
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solar electricity and fuel production with perylene monoimide dye-sensitised TiO2 in water†
Julien
Warnan
a,
Janina
Willkomm
a,
Yoann
Farré
b,
Yann
Pellegrin
b,
Mohammed
Boujtita
 *b,
Fabrice
Odobel
*b and
Erwin
Reisner
*a
*b,
Fabrice
Odobel
*b and
Erwin
Reisner
*a
aChristian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
bUniversité LUNAM, Université de Nantes, CNRS, Chimie et Interdisciplinarité: Synthèse, Analyse, Modélisation (CEISAM), UMR 6230, 2 rue de la Houssinière, 44322 Nantes cedex 3, France. E-mail: fabrice.odobel@univ-nantes.fr; hamada.boujtita@univ-nantes.fr
First published on 21st December 2018
Abstract
Dye-sensitisation of TiO2 and other metal oxides is an established strategy to couple solar light harvesting with efficient charge separation for the production of electricity in dye-sensitised solar cells (DSCs) or fuels in dye-sensitised semiconductor photocatalysis (DSP). Perylene monoimide (PMI) dyes have emerged as promising organic dyes, but they have not previously been used in a functional assembly with TiO2 in aqueous solution. Here, five novel PMI dyes bearing carboxylic acid, phosphonic acid, acetylacetone, hydroxyquinoline or dipicolinic acid anchoring groups for attachment onto TiO2 are reported. We identified functional DSC and DSP systems with PMI-sensitised TiO2 in aqueous solution, which permitted a side-by-side comparison with respect to performance between the two systems. Structure–activity relationships allowed us to suggest anchor-condition-system associations to suit specific anchoring groups at various pH values, and with different electron mediators (redox couple or sacrificial electron donor) and catalysts in DSC and DSP schemes. A DSC sensitised with the hydroxyquinoline-modified PMI dye reached the highest short-circuit current density (JSC ≈ 1.4 mA cm−2) in aqueous electrolyte solution during irradiation with simulated solar light. This dye also achieved a turnover number (TONPMI) of approximately 4900 for sacrificial proton reduction after 24 h irradiation in a DSP scheme with Pt as a H2-evolving co-catalyst at pH 4.5. This performance was only surpassed by the carboxylic acid-bearing dye, which reached a new benchmark turnover number (TONPMI ≈ 1.1 × 104 after 72 h) for an organic dye in nanoparticulate DSP for solar fuel production. At higher pH (8.5), our results showed that the phosphonic acid group allows for higher performance due to a stronger anchoring ability. This study provides a platform for aqueous PMI dye-sensitised TiO2 chemistry and gives valuable insights into the performance of different anchoring groups in DSC and DSP systems.
Introduction
Dye-sensitised semiconductor technology has emerged as a sustainable approach to produce renewable electricity in dye-sensitised solar cells (DSCs) and fuels in dye-sensitised photocatalysis (DSP) from abundant sunlight (Fig. 1).1–6 In a DSC, a photovoltage is produced through the photoexcitation of a sensitiser (S) anchored on a semiconductor electrode, followed by efficient charge injection into the semiconductor. These extracted charges are shuttled to a counter electrode (typically Pt), where they react with a diffusional redox mediator (M) that ultimately regenerates the ionised sensitiser. Analogously, solar irradiation of a DSP system results in accumulation of photoinjected electrons in the conduction band (CB) of the semiconducting particle. These electrons can subsequently be released to a co-immobilised catalyst to perform fuel synthesis (e.g. H2 production from proton reduction).4,6–8 The vast majority of DSP systems currently still rely on sacrificial electron donors (EDs) to regenerate the oxidised dye.4,9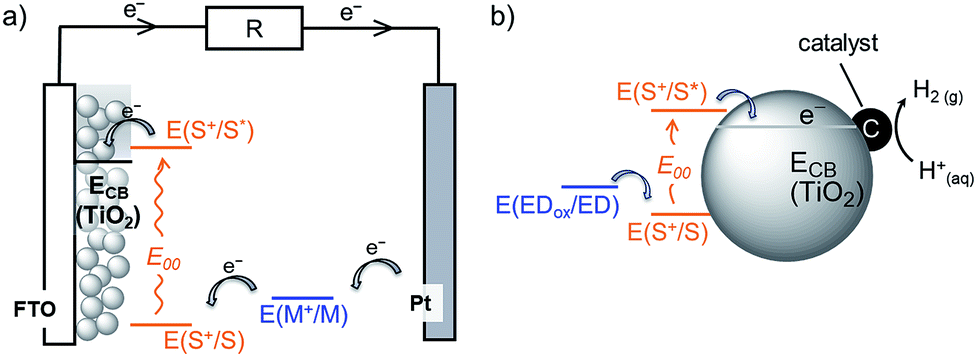 | ||
| Fig. 1 Schematic representation of solar energy conversion with dye-sensitised TiO2: (a) DSC3 and (b) DSP systems.4 See text for details. | ||
Operation in water is desirable for dye-sensitised technologies as this potentially reduces costs, flammability, instability and environmental incompatibility.10 Despite seminal reports on aqueous DSCs in the 1980s, organic solvents (e.g. acetonitrile) have been predominantly used during the past decades as they enable high efficiencies via dye and mediator engineering.3,11–14 However, increasing attention to fully green processes has driven growing efforts towards aqueous DSCs, notably targeting optimised electrolytes (e.g. redox mediator and additives), improved surface wettability or water-tolerant dyes.15–23 DSP for water splitting is naturally inclined towards an aqueous solution as water can act as both solvent and substrate for H2 and O2 evolution.4
Nanoparticulate TiO2 is the prototype semiconductor as it is inexpensive, stable, displays excellent charge transfer kinetics and has suitable energy levels for DSCs and DSP.24 The dye is the second pivotal component in DSC and DSP technologies as it collects photons and governs the kinetics of charge injection into the semiconductor, as well as dye regeneration and charge recombination. Nevertheless, dyes have rarely been optimised for efficient performance in an aqueous dye-semiconductor environment, and therefore typically show low device efficiencies under such conditions.10,17 In this context, organic chromophores have notable advantages over Ru-based dyes in terms of abundancy of their atoms, tunability and strong π–π* transitions, making them suitable candidates for dye-sensitised technologies.25–29
Here, we report the synthesis of five novel perylene monoimide (PMI) dyes and their incorporation into aqueous DSCs and H2-producing nanoparticulate DSP systems. PMI chromophores benefit from an established synthetic protocol, adjustable electronic and photophysical properties, good light stability and high photovoltaic performance.30–34 The synthesised PMI dyes were characterised by 1H NMR, 13C NMR, 31P NMR spectroscopy and HRMS, and their optoelectronic properties were assessed using electrochemistry and UV-vis spectroscopy. The five PMI dyes differ in their anchor functionality for binding to TiO2, bearing either a carboxylic acid (PMI-CO2H), a phosphonic acid (PMI-PO3H2), an acetylacetone (PMI-Acac), a hydroxyquinoline (PMI-HQui) or a dipicolinic acid (PMI-DPA) group (Chart 1). The anchor moiety affects not only the electronic properties of the PMI, but also modulates proximity, binding strength and electronic communication at the dye-semiconductor interface. In addition, the backbone of the chosen PMI dyes contain bulky hydrophobic units to limit deleterious dye aggregation and to minimise desorption in aqueous media.35,36
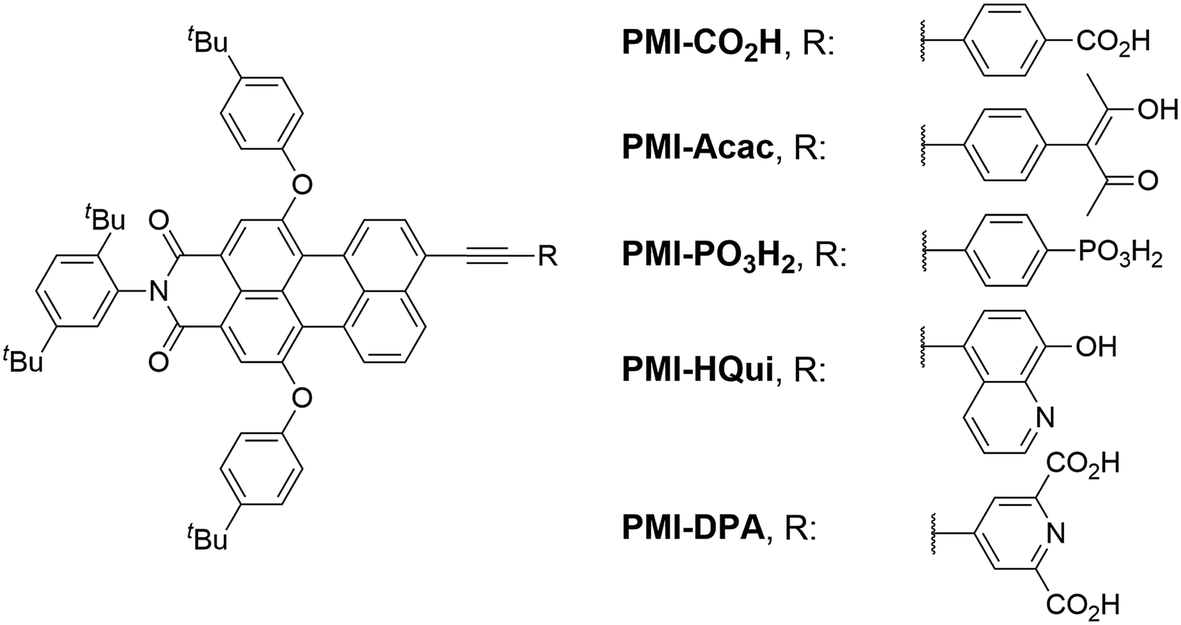 | ||
| Chart 1 Chemical structure of PMI dyes with different anchoring groups used in this study. | ||
Even though the nature of the anchor functionality is a vital part of the sensitiser in DSC and DSP, it has rarely been investigated in aqueous media, or using the same chromophore unit in DSC and DSP.37,38 The five PMI dyes were systematically studied side-by-side in DSC and DSP systems, at basic, neutral and acidic pH, in combination with I3−/I−, ascorbic acid (AA) or triethanolamine (TEOA) as mediators or sacrificial EDs. The diversity of studied systems, electron donors and pH values provides decisive information for the future design of dye-sensitised technology.
Results and discussion
Synthesis of PMI dyes
The preparation of the PMI dyes is based on a divergent modular synthetic approach starting from the brominated perylene monoimide PMI-Br (1) (Scheme 1).35 A Pd-catalysed Sonogashira cross-coupling reaction between PMI-Br and ethynyl derivatives bearing different anchoring groups generated the desired products. PMI-CO2H was obtained in 93% yield by coupling PMI-Br with 4-ethynylbenzoic acid (2). PMI-HQui and PMI-DPA were synthesised by reacting PMI-Br with 8-tert-butoxycarbonyloxy-5-ethynylquinoline (3) and 4-ethynyldipicolinic methyl ester (4), followed by cleavage of the Boc protecting group and ester hydrolysis in 46% and 33% overall yield, respectively. | ||
| Scheme 1 Synthesis of the PMI dyes with different anchoring groups. (i) Pd(PPh3)4, CuI, toluene, DIPEA or Et3N, [45 to 60] °C, [3 to 22] h; (ii) piperidine, DCM, r.t., 5 min; (iii) K2CO3, THF/H2O, 50 °C, 16 h; (iv) K2CO3, DCM/MeOH, r.t., 3 h; (v) 1,4-diiodobenzene, Pd(PPh3)4, CuI, toluene, Et3N, 70 °C, 2 h; (vi) DMIBE, Pd(PPh3)4, Cs2CO3, toluene/MeOH, 50 °C, 5 h; (vii) (a) Mo(CO)6, toluene/CH3CN/H2O, 90 °C, 3 h; (b) oxalic acid dihydrate, THF/H2O, 80 °C, 16 h; (viii) HPO3Et2, Et3N, Pd(PPh3)4, THF, 60 °C, 24 h; (ix) (a) Me3SiBr, Et3N, DCM, 50 °C, 5 h; (b) MeOH, r.t., 16 h. See ESI† for synthesis and characterisation of compounds 3 and 4. | ||
The synthesis of PMI-Acac and PMI-PO3H2 started from coupling of PMI-Br with ethynyltrimethylsilane followed by the cleavage of the trimethylsilyl group. A second Sonogashira reaction was subsequently performed between the resulting ethynyl-substituted PMI and an excess of 1,4-diiodobenzene to afford intermediate 5 in 81% yield. PMI-Acac was synthesised in two steps: a Suzuki–Miyaura reaction between 5 and 3,5-dimethylisoxazole-4-boronic acid pinacol ester (DMIBE) was followed by the opening of the isoxazole ring using [Mo(CO)6], and hydrolysis of the intermediate β-ketoenamine to give PMI-Acac (77% overall).39 The PMI-PO3H2 sensitiser was also obtained in two steps via a palladium-catalysed Hirao reaction from the iodo-PMI derivative 5 and diethyl phosphite, followed by hydrolysis of the formed phosphonate ester using Me3Si–Br and MeOH affording PMI-PO3H2 in an overall yield of 93%.
Electronic properties
The influence of the different anchoring groups on the electronic properties of the PMI photosensitisers was studied by electronic absorption spectroscopy in N,N-dimethylformamide (DMF) solution. All PMI dyes displayed a broad and intense (εmax > 3.5 × 104 M−1 cm−1) absorption in the visible part (from 450 to 650 nm) of the solar spectrum (Fig. S1†) with an absorption maximum at 535 nm and a shoulder at 500 nm. No significant difference was observed between the dyes' visible absorption, which suggests that the anchors do not directly affect the PMI transition. An exception is PMI-HQui, which displays a red-shifted absorption maximum (λmax = 545 nm) owing to the presence of a charge transfer band between the electron-rich quinoline unit and the PMI core. Consequently, this feature enables PMI-HQui to absorb photons from a wider range of the visible part of the solar spectrum.In order to obtain a better assessment on the light harvesting ability of the DSC and DSP systems, we recorded the absorption spectra of the PMI dyes (Fig. 2) upon immobilisation on 6 μm-thick, mesoporous anatase TiO2 electrodes. The TiO2 electrodes were prepared from an anatase paste, following previously published procedures, and sensitised by soaking the electrodes in a 0.25 mM solution of the PMI dye in DMF overnight (see ESI† for details). On TiO2, the dyes maintained a broad absorption in the visible spectrum, whereas λmax was generally observed at approximately 500 nm with a shoulder at 535 nm. Although also potentially affected by the protonation state of the anchoring group after immobilisation, this inversion behaviour of the two maxima intensities (at 500 and 535 nm) is in line with PMI aggregation affecting vibronic peak intensities.40,41
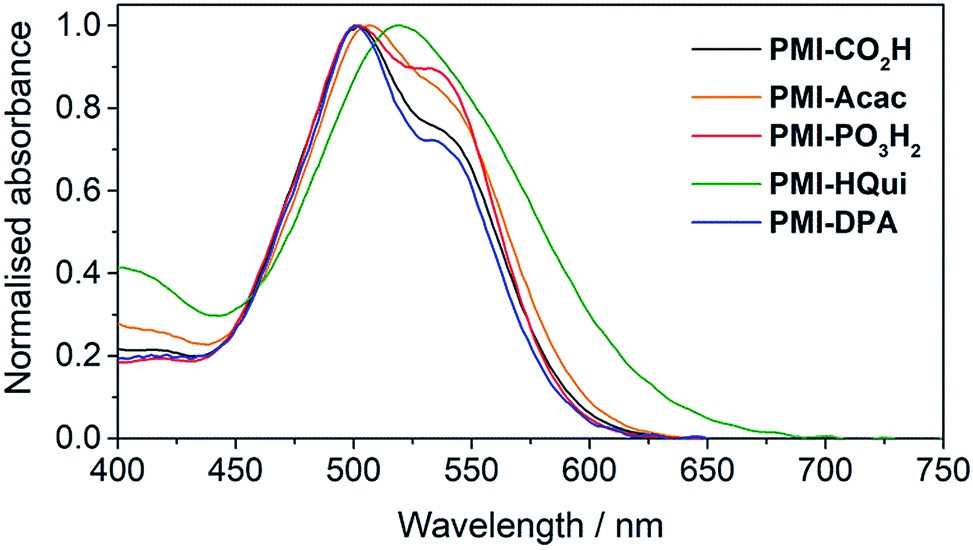 | ||
| Fig. 2 UV-Vis spectra of PMI dyes immobilised on a thin TiO2 film (6 μm thickness) recorded at room temperature. | ||
Electrochemical experiments were performed in a mixed dichloromethane (DCM)/DMF (95/5, v/v) solution – used instead of pure DMF conditions in order to reach higher dye concentration – with tetrabutylammonium hexafluorophosphate (TBAP, 0.1 M) as the supporting electrolyte. All cyclic voltammograms display an irreversible wave, located (onset potential) at approximately +1.4 V vs. normal hydrogen electrode (NHE) corresponding to the oxidation of the perylene core. This anodic wave is observed at more negative values in PMI-HQui (Eox(PMI-HQui) = +1.2 V vs. NHE), and at slightly more positive values in case of the PMI-DPA (Eox(PMI-DPA) ≈ +1.5 V vs. NHE). In the former case, this shift can be explained by the electron-rich hydroxyquinoline unit which destabilises the highest occupied molecular orbital energy level. The electron withdrawing effect of dipicolinic acid can explain the more difficult oxidation of PMI-DPA.
Fluorescence spectra of the PMI sensitisers were recorded in a diluted DMF solution, and the energy of the 0–0 transition values (E00) were estimated at approximately 2.22 eV by using the intersection between the normalised absorption and luminescence spectra (Fig. S2†). The different anchors do not strongly affect E00 values, giving oxidation potentials in the excited state (E(S+/S*)) between −1.01 and −0.75 V vs. NHE corresponding to PMI-HQui and PMI-DPA, respectively (Table 1).
| Dye | λ max (nm)a/(ε, M−1 cm−1) | E 00 (eV) | E(S+/S) (V vs. NHE) | E(S+/S*)b(V vs. NHE) |
|---|---|---|---|---|
| a In DMF. b E(S+/S*) = E(S+/S) − E00. S: ground state of PMI, S*: excited state, S+: oxidised state. | ||||
| PMI-CO2H | 536 (4.9 × 104) | 2.21 | 1.44 | −0.77 |
| PMI-Acac | 538 (5.4 × 104) | 2.22 | 1.34 | −0.88 |
| PMI-PO3H2 | 536 (3.8 × 104) | 2.21 | 1.43 | −0.78 |
| PMI-HQui | 545 (5.6 × 104) | 2.21 | 1.20 | −1.01 |
| PMI-DPA | 536 (4.0 × 104) | 2.24 | 1.49 | −0.75 |
The photoactivity mechanism of n-type DSC and DSP systems relies on an oxidative quenching of the sensitiser excited state (S*) by the conduction band of the semiconductor, followed by the regeneration of the dye ground state by the redox mediator or ED (Fig. 1).4 Considering the oxidation potentials of the redox mediator (E(I3−/I−) = 0.54 V vs. NHE) and sacrificial EDs (E(TEOA+/TEOA) = 0.82 V vs. NHE, pH 7.0; E(AA+/AA) < 0.20 V vs. NHE, pH 4.5) used in our study, dye regeneration is highly exergonic and therefore thermodynamically favourable.42,43 Similarly, a conduction band potential of TiO2, ECB(TiO2), of approximately −0.55 V vs. NHE (at pH 4.5) indicates a thermodynamically favourable electron injection.44 However, the driving force could become insufficient as the pH increases and ECB(TiO2) edge becomes more negative (shift of −59 mV per pH unit increase) (see Table S1†).45,46
Dye-sensitised electrodes
We investigated and compared the photo-conversion efficiency of PMI-sensitised DSCs, in water and acetonitrile (ACN) as an organic solvent benchmark, using sensitised TiO2 anodes (100% anatase, 0.25 cm2, thickness 16 μm) in combination with I3−/I− as the redox mediator and Pt as a counter electrode (Tables 2 and S2†). Digital photography of the sensitised electrodes and of an assembled DSC are available in Fig. S3.† Measurements were performed at room temperature without mask under simulated solar light (AM 1.5 G, 100 mW cm−2). Photovoltaic efficiencies (ηs) from <0.1% to 0.5% were recorded in aqueous electrolyte solution, highlighting a substantial difference in performance between the PMI dyes with different anchors (Table 2). The short-circuit photocurrents (JSCs) are in line with previously reported Ru complex-sensitised aqueous DSC systems.10| Dye | J SC (mA cm−2) | V OC (mV) | FF (%) | η (%) |
|---|---|---|---|---|
| a Conditions: aqueous electrolyte solution, redox mediator I3−/I−, 1 Sun, AM 1.5 G, after 9 d (see ESI for details). | ||||
| PMI-CO2H | 0.85 ± 0.40 | 470 ± 30 | 69 ± 6 | 0.28 ± 0.30 |
| PMI-Acac | 0.24 ± 0.10 | 380 ± 8 | 60 ± 3 | 0.06 ± 0.01 |
| PMI-PO3H2 | 0.70 ± 0.20 | 450 ± 30 | 35 ± 4 | 0.13 ± 0.10 |
| PMI-HQui | 1.37 ± 0.60 | 510 ± 5 | 68 ± 1 | 0.47 ± 0.30 |
| PMI-DPA | 1.30 ± 0.10 | 480 ± 10 | 69 ± 1 | 0.42 ± 0.10 |
The obtained ηs are still low compared to top performing organic systems, or to the highest η value recorded (∼4%) with an iodine based aqueous electrolyte and the organic dye D149.15 This can be partially attributed to the electron-withdrawing ability of the PMI core that would limit electron injection and promote charge recombination. Nevertheless, this work constitutes the first example of a PMI-based aqueous DSC, and further optimised electrolytes and TiO2 surfaces previously reported for aqueous DSCs are likely to enable higher performances in the future.10,46,47
The main discrepancies between the performance of the dyes originate from variations in JSC, which follow the order PMI-Acac ≪ PMI-PO3H2 ≈ PMI-CO2H < PMI-DPA ≤ PMI-HQui. Incident photon-to-current conversion efficiency (IPCE) measurements (Fig. S4†) also confirmed this order with PMI-HQui DSCs displaying the highest and broadest photon conversion efficiencies in line with the broader absorption of the dye (Fig. 2). As a result, the highest DSC efficiencies were obtained with PMI-HQui in both H2O and ACN conditions. The low JSC and IPCE obtained with the PMI-Acac-sensitised DSCs, in both water- and ACN-based electrolyte solution, are primarily accounted for by inefficient electron injection. This can be explained by the limited orbital overlap due to the perpendicular orientation of the anchor with respect to the phenyl plane, which impedes any electron-withdrawing effect from the anchor and ultimately reduces electron density close to the TiO2 surface.48,49
The similar JSC and open-circuit voltage (VOC) values obtained in water for the dyes bearing carboxylic, phosphonic and dipicolinic acids reflects their comparable photosensitising abilities. However, the low fill factors (FFs) recorded on PMI-PO3H2-based DSCs infer substantial charge recombination at the semiconductor–electrolyte interface. Interestingly, upon replacing H2O for ACN this photosensitiser produces high JSCs and FFs similar to the other PMI-dyes, revealing a specific deleterious impact/interaction of H2O with the phosphonic acid and the surface of TiO2 (Table S2†), leading to high recombination phenomena and series resistance.36
In order to obtain further insights on the potential of the anchors in water, we evaluated the performance of the dyes on the TiO2 photoanodes under an applied potential (in a three-electrode setup). Two commonly employed electron donors, AA and TEOA, were used instead of the DSC redox mediator I3−/I−.43 These experiments, ‘half way’ between DSC and DSP systems, allow for a preliminary evaluation of the sensitisers' capability to extract charge from EDs in a DSP system through application of a positive applied bias (Eapp), which limits geminate and non-geminate recombination.
The experiments were conducted using a dye-sensitised TiO2 film coated on a fluorine tin oxide-coated glass slide as a working electrode, a Hg/HgSO4 reference electrode (E0(vs. NHE) = E0(vs. Hg/HgSO4) + 0.64 V) and a platinum wire counter electrode. Linear sweep voltammetry conducted under chopped illumination (2 W m−2) showed constant J between 0.64 to 0.14 V vs. NHE for all dyes (Table 3, Fig. S5 and S6†). Photocurrents between 0.2 and 0.8 mA cm−2, displaying a similar trend to those obtained in the DSC configuration, were observed in aqueous AA solution (pH 4.5, Tables 2 and 3). This demonstrates the ability of AA to act as an ED for all sensitisers, although at a somewhat reduced performance compared to I3−/I−. Photocurrents attained with PMI-PO3H2-sensitised electrodes are significantly lower than those observed in a DSC configuration, which alludes to complications in the charge extraction process, in agreement with the low FF recorded in aqueous DSC (Table 2).
| Dye | J (mA cm−2) | |
|---|---|---|
| AA (pH 4.5) Eapp = 0.24 V vs. NHE | TEOA (pH 8.5) Eapp = 0.64 V vs. NHE | |
| a General conditions: aqueous electrolyte, ED = AA or TEOA, 2 W m−2 white light irradiation (see ESI for details). | ||
| PMI-CO2H | 0.38 | 0.025 |
| PMI-Acac | 0.24 | 0.009 |
| PMI-PO3H2 | 0.26 | 0.024 |
| PMI-HQui | 0.80 | 0.017 |
| PMI-DPA | 0.50 | 0.018 |
Substantially lower Js (≤0.025 mA cm−2) were recorded in the presence of TEOA as ED at pH 8.5 (Fig. S6†), in comparison to those obtained in AA-containing solutions at pH 4.5. This could be first explained by the lack of exergonicity in the electron injection process as illustrated by the small Gibbs free energy values (Table S1†) in basic conditions. Nevertheless, the order of J is not directly reflected by the Gibbs energy within the different dyes which could allude towards additional limiting factors. Interestingly, the Js of the cells level out at ∼0.020 mA cm−2 with very little discrepancies between the dyes (except PMI-Acac), which suggests the existence of a major kinetically limiting step, ascribed to slow dye regeneration by TEOA. As a result, the accurate evaluation of the impact of the anchors on performance is difficult under such conditions; however, it is clear that the acetylacetone anchor delivers lower photocurrents suggesting even further restrictions in the charge injection.
In order to study the differences in dye regeneration kinetics from AA and TEOA, we performed two additional measurements using PMI-CO2H as a model dye using AA as ED in neutral and basic conditions at Eapp = 0.54 V vs. NHE. Similar Js of 0.48 and 0.47 mA cm−2 were obtained at 7.0 and 8.5, respectively, which is close to the one recorded at pH 4.5, i.e. 0.6 mA cm−2. This contrast with the much lower value recorded with TEOA at pH 8.5 (J = 0.02 mA cm−2 at Eapp = 0.54 V vs. NHE) and suggest that the low regenerating ability of TEOA is a major limiting factor in our system rather than issues in electron injection.
Electrochemical impedance spectroscopy (EIS) measurements were performed on similar electrodes at open-circuit potential under white light illumination using AA and TEOA at pH 8.5. For both AA- and TEOA-based electrolytes, Bode plot traces revealed phase values close to zero for frequencies around 100 kHz. This indicates an ohmic resistive behaviour (Fig. S7a†), resulting from the sum of the electrolyte solution and defect of the photoelectrode resistances. In the middle frequencies region, phase values less than 90° were observed and attributed to charge transfer resistances (RCT) and a double layer capacitance at the PMI-CO2H dye/electrolyte interface. RCT were deduced from Nyquist plots (Fig. S7b†) at around 1 and ≫20 kΩ for AA and TEOA solutions, respectively. These results indicate a better regeneration of S+ by AA than by TEOA with the latter appearing responsible for the lower photocurrents recorded at pH 8.5.
We subsequently also performed applied bias incident photon-to-current efficiency (ABCE) experiments under optimal conditions using AA (pH 4.5) as ED, at Eapp = 0.24 V vs. NHE, in order to obtain insights on the wavelength-dependent efficiency of electron injection (Fig. S8†). The results confirmed that the cells sensitised with PMI-HQui and PMI-Acac/PMI-PO3H2 displayed the highest and lowest photon-to-electron efficiencies of 30 and 6–7% respectively at their respective maximum absorption. Compared to the other dyes, the former showed a broader light sensitivity over the solar spectrum, and demonstrated the highest conversion of photons. This observation is in line with the higher JSCs recorded in DSC configuration.
Assembly of DSP system
Pt was used as a H2-evolving catalyst in order to assess the dye performances in absence of catalyst-associated limitations, and was pre-deposited on TiO2 nanoparticles (P25, Evonik Industries) as previously described.37 The PMI-modified TiO2|Pt nanoparticles (PMI|TiO2|Pt) were assembled by sonicating the pre-platinised-TiO2 particles in a dilute solution of PMI dye in DMF (see ESI† for details). The PMI|TiO2|Pt particles were separated from solution after one hour via centrifugation, and the supernatant was analysed by UV-Vis spectroscopy to quantify the amount of immobilised dye on TiO2 (Table S3† and Fig. S9†). Approximately 85% of PMI-Acac, PMI-DPA and PMI-HQui (≈33 nmol) available in the original dyeing solution (ntot ≈ 39 nmol) were attached onto 2 mg of TiO2|Pt. This result indicates a strong binding ability of these anchors with a modest influence of the steric hindrance or footprint of the anchor. The loading increases and decreases in the case of PMI-PO3H2 (>95%) and PMI-CO2H (≈65%), respectively, demonstrating the superior anchoring ability of the phosphonic acid. The immobilisation of the PMI dyes was also confirmed by ATR-FTIR spectroscopy of the sensitised nanoparticles with the spectra revealing clear ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)amide and ν(CC)aromatic bands of the PMI-dyes at 1710 and 1650 cm−1, respectively (Fig. S9†).
O)amide and ν(CC)aromatic bands of the PMI-dyes at 1710 and 1650 cm−1, respectively (Fig. S9†).
Photocatalytic activity
Photocatalytic experiments were carried out in water at pH 4.5, 7.0 and 8.5 using either AA (pH 4.5, 0.1 M), or TEOA (pH 7.0 and 8.5, 0.1 M) as both buffer and ED.50–53 For photocatalytic experiments, the PMI|TiO2|Pt nanoparticles were dispersed in the aqueous ED solution (1.25 mg of pre-modified particles in 3 mL of ED solution) in a sealed photoreactor via sonication for 15 min, then purged with N2 (including 2% CH4 as internal gas chromatography standard), and irradiated with UV-filtered simulated solar light (AM 1.5 G, 100 mW cm−2, λ > 420 nm). Light-driven H2 evolution was monitored in regular time intervals by gas chromatography. Control experiments in absence of dye revealed no H2 or only negligible amounts of H2 are evolved.28The initial dye-based turnover frequencies (TOFPMI after 1 h) in aqueous DSP with PMI|TiO2|Pt in pH 4.5 AA (0.1 M) for solar H2-evolution agree well with trends observed in the aqueous DSCs (see above): PMI-Acac < PMI-PO3H2 < PMI-DPA ≈ PMI-CO2H < PMI-HQui (Table 4 and S4, and Fig. S10†). PMI-Acac shows the lowest performance, which is most likely due to the unfavourable orientation of the Acac anchoring group. PMI-HQui displays the highest initial TOFPMI, but it also exhibits the largest drop in photoactivity over time followed by PMI-DPA (Table S5†). The activity of the other PMI|TiO2|Pt systems remained generally constant during 7 h of irradiation (Fig. S10†). This indicates degradation of the PMI-HQui and PMI-DPA dye/anchor, or loss of dye molecules from the surface leading to a decrease in performance over time.
| Systema | TOFPMI/h−1 (1 h)b | n(H2)/μmol (24 h) | TONPMI (24 h)b |
|---|---|---|---|
| a Conditions: 1.25 mg PMI|TiO2|Pt in 3 mL ED solution (0.1 M of AA or TEOA), UV-filtered simulated solar irradiation (AM 1.5 G, 100 mW cm−2, λ > 420 nm, 25 °C). b TOFPMI (1 h) and TONPMI were calculated based on the loading of the TiO2|Pt nanoparticles (see Table S3). | |||
| pH 4.5 | |||
| PMI-CO2H | 344 ± 38 | 53.7 ± 6.2 | 6461 ± 749 |
| PMI-Acac | 112 ± 12 | 21.7 ± 2.2 | 2146 ± 203 |
| PMI-PO3H2 | 210 ± 27 | 42.5 ± 6.3 | 3546 ± 523 |
| PMI-HQui | 467 ± 72 | 53.3 ± 5.9 | 4928 ± 549 |
| PMI-DPA | 305 ± 59 | 41.4 ± 2.9 | 3943 ± 394 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| pH 7 | |||
| PMI-CO2H | 59.2 ± 5.9 | 3.9 ± 0.5 | 471 ± 63 |
| PMI-Acac | 10.9 ± 1.0 | 1.3 ± 0.1 | 133 ± 13 |
| PMI-PO3H2 | 27.4 ± 2.7 | 3.6 ± 0.4 | 303 ± 30 |
| PMI-HQui | 25.6 ± 2.6 | 2.5 ± 0.5 | 232 ± 26 |
| PMI-DPA | 27.5 ± 2.7 | 3.8 ± 0.2 | 366 ± 37 |
|
|||
| pH 8.5 | |||
| PMI-CO2H | 58.6 ± 14.5 | 4.1 ± 1.4 | 490 ± 170 |
| PMI-Acac | 23.4 ± 3.1 | 3.0 ± 0.7 | 294 ± 67 |
| PMI-PO3H2 | 60.9 ± 6.1 | 8.5 ± 1.3 | 708 ± 107 |
| PMI-HQui | 26.4 ± 2.8 | 2.8 ± 0.4 | 262 ± 36 |
| PMI-DPA | 32.8 ± 3.3 | 4.7 ± 0.7 | 444 ± 62 |
Up to 54 μmol of H2 were produced with PMI|TiO2|Pt, with PMI-CO2H giving an initial TOF of 344 h−1 and the highest TONPMI of approximately 6460 after 24 h of UV-filtered simulated solar light irradiation (Table 4, Fig. 3a). This is significantly higher than the TON obtained for our recently reported phosphonated diketopyrrolopyrrole (DPP) or Ru(2,2′-bipyridine)-based dye-sensitised TiO2|Pt systems, where a maximum TONDPP of 2660 and initial TOFDPP of 337 h−1 were obtained under the same experimental conditions.28 The similar initial TOFs of the two systems and the higher final TONPMI could reflect on superior stability of the PMI core compared to the DPP. A long-term experiment was performed with PMI-CO2H showing that the PMI|TiO2|Pt system, despite experiencing some dye degradation, remained active for over 72 h of light irradiation. A cumulative TONPMI of approximately 1.1 × 104 (Fig. 3b) was achieved, which corresponds to the highest TON obtained for an organic dye in an aqueous nanoparticulate DSP system.
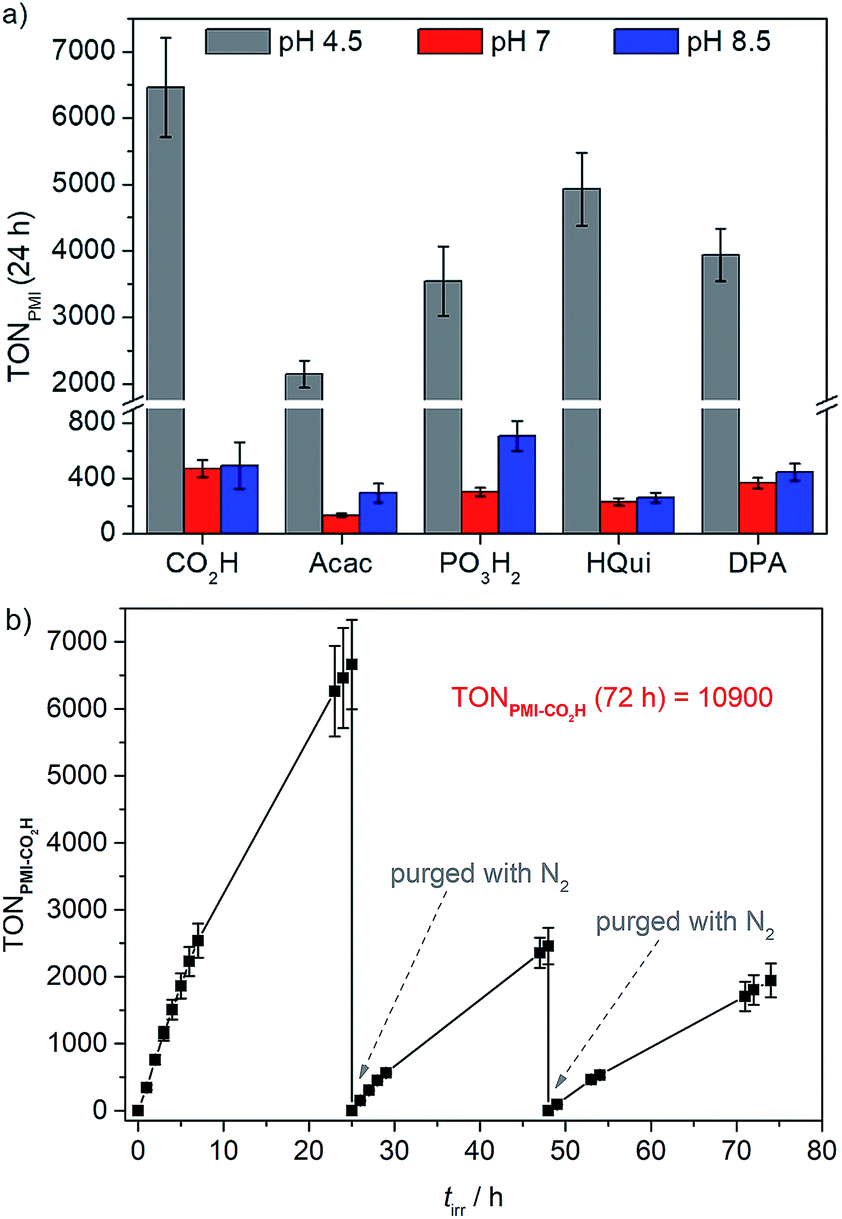 | ||
| Fig. 3 (a) Photocatalytic activity of PMI|TiO2|Pt expressed as TONPMI after 24 h of irradiation in pH 4.5 AA (grey), pH 7 TEOA (red), and pH 8.5 TEOA (blue) solution (0.1 M each); (b) long-term experiment (TONPMI-CO2Hvs. tirr) using PMI-CO2H in pH 4.5 AA solution. Samples were purged with N2 after 24 and 48 h. Conditions: UV-filtered simulated solar light irradiation (AM 1.5 G, 100 mW cm−2, λ > 420 nm) of 1.25 mg PMI|TiO2|Pt in 3 mL ED (AA or TEOA) solution. | ||
The compatibility of PMI dyes with a molecular catalyst was studied with a phosphonated DuBois-type nickel bis(diphosphine) complex (NiP, Fig. S11†).51NiP was added to Pt-free, PMI-CO2H-sensitised TiO2 nanoparticles, which were suspended in aqueous AA solution prior to addition. The deaerated PMI-CO2H|TiO2|NiP assembly achieved a TONNiP and TONPMI of approximately 110 and 170 after 24 h of irradiation, respectively (Fig. S12†). Although slightly lower, this performance for catalytic H2 production agrees well with previously reported TONs for DPP-based DSP systems using NiP as catalyst (TONDPP = 204 (21 h) vs. TONPMI = 170 (24 h)),28,51 and illustrates that Pt can be replaced to give a precious metal-free PMI|TiO2|catalyst system. The lower activity reached with the molecular catalyst compared to Pt was previously attributed to the slower kinetics of the former, that favours charge recombination from the e− in the TiO2 CB to the oxidised dye.28 When NiP is employed, the lower activity of the PMI-dyes compared to the DPP-dyes alludes towards faster charge recombination in the former case that can be attributed to the dye design as discussed above.
The photocatalytic performance of PMI|TiO2|Pt was subsequently studied with TEOA (pH 7 and 8.5) as an ED. The activity is significantly lower compared to the AA-containing systems (Table 4, S6 and S7,† and Fig. 3a, S13 and S14†). Although the order of efficiency does not clearly reflect the Gibbs energies (Table S1†), these results match the trend obtained from photocurrent measurements of PMI-sensitised TiO2 photoanodes and is probably due to more difficult dye regeneration by the ED and a more difficult electron injection at higher pH. The generally lower performance recorded at pH 7.0 compared to pH 8.5 has been ascribed to the coexistence of protonated and neutral forms of TEOA due to its pKa value within the pH range investigated (pKa = 7.9). As the pH of the solution decreases the equilibrium favours the existence of the protonated form of TEOA which is an inferior donor and therefore impedes the regeneration of the oxidised sensitiser.54
PMI dyes with acid-containing anchoring groups appear beneficial under these more basic conditions with PMI-CO2H and PMI-PO3H2 achieving a TONPMI of approximately 470 and 710 at pH 7.0 and pH 8.5, respectively. Specifically, the phosphonic acid anchoring group has been previously reported as a more stable anchor at higher pH values in comparison to carboxylic acids.4,37 This advantage is illustrated by the strong photoactivity drop during the first 7 h for PMI-CO2H (≈37%, Fig. S13 and S14†), whereas the other anchoring groups (Acac, DPA, PO3H2 (≈15–25% loss)) appear more robust under the pH neutral/alkaline conditions. The almost pH-independent stabilities (Table S5 and Fig. S15†) observed for the PMI-HQui and PMI-DPA dyes likely originate from dye degradation or from the system progressive aggregation (observed after experiments), rather than desorption or limitations from the regeneration of S+ by the EDs. The low activity observed for the PMI-HQui (TONPMI-HQui = 232 and 262 at pH 7.0 and 8.5, respectively) and generally fast deactivations highlight issues arising from fast desorption/degradation.
Conclusions
We report the synthesis of five novel PMI photosensitisers with different anchoring groups, and their successful integration into solar light-driven systems towards electricity and H2 production in pure aqueous solution. Varying the surface anchor group enabled a unique side-by-side comparison and highlights their advantages and weaknesses in aqueous DSC and DSP schemes. Despite high activities in acidic media, the hydroxyquinoline anchor is the most sensitive towards desorption/degradation under the chosen experimental conditions. Similarly, the carboxylic acid anchor delivers the highest photoactivity, and is a robust anchor in acidic and pH neutral media, but undergoes the fastest hydrolysis from the metal oxide surface in alkaline solution. Despite the hydrophobicity of the PMI dyes, a stronger anchor such as the phosphonic acid is required to allow for stable performance at alkaline pH.We show that the same dye design rules apply to aqueous DSC and DSP technologies, with the individual system performance being similarly affected by the anchor of the dye and by the variation of the pH value of the solution. Consequently, we achieved promising photocurrents and good fill factors in aqueous DSC, as well as record TONs and impressive stability towards H2 evolution in DSP with a molecular dye-sensitised TiO2. This first generation of PMI dyes reveals significant insights drawn from structure–activity relationships, which will be applicable to a broad range of dye-sensitised technology in aqueous media. As this study focused specifically on the nature of the anchor and the dye–TiO2 interface, this work leaves promising opportunities for further dye improvements towards high performance under aqueous media such as implementing directional push–pull architectures, extending π conjugation or increasing the wettability of the TiO2 surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support by the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), the OMV Group, Agence National de la Recherche (ANR) through the program POSITIF (ANR-12-PRGE-0016-01), and Région des Pays de la Loire for the project LUMOMAT. We thank Franziska Nousch for her help with initial photocatalysis experiments.Notes and references
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- P. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42–58 CrossRef CAS.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- K. L. Orchard, D. Hojo, K. P. Sokol, M.-J. Chan, N. Asao, T. Adschiri and E. Reisner, Chem. Commun., 2017, 53, 12638–12641 RSC.
- F. Bella, C. Gerbaldi, C. Barolo and M. Grätzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC.
- C. Law, S. C. Pathirana, X. Li, A. Y. Anderson, P. R. F. Barnes, A. Listorti, T. H. Ghaddar and B. C. O′Regan, Adv. Mater., 2010, 22, 4505–4509 CrossRef CAS PubMed.
- M. Matsumura, S. Matsudaira, H. Tsubomura, M. Takata and H. Yanagida, Ind. Eng. Chem. Prod. Res. Dev., 1980, 19, 415–421 CrossRef CAS.
- P. Liska, N. Vlachopoulos, M. K. Nazeeruddin, P. Comte and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 3686–3687 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- C. Law, O. Moudam, S. Villarroya-Lidon and B. O'Regan, J. Mater. Chem., 2012, 22, 23387–23394 RSC.
- T. Daeneke, Y. Uemura, N. W. Duffy, A. J. Mozer, N. Koumura, U. Bach and L. Spiccia, Adv. Mater., 2012, 24, 1222–1225 CrossRef CAS PubMed.
- C. Dong, W. Xiang, F. Huang, D. Fu, W. Huang, U. Bach, Y.-B. Cheng, X. Li and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 6933–6937 CrossRef CAS PubMed.
- R. Kato, F. Kato, K. Oyaizu and H. Nishide, Chem. Lett., 2014, 43, 480–482 CrossRef CAS.
- F. Bella, S. Galliano, M. Falco, G. Viscardi, C. Barolo, M. Grätzel and C. Gerbaldi, Chem. Sci., 2016, 7, 4880–4890 RSC.
- F. Bella, S. Galliano, M. Falco, G. Viscardi, C. Barolo, M. Grätzel and C. Gerbaldi, Green Chem., 2017, 19, 1043–1051 RSC.
- S. Husmann, L. F. Lima, L. S. Roman and A. J. G. Zarbin, ChemSusChem, 2018, 11, 1238–1245 CrossRef CAS PubMed.
- S. Galliano, F. Bella, G. Piana, G. Giacona, G. Viscardi, C. Gerbaldi, M. Grätzel and C. Barolo, Sol. Energy, 2018, 163, 251–255 CrossRef CAS.
- A. Glinka, M. Gierszewski and M. Ziółek, J. Phys. Chem. C, 2018, 122, 8147–8158 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V. Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1681–1686 CrossRef CAS PubMed.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- J. Warnan, J. Willkomm, J. N. Ng, R. Godin, S. Prantl, J. R. Durrant and E. Reisner, Chem. Sci., 2017, 8, 3070–3079 RSC.
- C. E. Creissen, J. Warnan and E. Reisner, Chem. Sci., 2018, 9, 1439–1447 RSC.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed.
- Z. Liu, S. P. Lau and F. Yan, Chem. Soc. Rev., 2015, 44, 5638–5679 RSC.
- R. J. Lindquist, B. T. Phelan, A. Reynal, E. A. Margulies, L. E. Shoer, J. R. Durrant and M. R. Wasielewski, J. Mater. Chem. A, 2016, 4, 2880–2893 RSC.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Chem. Sci., 2017, 8, 541–549 RSC.
- J. Boixel, E. Blart, Y. Pellegrin, F. Odobel, N. Perin, C. Chiorboli, S. Fracasso, M. Ravaglia and F. Scandola, Chem.–Eur. J., 2010, 16, 9140–9153 CrossRef CAS PubMed.
- S. Galliano, F. Bella, C. Gerbaldi, M. Falco, G. Viscardi, M. Grätzel and C. Barolo, Energy Technol., 2017, 5, 300–311 CrossRef CAS.
- E. Bae and W. Choi, J. Phys. Chem. B, 2006, 110, 14792–14799 CrossRef CAS PubMed.
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740–8749 CrossRef CAS PubMed.
- M. Nitta and T. Kobayashi, J. Chem. Soc., Chem. Commun., 1982, 877–878 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- X. Feng, Y. An, Z. Yao, C. Li and G. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 614–618 CrossRef CAS PubMed.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- B. Enright, G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1994, 98, 6195–6200 CrossRef CAS.
- H. Saito, S. Uegusa, T. N. Murakami, N. Kawashima and T. Miyasaka, Electrochemistry, 2004, 72, 310–316 CAS.
- C.-T. Li, R. Y.-Y. Lin and J. T. Lin, Chem.–Asian J., 2017, 12, 486–496 CrossRef CAS PubMed.
- J. Warnan, V.-M. Guerin, F. B. Anne, Y. Pellegrin, E. Blart, D. Jacquemin, T. Pauporté and F. Odobel, J. Phys. Chem. C, 2013, 117, 8652–8660 CrossRef CAS.
- J. Warnan, Y. Pellegrin, E. Blart, L. Zhang, A. Brown, L. Hammarström, D. Jacquemin and F. Odobel, Dyes Pigm., 2014, 105, 174–179 CrossRef CAS.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- J. Willkomm, N. M. Muresan and E. Reisner, Chem. Sci., 2015, 6, 2727–2736 RSC.
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 9402–9406 CrossRef CAS PubMed.
- K. Kalyanasundaram, J. Kiwi and M. Grätzel, Helv. Chim. Acta, 1978, 61, 2720–2730 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, supporting tables and figures. See DOI: 10.1039/c8sc05693e |
| This journal is © The Royal Society of Chemistry 2019 |