
- G. Matcham, M. Bhatia, W. Lang, C. Lewis, R. Nelson, A. Wang and W. Wu, Chim. Int. J. Chem., 1999, 53, 584–589 CAS.
- M. S. Weiß, I. V. Pavlidis, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Org. Biomol. Chem., 2016, 14, 10249–10254 RSC.
- M. S. Weiß, I. V. Pavlidis, C. Vickers, M. Hohne and U. T. Bornscheuer, Anal. Chem., 2014, 86, 11847–11853 CrossRef PubMed.
- S. C. Willies, J. L. Galman, I. Slabu and N. J. Turner, Philos. Trans. R. Soc. A Math. Phys. Eng. Sci., 2016, 374, 20150084 CrossRef PubMed.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- D. Baud, N. Ladkau, T. S. Moody, J. M. Ward and H. C. Hailes, Chem. Commun., 2015, 51, 17225–17228 RSC.
- L. Cerioli, M. Planchestainer, J. Cassidy, D. Tessaro and F. Paradisi, J. Mol. Catal. B Enzym., 2015, 120, 141–150 CrossRef CAS.
- A. Gomm, W. Lewis, A. P. Green and E. O'Reilly, Chem. – Eur. J., 2016, 22, 12692–12695 CrossRef CAS PubMed.
- X. Chen, J. Adrian, T. Cushing, H. DiMaio, L. Liang, V. Mayorga, S. Miao, M. G. Peterson, J. P. Powers, F. Spector, C. Stein, M. Wright, D. Xu, Q. Ye and J. Jaen, Bioorganic Med. Chem. Lett., 2007, 17, 2188–2192 CrossRef CAS PubMed.
- D. A. Drummond, B. L. Iverson, G. Georgiou and F. H. Arnold, J. Mol. Biol., 2005, 350, 806–816 CrossRef CAS PubMed.
- M. Wilding, T. S. Peat, S. Kalyaanamoorthy, J. Newman, C. Scott and L. S. Jermiin, Green Chem., 2017, 19, 5375–5380 RSC.
- L. Holm and P. Rosenström, Nucleic Acids Res., 2010, 38, 545–549 CrossRef PubMed.
- X. Robert and P. Gouet, Nucleic Acids Res., 2014, 42, W320–W324 CrossRef CAS PubMed.
- A. Nobili, Y. Tao, I. V. Pavlidis, T. Van Den Bergh, H. J. Joosten, T. Tan and U. T. Bornscheuer, ChemBioChem, 2015, 16, 805–810 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- T. Aleksiev, R. Potestio, F. Pontiggia, S. Cozzini and C. Micheletti, Bioinformatics, 2009, 25, 2743–2744 CrossRef CAS PubMed.
- B. C. Joseph, S. Pichaimuthu and S. Srimeenakshi, J. Cell Sci. Ther., 2015, 6, 221 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05712e |
| This journal is © The Royal Society of Chemistry 2019 |