
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral cyclopentadienyl RhIII-catalyzed enantioselective cyclopropanation of electron-deficient olefins enable rapid access to UPF-648 and oxylipin natural products†
Coralie
Duchemin
and
Nicolai
Cramer
 *
*
Laboratory of Asymmetric Catalysis and Synthesis, EPFL SB ISIC LCSA, BCH 4305, CH-1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
First published on 17th January 2019
Abstract
Chiral cyclopentadienyl RhIII complexes efficiently catalyze enantioselective cyclopropanations of electron-deficient olefins with N-enoxysuccinimides as the C1 unit. Excellent asymmetric inductions and high diastereoselectivities can be obtained for a wide range of substrate combinations. The reaction proceeds under mild conditions without precautions to exclude air and water. Moreover, the synthetic utility of the developed method is demonstrated by concise syntheses of members of the oxylipin natural products family and the KMO inhibitor UPF-648.
Introduction
Chiral cyclopropanes are important structural motifs frequently found in a diverse range of natural products and biologically active compounds.1 Cyclopropanes are attractive building blocks for drug discovery due to their rigid structure with defined three-dimensional vectors and their good metabolic stability.2 Moreover, they are versatile intermediates for synthesis as ring-opening reactions opens access to useful building blocks.3 Synthetically, the most practical strategy to build the cyclopropane motif consists of an enantioselective cycloaddition between an olefin and a suitable C1 unit.4 For instance, transition-metal catalyzed reactions5 – metal-carbenoid mediated transformations5a and the ring closure of π-allylpalladium species,5b Lewis-acid catalyzed Simmons–Smith reactions6 as well as radical processes7 have proven to be powerful methods for the asymmetric cyclopropanations of electron-rich olefins. Complementary, asymmetric Michael-initiated ring-closure (MIRC) reactions have been shown to be an attractive cyclopropanation method for electron-deficient olefins.8–10 Moreover, tailored transition-metal catalysts enable enantioselective cyclopropanations of electron-deficient olefins with diazo compounds.11 However, these transformations still have limitations in scope and frequently require potentially hazardous reactants. Therefore, the development of novel and efficient catalytic cyclopropanation strategies using complementary substrates remain an attractive and important task. In this respect, Rovis and co-workers recently reported a unique cyclopropanation using N-enoxyphthalimides and Michael acceptors as substrates (Fig. 1).12 Tailored achiral cyclopentadienyl RhIII catalysts enabled this transformation and moreover allowed to efficiently control its diastereoselectivity.12b Given our longstanding focus on the development of chiral cyclopentadienyl (Cpx) metal catalysts13 for challenging asymmetric transformations,14 we felt prompted to explore the feasibility of an enantioselective Rovis-cyclopropanation. This is a formidable challenge to expand the current boundaries of asymmetric CpxRhIII catalysis beyond functionalizations of aryl Csp2–H bonds.15 Despite ample precedence with achiral Cp*RhIII complexes,16 the corresponding asymmetric functionalization of alkenyl Csp2–H bonds with CpxRhIII catalysts remained so far elusive.17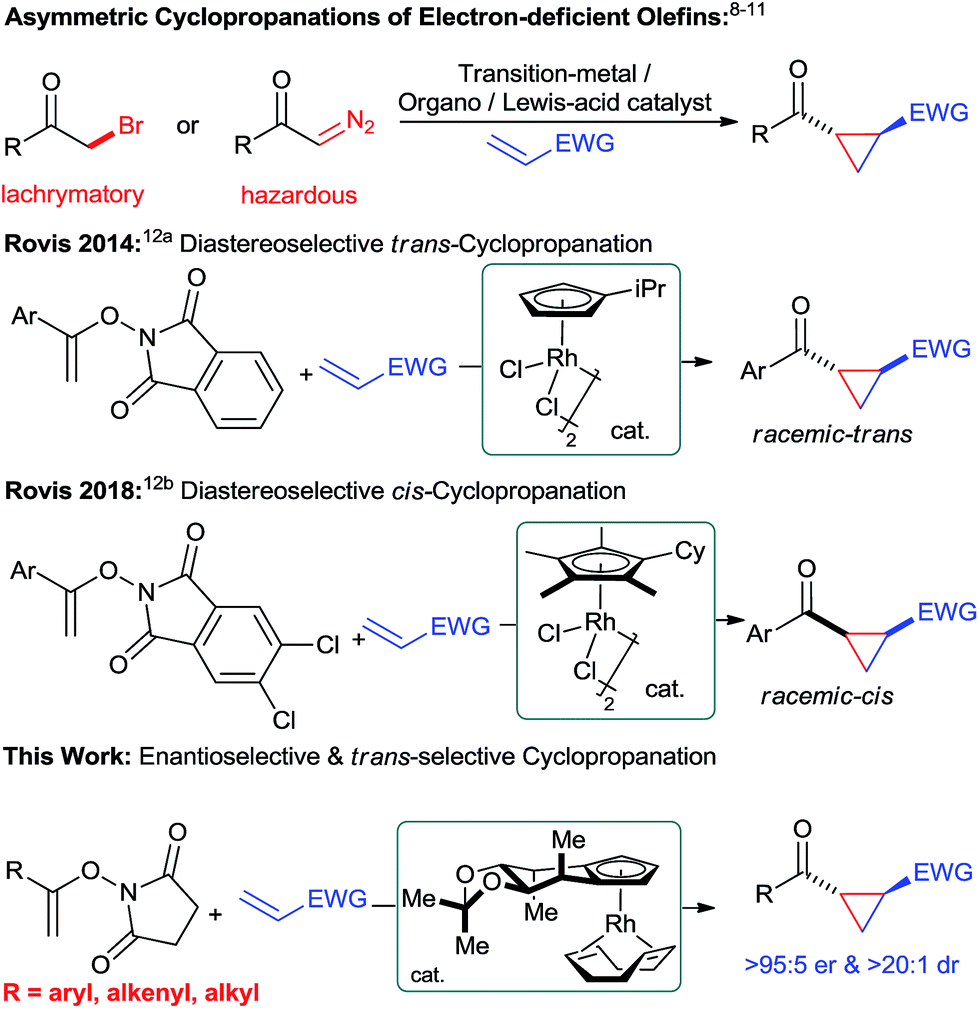 | ||
| Fig. 1 Catalytic methods for the selective cyclopropanation of electron-deficient olefins. | ||
Herein, we report a highly enantioselective alkenyl C–H bond functionalization providing access to chiral cyclopropanes under mild conditions.
Results and discussion
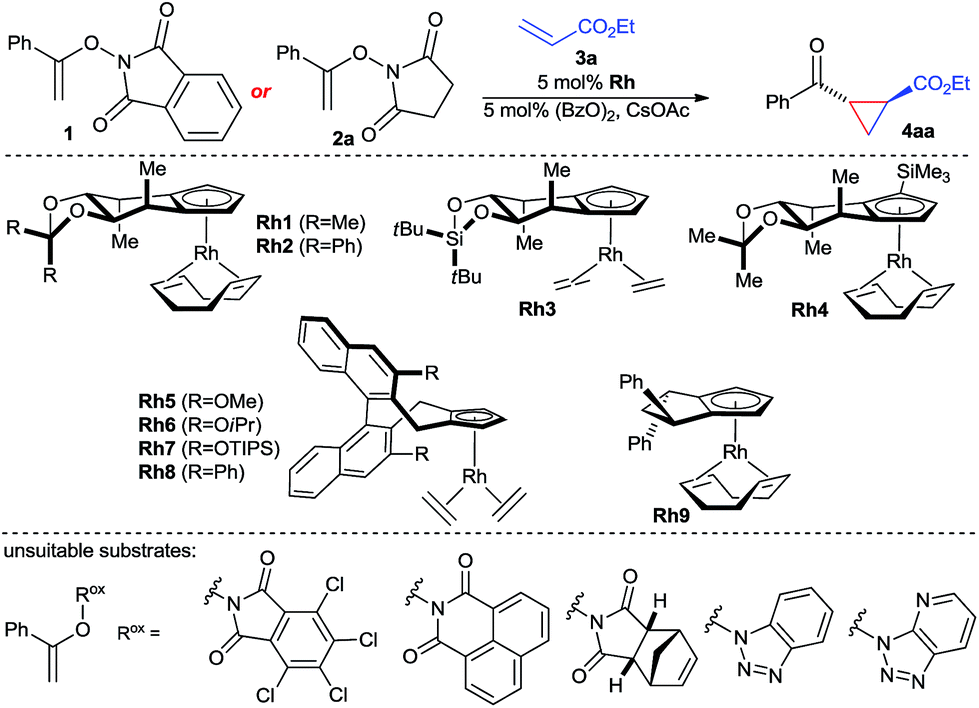
The envisioned enantioselective cyclopropanation was investigated with N-enoxyphthalimide 1 and ethyl acrylate (Table 1). Rh1 featuring our simplest first generation Cpx design13b provided desired cyclopropane 4aa in 71% yield, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trans/cis ratio and 93.5:6.5 er (entry 1). Increasing of the size of the backwall using a diphenyl acetal (Rh2) or a silyl bridge (Rh3) reduced the enantioselectivity (entries 2 and 3). Complex Rh4 with a trisubstituted TMS-bearing Cpx ligand13g was as well inferior (entry 4). Binaphthyl-derived ligands (Rh5–Rh8)13c are not suited and gave a general poor performance concerning yield, diastereo- and enantioselectivity (entries 5–8). Moreover, usage of Rh9 with a cyclopentyl-backbone Cpx ligand13f formed cyclopropane 4aa in negligible amounts (entry 9). The solvent has a large influence. Replacement of TFE by either ethanol or HFIP gave dramatically lower yields (entries 10 and 11). A lower reaction temperature (0 °C) caused a sluggish reaction with no discernible increase in enantioselectivity (entry 12), whereas heating to 50 °C triggered slight erosion in yield and selectivity (entry 13). A short premixing period between the rhodium catalyst and the oxidant increased the yield to 76% while maintaining an enantiomeric ratio of 93.5:6.5 (entry 14). The nature of the imide of the oxidizing directing group was important. A range of other oxidizing directing group Rox failed to provide the desired reactivity which was attributed to poor solubility. However, replacement of 1 by enoxysuccinimide 2a resulted in a cleaner and faster reaction, giving 4aa in 78% isolated yield with an improved excellent enantioselectivity of 97:3, although with a lower diastereoselectivity of 4:1 (entry 15).
:1 trans/cis ratio and 93.5:6.5 er (entry 1). Increasing of the size of the backwall using a diphenyl acetal (Rh2) or a silyl bridge (Rh3) reduced the enantioselectivity (entries 2 and 3). Complex Rh4 with a trisubstituted TMS-bearing Cpx ligand13g was as well inferior (entry 4). Binaphthyl-derived ligands (Rh5–Rh8)13c are not suited and gave a general poor performance concerning yield, diastereo- and enantioselectivity (entries 5–8). Moreover, usage of Rh9 with a cyclopentyl-backbone Cpx ligand13f formed cyclopropane 4aa in negligible amounts (entry 9). The solvent has a large influence. Replacement of TFE by either ethanol or HFIP gave dramatically lower yields (entries 10 and 11). A lower reaction temperature (0 °C) caused a sluggish reaction with no discernible increase in enantioselectivity (entry 12), whereas heating to 50 °C triggered slight erosion in yield and selectivity (entry 13). A short premixing period between the rhodium catalyst and the oxidant increased the yield to 76% while maintaining an enantiomeric ratio of 93.5:6.5 (entry 14). The nature of the imide of the oxidizing directing group was important. A range of other oxidizing directing group Rox failed to provide the desired reactivity which was attributed to poor solubility. However, replacement of 1 by enoxysuccinimide 2a resulted in a cleaner and faster reaction, giving 4aa in 78% isolated yield with an improved excellent enantioselectivity of 97:3, although with a lower diastereoselectivity of 4:1 (entry 15).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Rh | Solvent | T (°C) | % yieldb | Trans/cisc | erd |
| a 0.05 mmol 1, 0.055 mmol 3a, 2.5 μmol Rh, 2.5 μmol (BzO)2, 0.2 M in the indicated solvent and temperature for 16 h. b Isolated yield. c dr determined by 1H-NMR of the crude product. d er determined by HPLC analysis with a chiral stationary phase. e (BzO)2 and Rh were premixed for 2 min. f With 2a instead of 1. | ||||||
| 1 | Rh1 | TFE | 23 | 71 | >20:1 |
93.5:6.5 |
| 2 | Rh2 | TFE | 23 | 68 | >20:1 |
84:16 |
| 3 | Rh3 | TFE | 23 | 51 | >20:1 |
85:15 |
| 4 | Rh4 | TFE | 23 | 55 | >20:1 |
78.5:21.5 |
| 5 | Rh5 | TFE | 23 | 27 | 3.4:1 |
45:55 |
| 6 | Rh6 | TFE | 23 | 15 | 1.1:1 |
39:61 |
| 7 | Rh7 | TFE | 23 | 22 | 1:2.5 |
47:53 |
| 8 | Rh8 | TFE | 23 | 10 | 4.8:1 |
73:27 |
| 9 | Rh9 | TFE | 23 | <5 | — | — |
| 10 | Rh1 | HFIP | 23 | <5 | — | — |
| 11 | Rh1 | EtOH | 23 | 15 | >20:1 |
87:13 |
| 12 | Rh1 | TFE | 0 | 26 | >20:1 |
93.5:6.5 |
| 13 | Rh1 | TFE | 50 | 62 | 8:1 |
91.5:8.5 |
| 14e | Rh1 | TFE | 23 | 76 | >20:1 |
93.5:6.5 |
| 15e,f | Rh1 | TFE | 23 | 78 | 4:1 |
97:3 |
With the optimized conditions, the scope of the reaction was investigated (Scheme 1). A variety of acrylic esters were tested. Commonly used methyl, ethyl, butyl and benzyl esters gave the cyclopropane products with good yields, >95:5 er and useful diastereomeric ratios between 4:1 and 6:1. Notably, tert-butyl acrylate provided in all aspects superior results, giving 4ae in 85% yield with >20:1 dr and 97:3 er. Moreover, acrylamide derivatives, exemplified with morpholine 3f reacted smoothly, giving 4af in excellent dr and suitable yields and enantioselectivity. In particular, Weinreb acrylamide proved to be well suited, giving cyclopropane 4ag in 75% yield with >20:1 dr and 97:3 er. Surprisingly, both acrolein and MVK acceptors gave high yields of the corresponding cyclopropanes 4ah and 4ai, maintaining high levels of enantioselectivity. However, due to their small size, the diastereomeric ratio was with 1.6:1, respectively 2:1 lower. Interestingly, the cis-products were formed in approximately the same enantioselectivity. Besides MVK, similar reactivity was observed for longer chain vinyl ketone giving 4aj. Considering the dearth of methods for enantiopure cis-cyclopropanes from electron-poor olefins,18 this observation could be a starting point in the development of an enantioselective cis-selective variant. Heteroatom-based Michael acceptor such as phenyl vinyl sulfone/selenone or ethenesulfonyl fluoride did not undergo cyclopropanation. Acrylates with α or β-substitution were not reactive acceptors with the current catalytic system.
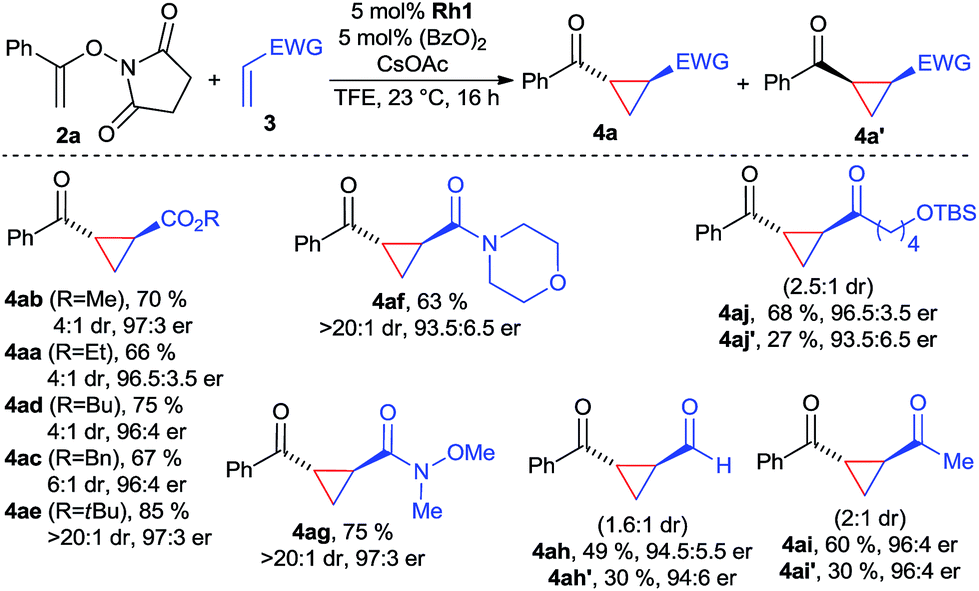 | ||
| Scheme 1 Suitable acceptors for the enantioselective cyclopropanation. Reaction conditions: 0.10 mmol 2a, 5.0 μmol Rh1, 5.0 μmol (BzO)2, 0.12 mmol 2, 0.2 M in TFE at 23 °C for 16 h; isolated yields; dr determined by 1H-NMR of the crude product; er determined by HPLC analysis with a chiral stationary phase. | ||
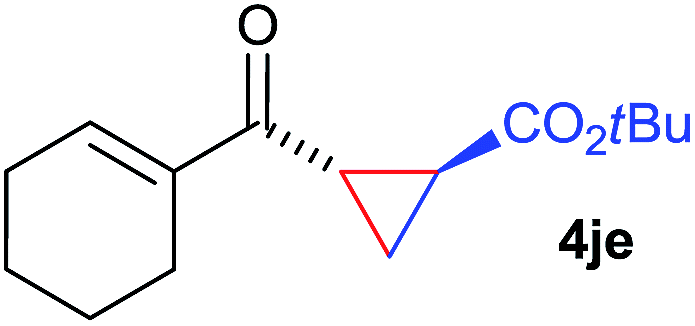
The range of suitable enoxy-succinimides was investigated (Table 2). We first evaluated variations of the steric and electronic properties of the aryl-substituted enoxy-succinimides. Electron-donating and withdrawing groups in the para position were found to have very little influence on the reaction outcome, providing high yields and enantioselectivities of the corresponding cyclopropanes 4 (entries 1–4). Along the same lines, meta- (2f) and ortho- (2g) substitution as well as heteroaryl (2i) and condensed aromatic substituent (2h) were tolerated well. Due to limited solubility in TFE, substrates having a naphthyl- (2h) or chloroarene substituent (2e) required longer reaction times. Attractively, besides aryl-substituted enoxy-succinimides, the cyclopropanation worked very well with dienenoxy substrates such as 2j and 2k giving enone products 4je and 4ke in an excellent er of 96:4. Notably, no competing Diels–Alder cycloaddition between the electron-rich diene and the acrylate acceptor was observed under the reaction conditions. Moreover, the reactivity, diastereo- and enantioselectivity were excellent for alkyl substituents, leading to functionalized cyclopropanes 4le and 4me (entries 11 and 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | 4 (R) | % yieldb | drc | erd |
| a 0.10 mmol 2, 5.0 μmol Rh1, 5.0 μmol (BzO)2, 0.12 mmol 3e, 0.20 mmol CsOAc, 0.2 M in TFE at 23 °C for 16 h. b Isolated yield. c Determined by 1H-NMR of the crude product. d Determined by HPLC analysis with a chiral stationary phase. e For 40 h. f For 56 h. | |||||
| 1 | 2b | 4be (4-Me-C6H4) | 85 | >20:1 |
97:3 |
| 2 | 2c | 4ce (4-OMe-C6H4) | 90 | >20:1 |
97.5:2.5 |
| 3 | 2d | 4de (4-F-C6H4) | 89 | >20:1 |
95:5 |
| 4e | 2e | 4ee (4-Cl-C6H4) | 81 | >20:1 |
97:3 |
| 5 | 2f | 4fe (3-MeO-C6H4) | 85 | >20:1 |
97:3 |
| 6 | 2g | 4ge (2-Me-C6H4) | 73 | >20:1 |
96:4 |
| 7f | 2h | 4he (2-naphthyl) | 83 | >20:1 |
93.5:6.5 |
| 8 | 2i | 4ie (3-thienyl) | 69 | >20:1 |
97.5:2.5 |
| 9 | 2j |
|
87 | >20:1 |
96:4 |
| 10 | 2k |
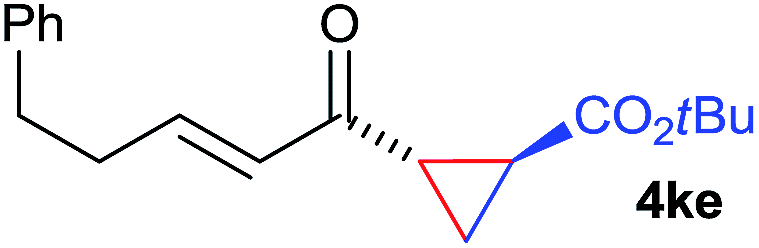
|
72 | >20:1 |
96:4 |
| 11 | 2l |
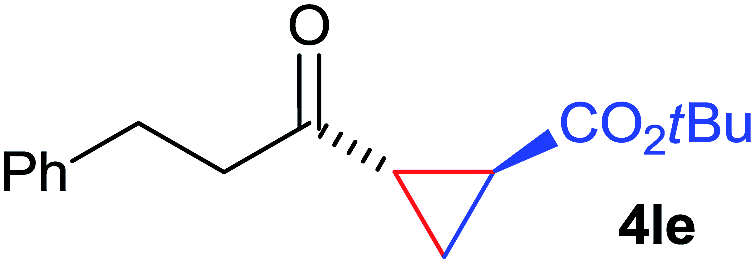
|
75 | >20:1 |
96:4 |
| 12 | 2m |
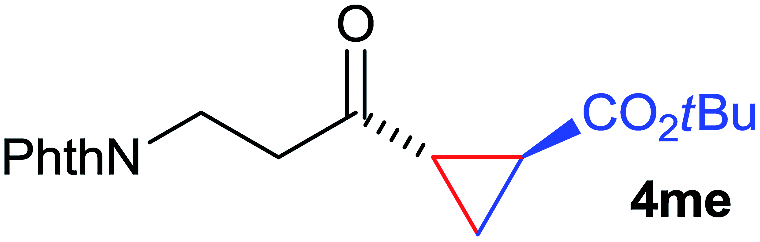
|
77 | >20:1 |
96:4 |
The synthetic utility of the method was demonstrated as key step in the synthesis of natural products and inhibitor UPC-648. Constanolactones19 and ent-eicosanoid 820 are marine oxylipins21 containing a trans-cyclopropane. Previous syntheses22,23 used lactone 7 as common intermediate which could be accessed in 623a or 13 steps.22c In a streamlined access to required N-enoxysuccinimide 2n, we developed a gold(I)-catalyzed addition of N-hydroxysuccinimide to terminal alkyne 5 which directly provided substrate 2n in 68% yield (Scheme 2).24 Subjecting 2n to the developed optimized enantioselective cyclopropanation conditions in the presence of Weinreb acryl amide 3g gave cyclopropane 4ng in 89% yield, 97:3 enantiomeric ratio and >20:1 dr. The transformation was efficient for gram-scale preparation giving 1.10 g of 4ng. Diastereoselective reduction of 4ng with Noyori's catalyst gave secondary alcohol 6 in 89% yield and 85:15 dr. Reduction of the Weinreb amide over the isopropyl ester of 6 and subsequent lactonization under acidic conditions yielded intermediate 7 in 50% yield over 2 steps. This intermediate can be elaborated either in a single step operation into constanolactone A and B,23a or by a two-step sequence into ent-eicosanoid 8.22c
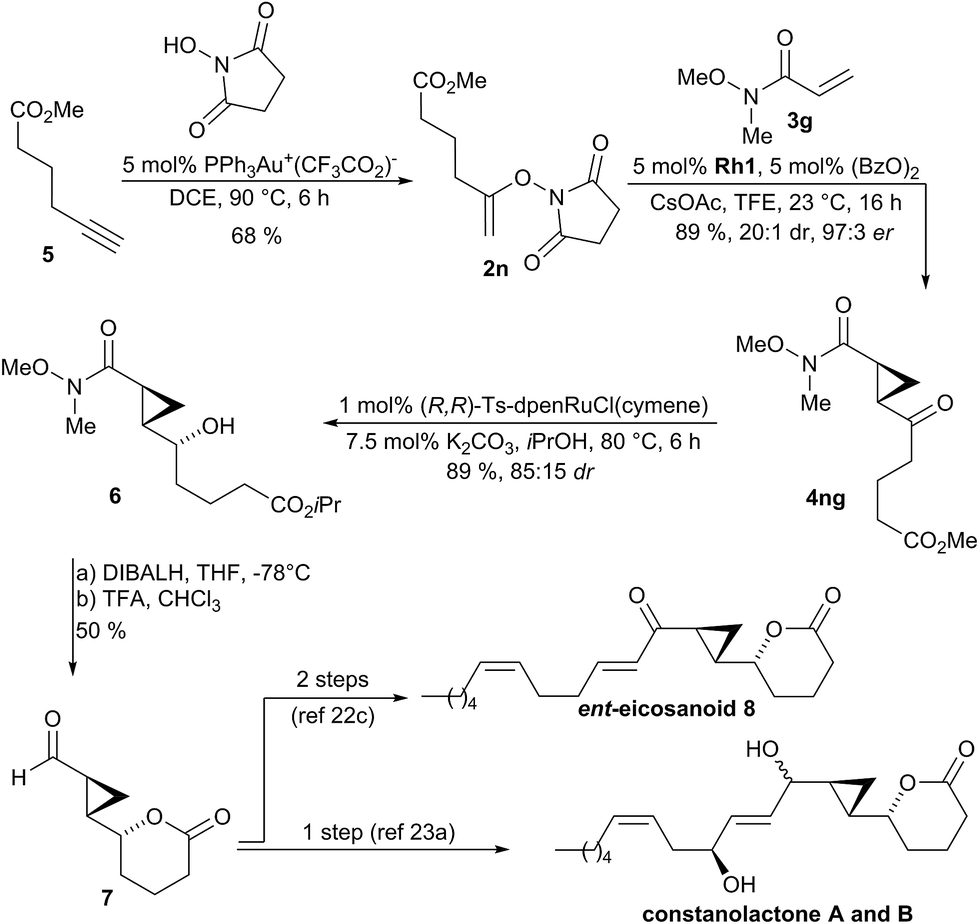 | ||
| Scheme 2 Synthetic application of the enantioselective cyclopropanation in the formal synthesis of members of oxylipin natural products family. | ||
UPF-648, a potent inhibitor (IC50 = 40 nM) for kynurenine 3-monooxygenase (KMO),25,26 was identified as another attractive target. Inhibition of KMO has therapeutic potential for several neurodegenerative disorders, including Huntington's disease.27 The two reported syntheses of UPF-648 are long and use a stoichiometric chiral auxiliary28 or involve a resolution.29 Therefore, a short catalytic enantioselective route represents significant synthetic value. Our synthesis starts with a gold-catalyzed addition of N-hydroxy succinimide to 3,4-dichloro phenyl acetylene (9) affording N-enoxysuccinimide 2o in 53% yield (Scheme 3). The enantioselective cyclopropanation was conducted without any precaution to exclude moisture or oxygen, giving cyclopropane 4oe in 80% yield and 95:5 er. Alternatively, application of our recently developed in situ CpxRh catalyst preparation13g provided 4oe in 76% yield and 94.5:4.5 er. Cleavage of the tert-butyl ester gave UPF-648 ester. A subsequent recrystallization increased its optical purity to 99:1 er. Overall, UPF-648 could be synthesized in 3 steps in a catalytic enantioselective fashion with an overall yield of 39%.
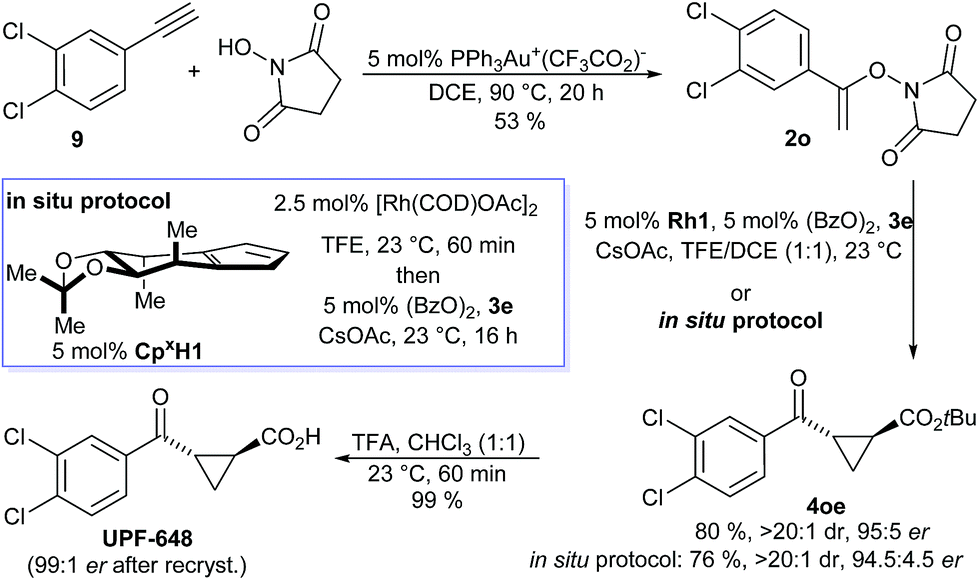 | ||
| Scheme 3 Synthetic application of the enantioselective cyclopropanation in the formal synthesis of the KMO inhibitor UPF-648. | ||
Conclusions
In summary, we have developed a highly enantioselective and diastereoselective cyclopropanation of electron-deficient olefins using enoxysuccinimides as the one-carbon component. The transformation is catalyzed by chiral CpxRhIII complexes and operates under mild and open-flask reaction conditions. We applied the transformation as a key step in the synthesis of the oxylipin family of natural products and the kynurenine 3-monooxygenase inhibitor UPF-648, showcasing its synthetic utility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Swiss National Science Foundation (no. 157741).Notes and references
- (a) A. de Meijere, S. I. Kozhushkov and H. Schill, Chem. Rev., 2006, 106, 4926 CrossRef CAS PubMed; (b) J. Pietruszka, Chem. Rev., 2003, 103, 1051 CrossRef CAS PubMed.
- (a) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625 CrossRef CAS PubMed; (b) T. T. Talele, J. Med. Chem., 2016, 59, 8712 CrossRef CAS PubMed.
- (a) D. C. Nonhebel, Chem. Soc. Rev., 1993, 22, 347 RSC; (b) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651 CrossRef CAS PubMed.
- (a) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (b) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS; (c) W. Wu, Z. Lin and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315 RSC.
- (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (b) J.-Q. Huang, W. Liu, B.-H. Zheng, X. Y. Liu, Z. Yang, C.-H. Ding, H. Li, Q. Peng and X.-L. Hou, ACS Catal., 2018, 8, 1964 CrossRef CAS.
- A. B. Charette, C. Molinaro and C. Brochu, J. Am. Chem. Soc., 2001, 123, 12168 CrossRef CAS.
- L. Ye, Q.-S. Gu, Y. Tian, X. Meng, G.-C. Chen and X.-Y. Liu, Nat. Commun., 2018, 9, 227 CrossRef PubMed.
- (a) C. D. Papageorgiou, M. A. Cubillo de Dios, S. V. Ley and M. J. Gaunt, Angew. Chem., Int. Ed., 2004, 43, 4641 CrossRef CAS PubMed; (b) C. C. C. Johansson, N. Bremeyer, S. V. Ley, D. R. Owen, S. C. Smith and M. J. Gaunt, Angew. Chem., Int. Ed., 2006, 45, 6024 CrossRef CAS PubMed.
- (a) V. K. Aggarwal, H. W. Smith, R. V. H. Jones and R. Fieldhouse, Chem. Commun., 1997, 1785 RSC; (b) V. K. Aggarwal, H. W. Smith, G. Hynd, R. V. H. Jones, R. Fieldhouse and S. E. Spey, J. Chem. Soc., Perkin Trans. 1, 2000, 3267 RSC; (c) V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcelloni, Angew. Chem., Int. Ed., 2001, 40, 1433 CrossRef CAS.
- (a) L. Gao, G.-S. Hwang and D. H. Ryu, J. Am. Chem. Soc., 2011, 133, 20708 CrossRef CAS PubMed; (b) S. Y. Shim, J. Y. Kim, M. Nam, G.-S. Hwang and D. H. Ryu, Org. Lett., 2016, 18, 160 CrossRef CAS PubMed; (c) G.-J. Sun, J. Gong and Q. Kang, J. Org. Chem., 2017, 82, 796 CrossRef CAS PubMed; (d) X. Zhong, J. Lv and S. Luo, Org. Lett., 2017, 19, 3331 CrossRef CAS PubMed.
- (a) J. A. Miller, W. Jin and S. T. Nguyen, Angew. Chem., Int. Ed., 2002, 41, 2953 CrossRef CAS; (b) Y. Chen, J. V. Ruppel and X. P. Zhang, J. Am. Chem. Soc., 2007, 129, 12074 CrossRef CAS PubMed; (c) S. Zhu, X. Xu, J. A. Perman and X. P. Zhang, J. Am. Chem. Soc., 2010, 132, 12796 CrossRef CAS PubMed; (d) H. Wang, D. M. Guptill, A. Varela-Alvarez, D. G. Musaev and H. M. L. Davies, Chem. Sci., 2013, 4, 2844 RSC; (e) X. Xu, Y. Wang, X. Cui, L. Wojtas and X. P. Zhang, Chem. Sci., 2017, 8, 4347 RSC.
- (a) T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292 CrossRef CAS PubMed; (b) T. Piou, F. Romanov-Michailidis, M. A. Ashley, M. Romanova-Michaelides and T. Rovis, J. Am. Chem. Soc., 2018, 140, 9587 CrossRef CAS PubMed; (c) T. Piou and T. Rovis, Acc. Chem. Res., 2018, 51, 170 CrossRef CAS PubMed; (d) S. Vásquez-Céspedes, X. Wang and F. Glorius, ACS Catal., 2018, 8, 242 CrossRef.
- (a) C. G. Newton, D. Kossler and N. Cramer, J. Am. Chem. Soc., 2016, 138, 3935 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (c) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636 CrossRef CAS PubMed; (d) D. Kossler and N. Cramer, J. Am. Chem. Soc., 2015, 137, 12478 CrossRef CAS PubMed; (e) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed; (f) S.-G. Wang, S. H. Park and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 5459 CrossRef CAS PubMed; (g) B. Audic, M. D. Wodrich and N. Cramer, Chem. Sci., 2019, 10, 781 RSC.
- (a) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (b) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed; (c) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, 759 CrossRef CAS PubMed.
- (a) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed; (b) B. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2013, 53, 507 CrossRef PubMed; (c) B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896 CrossRef CAS PubMed; (d) J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244 CrossRef CAS PubMed; (e) B. Ye and N. Cramer, Synlett, 2015, 26, 1490 CrossRef CAS; (f) J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2015, 137, 4880 CrossRef CAS PubMed; (g) S. Reddy Chidipudi, D. J. Burns, I. Khan and H. W. Lam, Angew. Chem., Int. Ed., 2015, 54, 13975 CrossRef CAS PubMed; (h) M. V. Pham and N. Cramer, Chem.–Eur. J., 2015, 22, 2270 CrossRef PubMed; (i) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242 CrossRef CAS PubMed; (j) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2016, 56, 364 CrossRef PubMed; (k) T. J. Potter, D. N. Kamber, B. Q. Mercado and J. A. Ellman, ACS Catal., 2017, 7, 150 CrossRef CAS PubMed; (l) Z.-J. Jia, C. Merten, R. Gontla, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2429 CrossRef CAS PubMed; (m) J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 4540 CrossRef CAS PubMed; (n) Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981 RSC; (o) E. A. Trifonova, N. M. Ankudinov, A. A. Mikhaylov, D. A. Chusov, Y. V. Nelyubina and D. S. Perekalin, Angew. Chem., Int. Ed., 2018, 57, 7714 CrossRef CAS PubMed; (p) G. Shan, J. Flegel, H. Li, C. Merten, S. Ziegler, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2018, 57, 14250 CrossRef CAS PubMed; (q) B. Shen, B. Wan and X. Li, Angew. Chem., Int. Ed., 2018, 57, 15534 CrossRef CAS PubMed; (r) N. Cramer and Y. Sun, Angew. Chem., Int. Ed., 2018, 57, 15539 CrossRef PubMed.
- (a) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed; (b) T. Piou and T. Rovis, Nature, 2015, 527, 86 CrossRef CAS PubMed; (c) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780 CrossRef CAS PubMed; (d) M. Boultadakis-Arapinis, M. N. Hopkinson and F. Glorius, Org. Lett., 2014, 16, 1630 CrossRef CAS PubMed; (e) K. D. Collins, F. Lied and F. Glorius, Chem. Commun., 2014, 50, 4459 RSC; (f) C. Feng, D. Feng and T.-P. Loh, Chem. Commun., 2014, 50, 9865 RSC; (g) C. Feng, D. Feng, Y. Luo and T.-P. Loh, Org. Lett., 2014, 16, 5956 CrossRef CAS PubMed.
- (a) N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Chem. Lett., 1997, 26, 425 CrossRef; (b) K. Tsuchikama, Y. Kuwata, Y.-k. Tahara, Y. Yoshinami and T. Shibata, Org. Lett., 2007, 9, 3097 CrossRef CAS PubMed; (c) D. F. Fernández, M. Gulías, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2017, 56, 9541 CrossRef PubMed.
- (a) S. Bachmann, M. Furler and A. Mezzetti, Organometallics, 2001, 20, 2102 CrossRef CAS; (b) W. Hu, D. J. Timmons and M. P. Doyle, Org. Lett., 2002, 4, 901 CrossRef CAS PubMed; (c) M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 18002 CrossRef CAS PubMed; (d) H. Suematsu, S. Kanchiku, T. Uchida and T. Katsuki, J. Am. Chem. Soc., 2008, 130, 10327 CrossRef CAS PubMed; (e) B. Herlé, P. M. Holstein and A. M. Echavarren, ACS Catal., 2017, 7, 3668 CrossRef PubMed; (f) O. F. Brandenberg, C. K. Prier, K. Chen, A. M. Knight, Z. Wu and F. H. Arnold, ACS Catal., 2018, 8, 2629 CrossRef CAS.
- D. G. Nagle and W. H. Gerwick, Tetrahedron Lett., 1990, 31, 2995 CrossRef CAS.
- S. W. Baertschi, A. R. Brash and T. M. Harris, J. Am. Chem. Soc., 1989, 111, 5003 CrossRef CAS.
- W. H. Gerwick, Chem. Rev., 1993, 93, 1807 CrossRef CAS.
- (a) J. D. White and M. S. Jensen, J. Am. Chem. Soc., 1993, 115, 2970 CrossRef CAS; (b) T. Nagasawa, Y. Handa, Y. Onoguchi and K. Suzuki, Bull. Chem. Soc. Jpn., 1996, 69, 31 CrossRef CAS; (c) G. Kumaraswamy and M. Padmaja, J. Org. Chem., 2008, 73, 5198 CrossRef CAS PubMed; (d) V. Angamuthu, W.-J. Chang and D.-R. Hou, ACS Omega, 2017, 2, 4088 CrossRef CAS.
- (a) J. D. White and M. S. Jensen, J. Am. Chem. Soc., 1995, 117, 6224 CrossRef CAS; (b) J. Yu, J.-Y. Lai, J. Ye, N. Balu, L. M. Reddy, W. Duan, E. R. Fogel, J. H. Capdevila and J. R. Falck, Tetrahedron Lett., 2002, 43, 3939 CrossRef CAS; (c) J. Pietruszka and T. Wilhelm, Synlett, 2003, 1698 CrossRef CAS.
- C. Duchemin and N. Cramer, Org. Chem. Front., 2019, 6, 209 RSC.
- L. Vécsei, L. Szalárdy, F. Fülöp and J. Toldi, Nat. Rev. Drug Discovery, 2012, 12, 64 CrossRef PubMed.
- M. Amaral, C. Levy, D. J. Heyes, P. Lafite, T. F. Outeiro, F. Giorgini, D. Leys and N. S. Scrutton, Nature, 2013, 496, 382 CrossRef CAS PubMed.
- M. T. Sapko, P. Guidetti, P. Yu, D. A. Tagle, R. Pellicciari and R. Schwarcz, Exp. Neurol., 2006, 197, 31 CrossRef CAS PubMed.
- L. Benatti, R. Fariello, P. Salvati, R. Pellicciari and C. Caccia, EP1475385 A1, 2004.
- M. Varasi, A. Giordani, P. Pevarello, R. Pellicciari and C. Speciale, WO 9840344, 1991.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds. See DOI: 10.1039/c8sc05702h |
| This journal is © The Royal Society of Chemistry 2019 |