
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric allylation of aldehydes with alkenes through allylic C(sp3)–H functionalization mediated by organophotoredox and chiral chromium hybrid catalysis†
Harunobu
Mitsunuma
 *a,
Shun
Tanabe
a,
Hiromu
Fuse
a,
Kei
Ohkubo
b and
Motomu
Kanai
*a
*a,
Shun
Tanabe
a,
Hiromu
Fuse
a,
Kei
Ohkubo
b and
Motomu
Kanai
*a
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: h-mitsunuma@mol.f.u-tokyo.ac.jp; kanai@mol.f.u-tokyo.ac.jp
bInstitute for Advanced Co-Creation Studies, Open and Transdisciplinary Research Initiatives, Osaka University, Osaka 565-0871, Japan
First published on 17th January 2019
Abstract
We describe a hybrid system that realizes cooperativity between an organophotoredox acridinium catalyst and a chiral chromium complex catalyst, thereby enabling unprecedented exploitation of unactivated hydrocarbon alkenes as precursors to chiral allylchromium nucleophiles for asymmetric allylation of aldehydes. The reaction proceeds under visible light irradiation at room temperature, affording the corresponding homoallylic alcohols with a diastereomeric ratio >20/1 and up to 99% ee. The addition of Mg(ClO4)2 markedly enhanced both the reactivity and enantioselectivity.
Introduction
Catalytic asymmetric C(sp3)–H bond functionalization is an emerging synthetic method affording direct access to useful chiral building blocks from stable organic molecules.1 For example, catalytic asymmetric allylation of aldehydes using unactivated alkenes as pronucleophiles produces enantiomerically-enriched homoallylic alcohols, which act as versatile synthetic intermediates for numerous functional molecules, including various drug leads.2 The catalytic asymmetric carbonyl ene reaction is a representative example (Fig. 1(a)).3 The electrophile scope of catalytic asymmetric carbonyl ene reactions, however, is limited to highly reactive aldehydes or ketones, such as glyoxylic esters, formaldehyde, fluoral, or ketoesters. In an effort to expand the substrate scope, we envisioned that a reaction mechanism dissected by chiral nucleophilic allylmetal species, which are generated from alkenes via in situ allylic C(sp3)–H bond activation, would be feasible.4 Some previous examples related to this strategy are reported. Gong presented a one-pot procedure for asymmetric nucleophilic allylation of an aldehyde with an alkene involving palladium-catalyzed allylic C(sp3)–H borylation of the alkene to generate an allylboronate and chiral phosphoric acid-catalyzed asymmetric allylation of the aldehyde with the thus-generated allylboronate (Fig. 1(b)).5 Mita and Sato reported a cobalt-catalyzed enantioselective allylation between acetone and an allylarene via nucleophilic chiral allylcobalt(I) species generated through oxidative addition of an allylic C(sp3)–H bond to a low valent cobalt complex (Fig. 1(c)).6 During our study, Glorius' group reported an asymmetric allylation between an aldehyde and an allylamine mediated by combining an iridium photoredox catalyst and a chiral chromium catalyst (Fig. 1(d)).7 Considering the attractive feature of this reaction to generate versatile chiral homoallylic alcohols in a single operation from aldehydes and alkenes, further investigation of this reaction type are highly desirable.8,9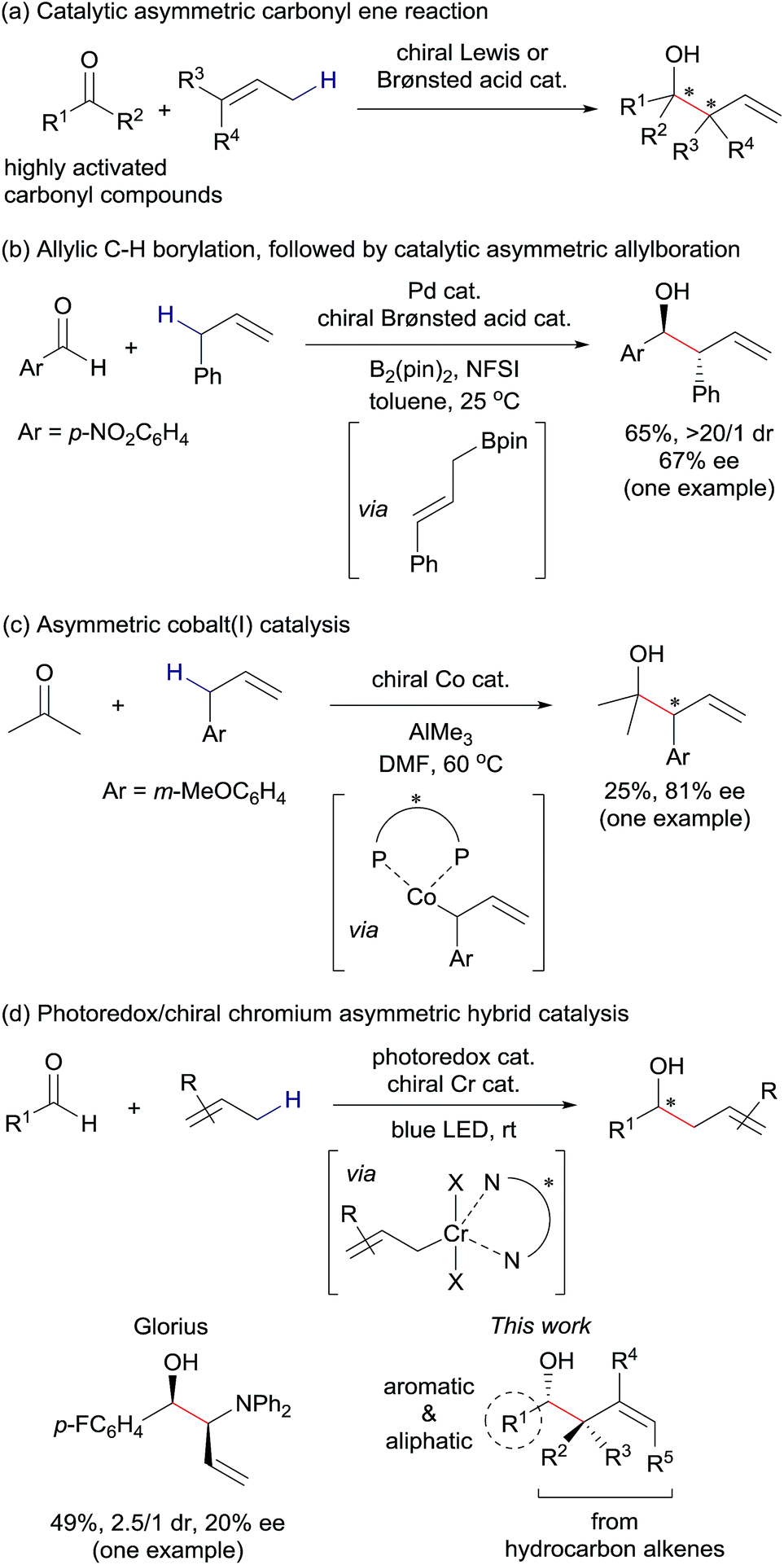 | ||
| Fig. 1 Methods for catalytic asymmetric allylation of carbonyl compounds through C(sp3)–H functionalization of unactivated alkenes as pronucleophiles. (a) Chiral Lewis acid or Brønsted acid-catalyzed carbonyl ene reactions.3 (b) One-pot, stepwise generation of allylboronate followed by chiral Brønsted acid-catalyzed allylboration.5 (c) Through chiral allylcobalt species.6 (d) Through chiral allylchromium species by Glorius7 and our group (this work). | ||
Photocatalyzed C(sp3)–H bond activation followed by oxidative interception of the resulting carbon-centered radical by a metal complex catalyst is a groundbreaking concept for generating organometallic intermediates from substrates traditionally considered inert.10–13 Application of organometallic intermediates generated by this method, however, has mainly been limited to cross-coupling reactions. Extension of the chemistry to facilitate the addition of these nucleophiles to polar moieties, such as carbonyl groups, has yet to be explored, except for the recent example by Glorius using electron-rich aromatic- or amine-substituted alkenes.7 Herein we report an asymmetric hybrid catalyst system comprising an organophotoredox catalyst and a chiral chromium complex catalyst, which enables asymmetric allylation of aldehydes by nucleophilic chiral allylchromium species generated in situ from hydrocarbon feedstock alkenes by C(sp3)–H bond activation (Fig. 1(d)).
Results and discussion
Optimization of reaction conditions
Our mechanistic rationale for this transformation is illustrated in Fig. 2. Based on an earlier report by the Wu laboratory,14 allyl radical 4 should be accessible from alkene 1avia electron-transfer oxidation of the π-bond by a photoexcited electron-donor substituted acridinium catalyst (D˙+–Acr˙; D = 2,6-xylyl or mesityl) to generate radical cation 3, followed by deprotonation. A reduced form of the chiral chromium(II) catalyst 5 would then intercept the thus-formed allyl radical 4 to give chiral allyl chromium(III) complex 6. We anticipated that this species would react with aldehydes 2via a six-membered chair transition state to produce enantiomerically-enriched chromium alkoxide 7 in a syn-selective manner. Protonolysis of 7 would then afford the target homoallylic alcohol 8 and an oxidized chromium(III) complex 9. Finally, electron-transfer reduction of 9 by the reduced form of the photocatalyst (D–Acr˙) would regenerate 5 and the oxidized form of the photocatalyst (D–Acr+), thus closing the catalytic cycle.15 | ||
| Fig. 2 Proposed catalytic cycle. | ||
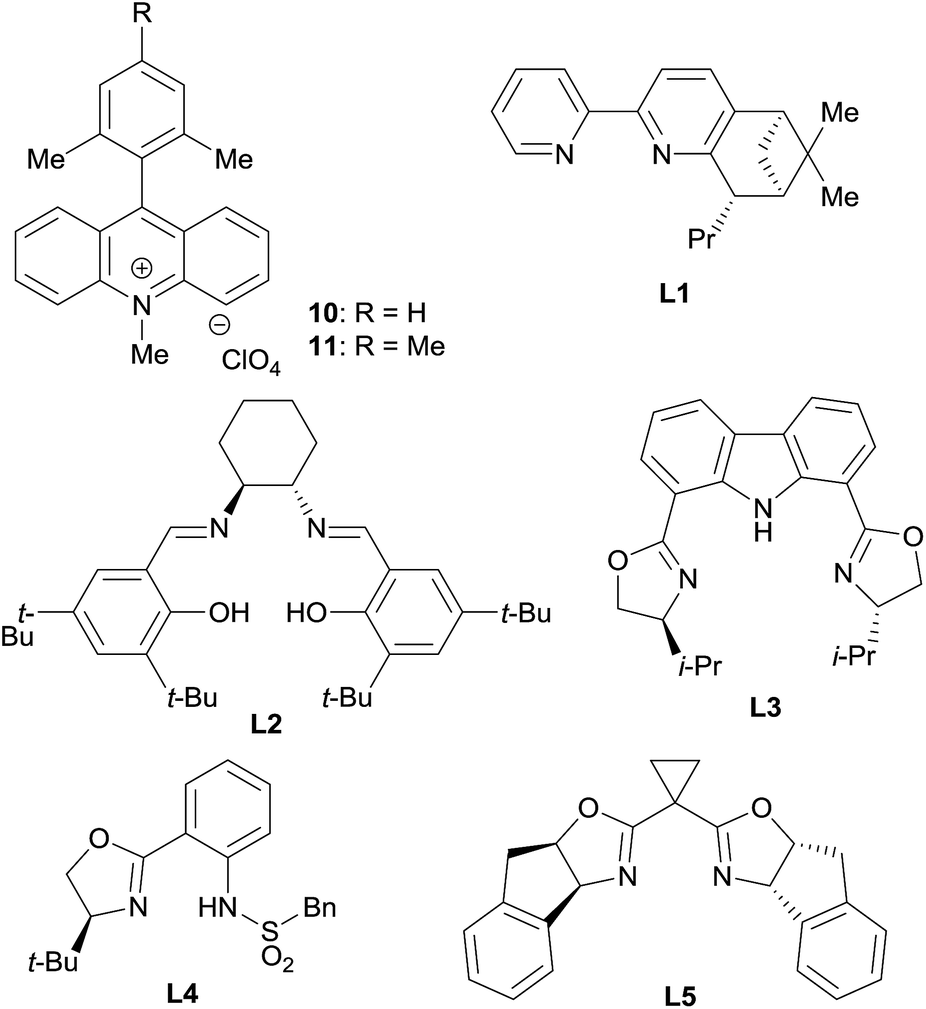
Based on this hypothesis, we began optimizing the reaction conditions using benzaldehyde (2a) and cyclohexene (1a: 20 equiv.) as model substrates, and a combination of 5 mol% CrCl2 and 2.5 mol% acridinium photoredox catalysts (2,6-Xyl–Acr+·ClO4−; 10),16 under 430 nm visible light irradiation at room temperature (Table 1). As expected, the desired reaction did not proceed at all in the absence of the chromium complex (entry 1). In the presence of CrCl2, however, 8a was obtained in 36% yield with an excellent diastereomeric ratio (dr) of >20/1 (entry 2). Encouraged by this finding, we then screened various chiral ligands for the chromium catalysts that were previously shown to be effective for asymmetric Nozaki–Hiyama–Kishi reactions (entries 3–6).17 The chiral catalysts strongly retarded the reaction, however, with only L1 (ref. 18) affording 8a with diminished yield (12%) and low enantioselectivity (20% ee). Through extensive screening of other chiral ligands, we identified an indane-BOX ligand (L5)19 that effectively induced good enantioselectivity (74% ee), although the yield of 8a remained unsatisfactory (8%, entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Additive | Yield (%) | dr | ee (%) |
| a General reaction conditions: 2a (0.25 mmol), 1a (5.0 mmol), CrCl2 (0.0125 mmol), ligand (0.0125 mmol), 10 (0.00625 mmol), and additive (0.25 mmol) were reacted in dichloromethane (DCM; 2.5 mL) at room temperature under 430 nm LED irradiation for 12 h. Yield and diastereomeric ratio were determined by 1H NMR analysis of the crude mixture using 1,1,2,2-tetrachloroethane as an internal standard. The enantioselectivity of 8a was determined by chiral stationary HPLC analysis after isolation. n.d. = not determined. b Without CrCl2. c 10 mol% Et3N was added. d 5 mol% Et3N was added. e Mes–Acr+·ClO4−11 was used as a photocatalyst. | |||||
| 1b | None | None | 0 | n.d. | n.d. |
| 2 | None | None | 36 | >20/1 | n.d. |
| 3 | L1 | None | 12 | >20/1 | 20 |
| 4c | L2 | None | 0 | n.d. | n.d. |
| 5d | L3 | None | 0 | n.d. | n.d. |
| 6d | L4 | None | 0 | n.d. | n.d. |
| 7 | L5 | None | 8 | >20/1 | 74 |
| 8 | L5 | LiBF4 | 40 | >20/1 | 63 |
| 9 | L5 | LiClO4 | 44 | >20/1 | 99 |
| 10 | L5 | NaClO4 | 14 | >20/1 | 99 |
| 11 | L5 | Ca(ClO4)2·xH2O | 8 | >20/1 | 98 |
| 12 | L5 | Mg(ClO4)2 | 68 | >20/1 | 99 |
| 13e | L5 | Mg(ClO4)2 | 63 | >20/1 | 99 |
|
|||||
We supposed that the low reactivity was due to the high oxidation potential of 1a. Due to the small oxidation potential difference between substrate 1a and photoredox catalyst 10 (see ESI†), only low concentration of cation radical 3 were generated. To improve the reactivity, we investigated the effects of salt additives to stabilize cation radical 3, which would accelerate the overall reaction rate.20 Screening of several electrolytes revealed that adding LiBF4 dramatically enhanced the reactivity; 8a was obtained in 40% yield with 63% ee (entry 8). Moreover, the use of LiClO4 increased the enantioselectivity up to 99% (entry 9). Further exploration of alkali and alkali-earth metal perchlorates (entries 10–12) identified Mg(ClO4)2 as the optimal additive; 8a was obtained in 68% yield with >20/1 dr and 99% ee (entry 12). Additionally, the use of photocatalyst 11, bearing a mesityl group instead of a xylyl group, did not negatively affect these results (entry 13). It is noteworthy that compared to traditional catalytic asymmetric carbonyl allylations,2 except for the Krische's method,2i,p this reaction can bypass the preactivation step of the nucleophile using stoichiometric metal species.
Substrate scope
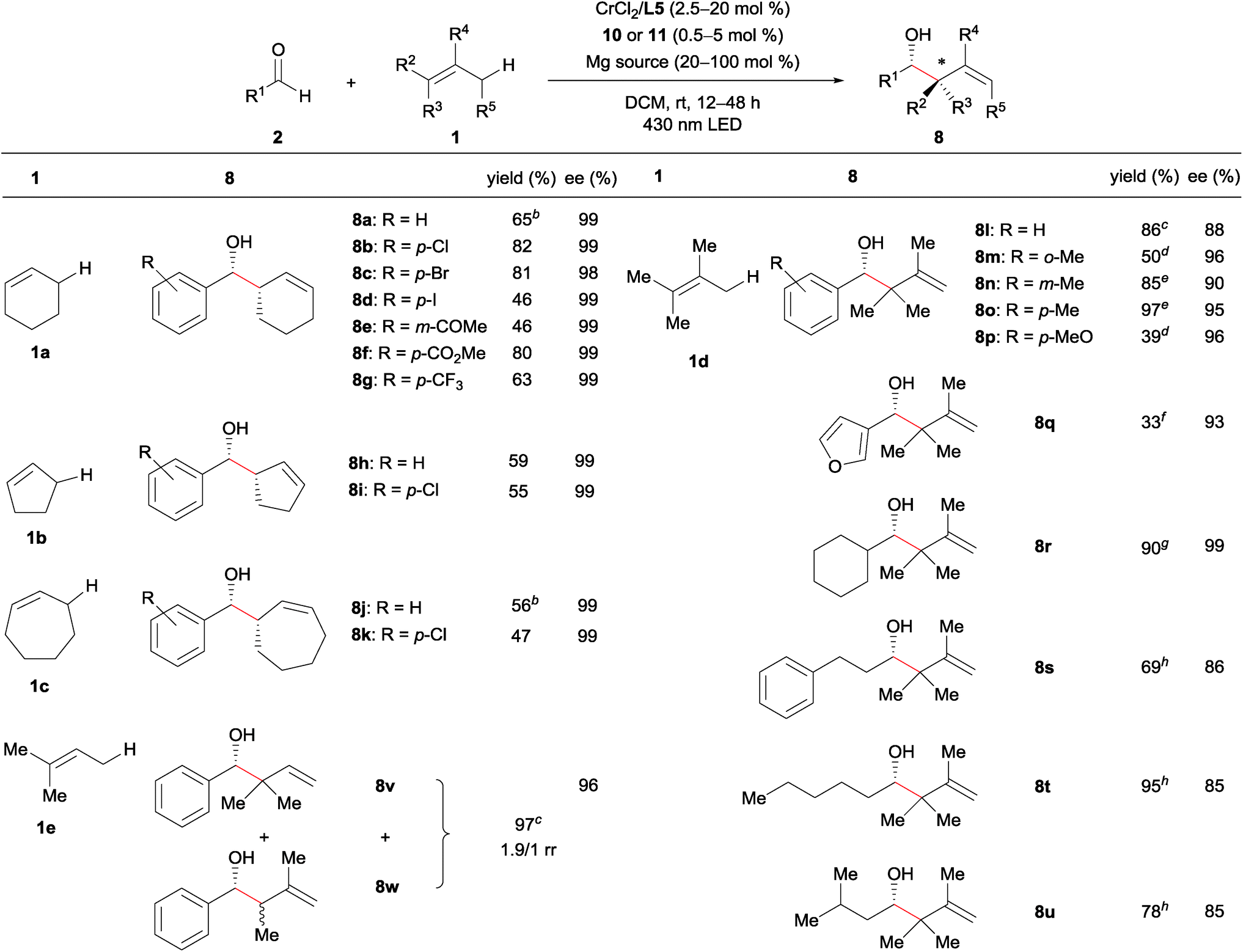
Under these optimized conditions, we next evaluated the substrate scope (Table 2). The reaction of cyclohexene (1a) with substituted benzaldehydes afforded products 8a–8g with almost complete diastereo- and enantioselectivity (up to >20/1 dr, 99% ee). The reaction well tolerated aryl halide moieties (8b–8d), and proceeded chemoselectively at the aldehyde functional group in the presence of a ketone (8e) or an ester (8f) functional group. The method was also easily extended to other cyclic alkenes, with both cyclopentene (1b) and cycloheptene (1c) reacting with excellent stereoselectivity (8h–8k).| a General reaction conditions: aldehyde 2 (0.25 mmol), alkene 1 (5.0 mmol), CrCl2 (0.0125 mmol), L5 (0.0125 mmol), 10 (0.00625 mmol), and Mg(ClO4)2 (0.25 mmol) were reacted in DCM (2.5 mL) at room temperature under 430 nm LED irradiation for 12 h. Yield is isolated yield. The diastereomeric ratio was >20/1 in each case (8a–8k), as determined by 1H NMR analysis of the crude mixture. The enantioselectivity was determined by chiral stationary HPLC analysis after isolation. b CrCl2 (10 mol%) and L5 (10 mol%) were used. c Alkene (2 equiv.), CrCl2 (2.5 mol%), L5 (2.5 mol%), 11 (0.5 mol%), Mg(ClO4)2 (1 equiv), and DCM (0.125 M) were used. d Alkene (20 equiv.), CrCl3·3THF (10 mol%), NaOt-Bu (30 mol%), L5 (10 mol%), 11 (1.25 mol%), Mg(ClO4)2 (1 equiv.), and DCM (0.0625 M) were used. e Alkene (5 equiv.), CrCl2 (10 mol%), L5 (10 mol%), 11 (1.25 mol%), Mg(ClO4)2 (1 equiv.), and DCM (0.0625 M) were used. f Alkene (20 equiv.), CrCl2 (20 mol%), L5 (20 mol%), 11 (5 mol%), Mg(ClO4)2 (1 equiv.), and DCM (0.0625 M) were used. g Alkene (5 equiv.), CrCl2 (10 mol%), L5 (10 mol%), 11 (5 mol%), Mg(ClO4)2 (1 equiv.), and DCE (0.05 M) were used. h Alkene (20 equiv.), CrCl2 (20 mol%), L5 (20 mol%), 11 (5 mol%), MgPhPO3 (20 mol%), and DCE (0.1 M) were used. Reaction time was 48 h. |
|---|
|
Linear alkenes were also competent substrates. Tetrasubstituted alkene 1d reacted with various aldehydes, including ortho-, meta-, and para-substituted benzaldehydes, an electron-rich benzaldehyde, and a heteroaromatic aldehyde, affording the corresponding products 8l–8q (containing an allylic quaternary carbon) with excellent enantioselectivity. The loading of alkene 1d could be reduced to 2 equiv., likely due to the lower oxidation potential of 1d relative to 1a–1c. For less reactive aldehydes such as o-tolualdehyde and p-methoxy benzaldehyde, the chiral chromium alkoxide complex generated from CrCl3·3THF and NaOt-Bu21 exhibited higher catalytic activity than the CrCl2-derived species (8m and 8p). We postulate that this is as a result of allylchromium species 6 bearing alkoxide ligands (X = OR) with higher nucleophilicity than those bearing electron-withdrawing chloride ligands (X = Cl).22,23 The reaction of aliphatic aldehydes also proceeded with high enantioselectivity (8r–8u) following minor modifications of the reaction conditions (1,2-dichloroethane [DCE] as the solvent, 20 mol% MgPhPO3 additive). In the case of asymmetric trisubstituted alkene 1e, an inseparable mixture of 8v and 8w (itself a diastereomixture) was produced with moderate regioselectivity (regioisomeric ratio; rr = 8v/8w = 1.9/1). Nevertheless, both the reactivity and enantioselectivity of 8v were very high: use of 2.5 mol% and 0.5 mol% loadings of the chromium catalyst and photocatalyst 11, respectively, led to the products in 97% combined yield, with 8v in 96% ee. Major isomer 8v presumably derives from prenylchromium species with the chromium atom at the terminal carbon, while minor isomer 8w originates from 2-methyl but-2-enylchromium species with chromium at the terminal carbon. We anticipate that improving the regioselectivity so that the carbon-centered radical can be intercepted by the metal complex in the case of asymmetric alkenes will be a very important avenue for future research. In addition, linear terminal alkenes and disubstituted internal alkenes (e.g. 1-hexene and 2-butene) were unreactive under the current optimal conditions probably due to their high oxidation potentials.
The following experimental results provide key insights into the reaction mechanism (see ESI† for details). First, the addition of TEMPO (2,2,6,6-tetramethylpiperidinyloxyl) as a radical trapping agent to the reaction between 1a and 2a under otherwise optimized conditions completely inhibited the desired reaction. A TEMPO adduct of 1a at the terminal carbon was detected by 1H NMR analysis of the crude mixture after the workup. This result supports our hypothesis that the reaction proceeds through carbon-centered radicals derived from alkene 1. Second, we performed a radical clock experiment using 2-phenylcyclopropylcarbaldehyde and 1d. The reaction proceeded with 77% yield without any cyclopropane ring-opening, indicating that ketyl radicals derived from aldehydes are not involved in the catalytic cycle. These findings, together with the observation that the presence of the chromium complex was essential for the reaction (Table 1, entry 1), are consistent with our working hypothesis for the reaction mechanism depicted in Fig. 2.
To clarify the electron-transfer dynamics between 1a and the photoexcited 10, we examined the transient absorption measurements as shown in Fig. 3(a). Laser flash irradiation (Λ = 355 nm) of 10 gave the electron-transfer state (Acr˙–Xyl˙+). The spectrum furnished absorption peaks at Λmax = 520 nm (ref. 24) and 700 nm,25 which attributed to the acridinyl radical moiety (Acr˙) and the xylyl radical cation moiety (Xyl˙+), respectively (Fig. 3(a)). The time profiles of the decay at 700 nm due to the Xyl˙+ moiety obeyed pseudo-first-order kinetics. The decay rate constant (kobs) linearly increased with the concentration of 1a (Fig. 3(b)). The rate constant of electron transfer from 1a to the acridinyl radical moiety of Acr˙–Xyl˙+ was determined from the linear plot to be 2.2 × 105 M−1 s−1. Thus, electron transfer efficiently occurred to yield 3 and the reduced 10 (Acr˙–Xyl) as the initial step of the photocatalytic redox process.
 | ||
| Fig. 3 Detection of radical intermediates in the photocatalytic redox cycle. (a) Transient absorption spectrum of 10 (50 μM) measured in DCM at 200 μs after laser excitation at 355 nm. (b) Relationship between the decay rate constant (kobs) of the Xyl˙+ moiety observed at 700 nm and the concentration of 1a. (c) Time profiles at 520 nm of Acr˙–Xyl generated by photoexcitation of Acr+–Xyl (50 μM) with cyclohexene (300 mM) in the absence (black) and presence (red) of Mg(ClO4)2. | ||
Furthermore, to confirm that the dramatically increased reactivity in the presence of additive Mg(ClO4)2 was due to the increased concentration of the radical pair (3 and Acr˙–Xyl) generated by electron transfer from 1a to the electron-transfer state of 10 (Acr˙–Xyl˙+), we monitored the initial transient absorption intensities due to Acr˙–Xyl (Λmax = 520 nm) generated by laser irradiation of 10 with a large excess of 1a (300 mM) in the absence and presence of Mg(ClO4)2 (Fig. 3(c)). The initial intensity in the presence of Mg(ClO4)2 was 1.9 times higher than that in the absence of Mg(ClO4)2 (Fig. 3(c)). The radical pair is efficiently stabilized in the presence of Mg(ClO4)2 salt by the electrostatic interaction of 3 with ClO4−.20 Therefore, the concentration of cation radical 3 is enhanced by the presence of Mg(ClO4)2, and this is likely the reason for the dramatically higher product yield in the presence of Mg(ClO4)2 (Table 1, entry 7 vs. 12).26
Conclusions
In conclusion, we developed the first catalytic asymmetric allylation of aldehydes using unactivated hydrocarbon alkenes as pronucleophiles. The reaction enabled direct access to enantiomerically and diastereomerically-enriched homoallylic alcohols, starting from readily available and stable substrates. Critical to the success of the reaction was the development of an asymmetric hybrid catalyst system comprising an acridinium photoredox catalyst and a chiral chromium complex catalyst. The hybrid catalysis enabled a key radical–polar crossover process involving the catalytic generation of chiral and nucleophilic (i.e., polar) organometallic species from simple alkenes via allylic C(sp3)–H activation. Further studies to improve the efficiency of the process, fully elucidate the reaction mechanism, and expand the substrate scope are ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Numbers JP17H06442 (M. K.) (Hybrid Catalysis), 17H01522 (M. K.), 17K19479 (M. K.), 18H05969 (H. M.), 17H0310 (K. O.), 16K13964 (K. O.), and 18H04650 (K. O.) (Hybrid Catalysis). Authors deeply thanks to Prof. Shunichi Fukuzumi (Ewha Womans University, Korea) for the useful discussion and transient absorption measurements.Notes and references
- For selected recent reviews on catalytic enantioselective C(sp3)–H bond functionalization, see: (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugelc and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) Q. Lu and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 49 CrossRef CAS PubMed; (c) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaa04798 CrossRef PubMed.
- For selected recent reviews on stereoselective allylation of carbonyl compounds, see: (a) P. V. Ramachandran, Aldrichimica Acta, 2002, 35, 23 CAS; (b) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed; (c) C.-M. Yu, J. Youn and H.-K. Jung, Bull. Korean Chem. Soc., 2006, 27, 463 CrossRef CAS; (d) I. Marek and G. Sklute, Chem. Commun., 2007, 1683 RSC; (e) D. G. Hall, Synlett, 2007, 11, 1644 CrossRef; (f) G. C. Hargaden and P. J. Guiry, Adv. Synth. Catal., 2007, 349, 2407 CrossRef CAS; (g) H. Yamamoto and M. Wadamoto, Chem.–Asian J., 2007, 2, 692 CrossRef CAS PubMed; (h) H. Lachance and D. G. Hall, Org. React., 2009, 73, 1 Search PubMed; (i) S. B. Han, I. S. Kim and M. J. Krische, Chem. Commun., 2009, 7278 RSC; (j) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed; (k) J. Moran and M. J. Krische, in Asymmetric Synthesis: More Methods and Applications, ed. M. Christmann and S. Bräse, Wiley-VCH, Weinheim, Germany, 2012, p. 187 Search PubMed; (l) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed; (m) H.-X. Huo, J. R. Duvall, M.-Y. Huang and R. Hong, Org. Chem. Front., 2014, 1, 303 RSC; (n) Q. Tian and G. Zhang, Synthesis, 2016, 48, 4038 CrossRef CAS; (o) P. Kumar, D. Tripathi, B. M. Sharma and N. Dwivedi, Org. Biomol. Chem., 2017, 15, 733 RSC; (p) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371 CrossRef CAS; (q) P.-S. Wang, M.-L. Shen and L.-Z. Gong, Synthesis, 2018, 50, 956 CrossRef CAS; (r) D. M. Sedgwlck, M. N. Grayson, S. Fustero and P. Barrlo, Synthesis, 2018, 50, 1935 CrossRef; (s) K. Spielmann, G. Niel, R. M. de Figueiredo and J.-M. Campagne, Chem. Soc. Rev., 2018, 47, 1159 RSC; (t) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026 CrossRef CAS PubMed.
- For selected reviews on catalytic asymmetric carbonyl ene reactions, see: (a) K. Mikami and M. Shimizu, Chem. Rev., 1992, 92, 1021 CrossRef CAS; (b) D. J. Berrisford and C. Bolm, Angew. Chem., Int. Ed., 1995, 34, 1717 CrossRef CAS; (c) Comprehensive Asymmetric Catalysis, ed.K. Mikami, M. Terada, E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, Heidelberg, 1999, p. 1143 Search PubMed; (d) L. C. Dias, Curr. Org. Chem., 2000, 4, 305 CrossRef CAS; (e) K. Mikami and T. Nakai, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2000, p. 543 Search PubMed; (f) M. L. Clarke and M. B. France, Tetrahedron, 2008, 64, 9003 CrossRef CAS.
- Catalytic allylic C(sp3)–H activation reported so far mainly generates electrophilic π-allylmetal complexes. For selected reviews of allylic C(sp3)–H activation, see: (a) T. Jensen and P. Fristrup, Chem.–Eur. J., 2009, 15, 9632 CrossRef CAS PubMed; (b) G. Liu and Y. Wu, Top. Curr. Chem., 2010, 292, 195 CrossRef CAS; (c) C. J. Engelin and P. Fristrup, Molecules, 2011, 16, 951 CrossRef CAS PubMed; (d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (f) B. J. Li and Z. J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (g) F. Liron, J. Oble, M. M. Lorion and G. Poli, Eur. J. Org. Chem., 2014, 5863 CrossRef CAS; (h) C. Zheng and S. L. You, RSC Adv., 2014, 4, 6173 RSC; (i) C. G. Newton, S. G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed.
- Z.-L. Tao, X.-H. Li, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 4054 CrossRef CAS.
- T. Mita, S. Hanagata, K. Michigami and Y. Sato, Org. Lett., 2017, 19, 5876 CrossRef CAS PubMed.
- J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705 CrossRef CAS PubMed . The racemic variant exhibited broad substrate scope and excellent diastereoselectivity..
- For chiral Brønsted base-catalyzed enantioselective allylation of ketones with special hydrocarbon alkenes (i.e., skipped enynes), see: X.-F. Wei, X.-W. Xie, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2017, 139, 4647 CrossRef CAS PubMed.
- For Brønsted base-catalyzed racemic allylation of imines with hydrocarbon alkenes, see: W. Bao, H. Kossen and U. Schneider, J. Am. Chem. Soc., 2017, 139, 4362 CrossRef CAS PubMed.
- For related methods for the generation of organonickel species, see: (a) C. L. Joe and A. G. Doyle, Angew. Chem., Int. Ed., 2016, 55, 4040 CrossRef CAS PubMed; (b) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002 RSC; (c) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715 CrossRef CAS PubMed; (d) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719 CrossRef CAS PubMed; (e) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304 CrossRef CAS PubMed; (f) X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353 CrossRef CAS PubMed; (g) Y.-Y. Gui, L.-L. Liao, L. Sun, Z. Zhang, J.-H. Ye, G. Shen, Z.-P. Lu, W.-J. Zhou and D.-G. Yu, Chem. Commun., 2017, 53, 1192 RSC; (h) Y.-Y. Gui, X.-W. Chen, W.-J. Zhou and D.-G. Yu, Synlett, 2017, 28, 2581 CrossRef CAS; (i) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef CAS PubMed; (j) H.-P. Deng, X.-Z. Fan, Z.-H. Chen, Q.-H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579 CrossRef CAS PubMed; (k) Y.-Y. Gui, Z.-X. Wang, W.-J. Zhou, L.-L. Liao, L. Song, Z.-B. Yin, J. Li and D.-G. Yu, Asian J. Org. Chem., 2018, 7, 537 CrossRef CAS; (l) H. Fuse, M. Kojima, H. Mitsunuma and M. Kanai, Org. Lett., 2018, 20, 2042 CrossRef CAS; (m) S. Y. Go, G. S. Lee and S. H. Hong, Org. Lett., 2018, 20, 4691 CrossRef CAS PubMed; (n) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369 CrossRef CAS PubMed; (o) L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333 CrossRef CAS PubMed; (p) I. B. Perry, T. W. Brewer, P. J. Sarver, D. M. Schlutz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70 CrossRef CAS PubMed; (q) Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200 CrossRef CAS PubMed; (r) L. K. G. Ackerman, J. I. M. Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059 CrossRef CAS PubMed; (s) D.-M. Yan, C. Xiao and J.-R. Chen, Chem, 2018, 4, 2496 CrossRef CAS; (t) L. Zhang, X. Si, Y. Yang, M. Zimmer, S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57 DOI:10.1002/anie.201810526.
- For related methods for the generation of organopalladium species, see: (a) S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204 CrossRef CAS PubMed; (b) U. K. Sharma, H. P. L. Gemoets, F. Schröder, T. Noël and E. V. Van der Eycken, ACS Catal., 2017, 7, 3818 CrossRef CAS.
- For a related method for the generation of organocobalt species, see: S. M. Thullen and T. Rovis, J. Am. Chem. Soc., 2017, 139, 15504 CrossRef CAS PubMed.
- For the generation of chiral organometallic species through homolytic cleavage of a stable C(sp3)–H bond and subsequent oxidative trap by a chiral metal complex, which was applied to a catalytic asymmetric coupling reaction (oxidative benzylic C(sp3)–H cyanation), see: W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014 CrossRef CAS PubMed . One preliminary example (54% ee) with this reaction pattern was also described in ref. 10q..
- R. Zhou, H. Liu, H. Tao, X. Yu and J. Wu, Chem. Sci., 2017, 8, 4654 RSC.
- For a discussion of the feasibility of the working hypothesis shown in Fig. 2 based on the redox potential of each intermediate, see ESI.†.
- T. Tsudaka, H. Kotani, K. Ohkubo, T. Nakagawa, N. V. Tkachenko, H. Lemmetyinen and S. Fukuzumi, Chem.–Eur. J., 2017, 23, 1306 CrossRef CAS PubMed.
- For selected examples of catalytic asymmetric Nozaki–Hiyama–Kishi allylation reaction, see: (a) M. Bandini, P. G. Cozzi, P. Melchiorre and A. Umani-Ronchi, Angew. Chem., Int. Ed., 1999, 38, 3357 CrossRef CAS; (b) A. Berkessel, D. Menche, C. A. Sklorz, M. Schröder and I. Paterson, Angew. Chem., Int. Ed., 2003, 42, 1032 CrossRef CAS PubMed; (c) M. Inoue, T. Suzuki and M. Nakada, J. Am. Chem. Soc., 2003, 125, 1140 CrossRef CAS PubMed; (d) M. Kurosu, M.-H. Lin and Y. Kishi, J. Am. Chem. Soc., 2004, 126, 12248 CrossRef CAS PubMed; (e) J.-Y. Lee, J. J. Miller, S. S. Hamilton and M. S. Sigman, Org. Lett., 2005, 7, 1837 CrossRef CAS PubMed; (f) G. Xia and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 2554 CrossRef CAS PubMed; (g) J. J. Miller and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 2752 CrossRef CAS PubMed; (h) Z. Zhang, J. Huang, B. Ma and Y. Kishi, Org. Lett., 2008, 10, 3073 CrossRef CAS PubMed; (i) Q.-H. Deng, H. Wadepohl and L. H. Gade, Chem.–Eur. J., 2011, 17, 14922 CrossRef PubMed.
- C. Chen, K. Tagami and Y. Kishi, J. Org. Chem., 1995, 60, 5386 CrossRef CAS.
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS PubMed.
- (a) P. A. Thompson and J. D. Simon, J. Am. Chem. Soc., 1993, 115, 5657 CrossRef CAS; (b) T. M. Bockman, S. M. Hubig and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 2826 CrossRef CAS; (c) T. Yabe and J. K. Kochi, J. Am. Chem. Soc., 1992, 114, 4491 CrossRef CAS; (d) V. N. Grosso, C. M. Previtali and C. A. Chesta, Photochem. Photobiol., 1998, 68, 481 CrossRef CAS; (e) R. A. Marcus, J. Phys. Chem. B, 1998, 102, 10071 CrossRef CAS; (f) T. Kluge and H. Knoll, J. Photochem. Photobiol., A, 2000, 130, 95 CrossRef CAS; (g) E. V. Vakarin, M. F. Holovko and P. Piotrowiak, Chem. Phys. Lett., 2002, 363, 7 CrossRef CAS; (h) K. Okamoto, K. Ohkubo, K. M. Kadish and S. Fukuzumi, J. Phys. Chem. A, 2004, 108, 10405 CrossRef CAS.
- K. N. Mahendra and R. C. Mehrotra, Synth. React. Inorg. Met.-Org. Chem., 1990, 20, 963 CrossRef CAS.
- K. Sugimoto, S. Aoyagi and C. Kibayashi, J. Org. Chem., 1997, 62, 2322 CrossRef CAS PubMed.
- In the reaction of p-methoxy benzaldehyde and 1d, NMR yields of 8p were 33% and 62% by using CrCl2 and CrCl3·3THF–NaOt-Bu catalysts, respectively..
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600 CrossRef CAS PubMed.
- K. Ohkubo, K. Suga, K. Morikawa and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 12850 CrossRef CAS PubMed.
- On the other hand, the role of Mg(ClO4)2 (or perchlorate in general, see Table 1, entry 7 vs. 9–12) in enhancing the enantioselectivity is still unclear. We assume that chiral ligand L5 remains coordinated to the chromium atom during the reaction in the presence of stoichiometric Mg(ClO4)2; the reaction between 1a and 2a did not proceed at all by premixing of L5 with Mg(ClO4)2. See ESI† for details..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05677c |
| This journal is © The Royal Society of Chemistry 2019 |