
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of acyl(chloro)phosphines enabled by phosphinidene transfer†
Kevin M.
Szkop
 ab,
Michael B.
Geeson
b,
Douglas W.
Stephan
*a and
Christopher C.
Cummins
*b
ab,
Michael B.
Geeson
b,
Douglas W.
Stephan
*a and
Christopher C.
Cummins
*b
aDepartment of Chemistry, University of Toronto, 80 St George St, Toronto, Ontario M5S3H6, Canada. E-mail: dstephan@chem.utoronto.ca; Tel: +1 416 946 3294
bDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA. E-mail: ccummins@mit.edu; Tel: +1 617 253 5332
First published on 7th February 2019
Abstract
Acyl(chloro)phosphines RC(O)P(Cl)(t-Bu) have been prepared by formal insertion of tert-butyl phosphinidene (t-Bu–P) from t-BuPA (A = C14H10 or anthracene) into the C–Cl bond of acyl chlorides. We show that the under-explored acyl(chloro)phosphine functional group provides an efficient method to prepare bis(acyl)phosphines, which are important precursors to compounds used industrially as radical polymerization initiators. Experimental and computational investigations into the mechanism of formation of acyl(chloro)phosphines by our synthetic method reveal a pathway in which chloride attacks a phosphonium intermediate and leads to the reductive loss of anthracene from the phosphorus center in a P(V) to P(III) process. The synthetic applicability of the acyl(chloro)phosphine functional group has been demonstrated by reduction to an acylphosphide anion, which can in turn be treated with an acyl chloride to furnish dissymmetric bis(acyl)phosphines.
Acylphosphines and their oxides are used as photoinitiators for radical-initiated polymerization1–4 reactions used in the manufacture of consumer goods such as automotive coatings, adhesives, latex composition kits, and various dental and orthodontic materials.5–14 Known methodologies for the synthesis of acylphosphines employ nucleophilic sources of phosphorus such as tris(trimethylsilyl)phosphine (P(TMS)3, TMS = trimethylsilyl),15–17 which is volatile, pyrophoric, toxic, expensive, and difficult to handle.18 Transition metal-supported phosphines and phosphides have been used as a platform in niche syntheses of acylphosphines, though scope is limited and products remain bound to the metal center.19–23 Perhaps the most versatile synthetic approach has emerged from the groups of Liotta, Becker, Grützmacher and Mézailles who employ PH3 and MPH2 (M = Li, Na, K) as nucleophilic sources of phosphorus en route to a wide variety of mono-, bis-, and tris(acyl)phosphines.24–31 More recently, salts of the 2-phosphaethynolate anion ([PCO]−)have been used in the synthesis of functional groups related to acylphosphines.32 Despite these advances, enlarging the scope of methods for accessing valuable acylphosphines with different substitution patterns remains an attractive goal.33
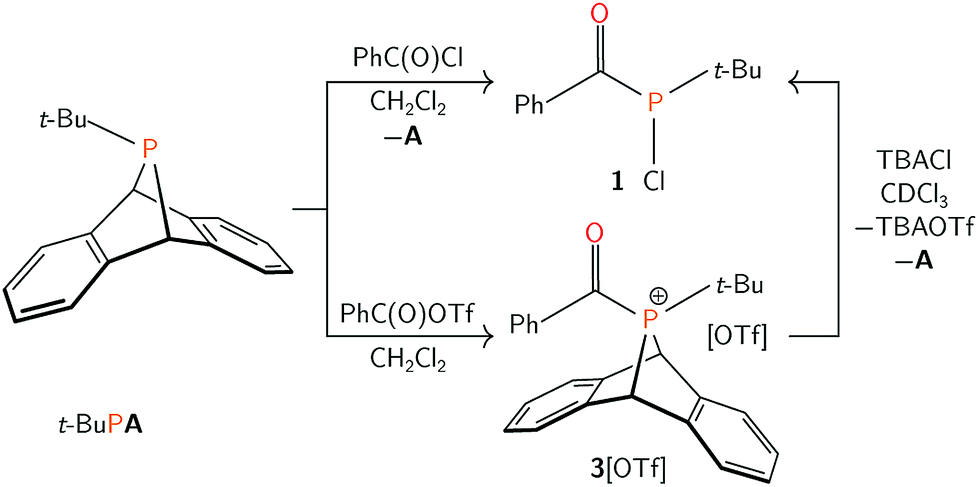
We have identified dibenzo-7λ3-phosphanorbornadienes (RPA, A = anthracene or C14H10) as efficient compounds to generate and transfer metastable singlet phosphinidene species.34,35 Herein, the tert-butyl phosphinidene group of t-BuPA is shown to formally insert into the carbon–chlorine bond of acyl chlorides to generate the rare acyl(chloro)phosphine functional group.36,37 This product is subsequently exploited for further functionalization.
Treatment of t-BuPA with benzoyl chloride (PhC(O)Cl) in dichloromethane at 23 °C (Scheme 1) led to an instant colour change from colourless to bright yellow, and the quantitative formation of 1 as a yellow powder, isolated in 82% yield. The species exhibited a singlet resonance at δ 114 ppm in the 31P{1H} NMR spectrum. The 13C{1H} NMR spectrum of 1 shows a diagnostic doublet resonance in the carbonyl region at δ 210 ppm (JPC = 67 Hz) while the IR spectrum of 1 displays a complementary C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1648 cm−1. Collectively, these data support the formulation of 1 as (tert-butylchlorophosphanyl)(phenyl)methanone, PCl(t-Bu)(C(O)Ph).
O stretching band at 1648 cm−1. Collectively, these data support the formulation of 1 as (tert-butylchlorophosphanyl)(phenyl)methanone, PCl(t-Bu)(C(O)Ph).
 | ||
| Scheme 1 Synthesis of 1 and 3[OTf]. | ||
As 1 could not be isolated as single crystals suitable for structural analysis, a transition metal complex of this phosphine was prepared. Treatment of 1 with half an equivalent of [Ru(p-cymene)Cl2]2 in tetrahydrofuran led to the formation of coordination complex 2. Layering the reaction mixture with pentane at 23 °C gave 2 as a red-orange crystalline material in 77% yield. Multinuclear NMR spectroscopy studies, in addition to a single crystal X-ray diffraction study confirmed the formulation of 2 as Ru(p-cymene)Cl2(PCl(t-Bu)(C(O)Ph)) (Fig. 1). Racemic 2 crystallizes in the P21/c space group, and the solid state structure showed C–O, P–C, and P–Ru bond lengths of 1.221(4), 1.875(4) and 2.3178(8) Å respectively, which are in line with the expected values.38 Ru–P binding of acylphosphines to ruthenium has been observed in the coordination chemistry of related P(III) ligands.39,40
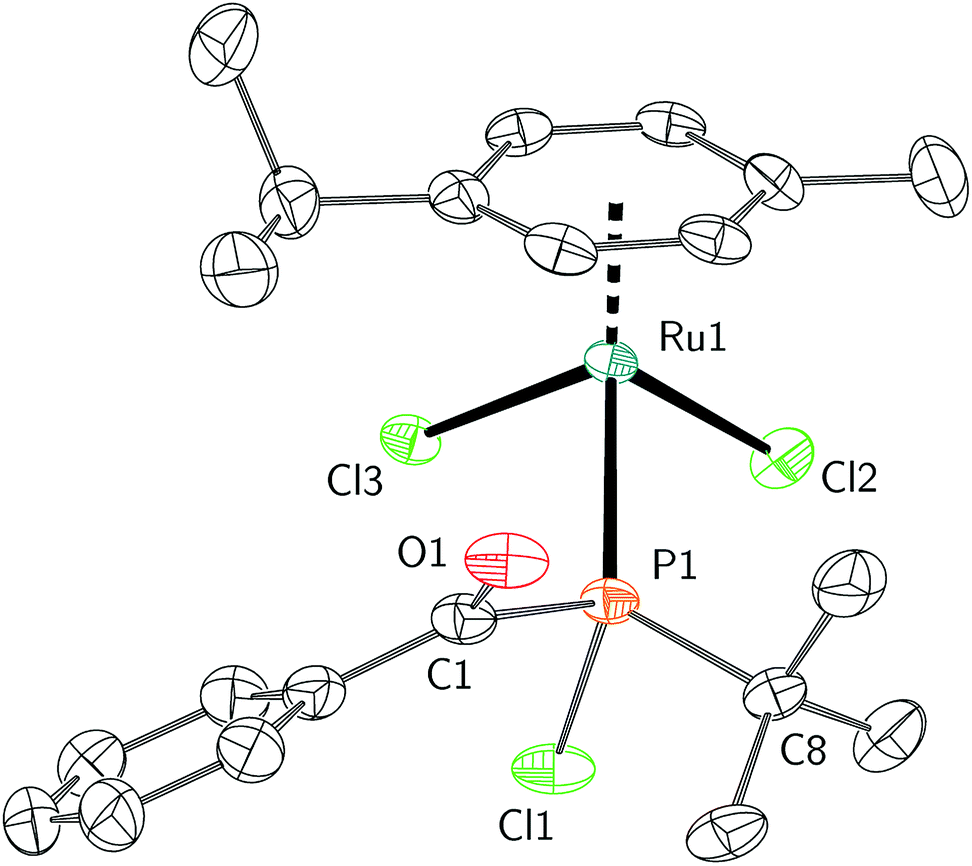 | ||
| Fig. 1 Molecular structure of 2 with ellipsoids set at the 50% probability level and hydrogen atoms omitted for clarity. | ||
Previous studies on thermal phosphinidene transfer have established that thermal loss of a phosphinidene does not occur when the R-group of an RPA compound is not π-donating.35 Therefore, the mechanism of acylphosphine formation is more likely to proceed associatively via nucleophilic attack by phosphorus at the electrophilic carbonyl group of benzoyl chloride. We envisioned that replacement of the chloride for a less nucleophilic anion may lead to the isolation of the putative cationic acylphosphonium intermediate. To this end, the reaction of equimolar solutions of t-BuPA and benzoyl triflate (PhC(O)OTf, OTf = CF3SO3−)41 led to the formation of 3[OTf] (Scheme 1) as a white powder in 75% yield. A singlet resonance is observed at δ 127 ppm in the 31P{1H} NMR spectrum of 3[OTf]. The 13C{1H} NMR spectrum of 3[OTf] showed a diagnostic carbonyl resonance at 208 ppm and the IR spectrum displayed a CO absorption peak at 1636 cm−1. These data are consistent with the formulation of 3[OTf] as [AP(t-Bu)(C(O)Ph)][OTf].
Compound 3[OTf] decomposes slowly in solution at ambient temperature, yielding an anthracenyl(acyl)hydridophosphonium triflate salt as the major product (see ESI†). Similar C–H activation of arenes by phosphorus-containing electrophiles has been described for metal-stabilized phosphenium triflates.42–44 Despite its instability in solution, 3[OTf] can be stored in the solid state at −35 °C for weeks without decomposition and purified by recrystallization from saturated dichloromethane/pentane solutions, also at −35 °C. Treatment of 3[OTf] with a chloride source (tetra-n-butylammonium chloride) in chloroform at 23 °C quantitatively generated 1, [TBA][OTf] and anthracene (Scheme 1). This reactivity is consistent with nucleophilic attack of the phosphonium center in 3[OTf] by chloride, inducing reductive loss of anthracene.
The mechanism leading to the formation of 1 was probed via competition experiments involving the addition of two different para-substituted benzoyl chloride substrates in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to t-BuPA. The product ratio was determined by quantitative 31P{1H} NMR spectroscopy and a series of such experiments is summarised in the Hammett plot (Fig. 2). A small negative value for ρ of −2.36 is consistent with build-up of positive charge in the transition state.45 This is particularly evident for electron withdrawing groups, such as CF3, where the reaction is accelerated compared to electron rich para-substituted benzoyl chlorides. These electronic effects are further evidence of phosphine attack at the electrophilic carbon atom of the acylphosphine functional group in the rate limiting step of the reaction.
:1 ratio to t-BuPA. The product ratio was determined by quantitative 31P{1H} NMR spectroscopy and a series of such experiments is summarised in the Hammett plot (Fig. 2). A small negative value for ρ of −2.36 is consistent with build-up of positive charge in the transition state.45 This is particularly evident for electron withdrawing groups, such as CF3, where the reaction is accelerated compared to electron rich para-substituted benzoyl chlorides. These electronic effects are further evidence of phosphine attack at the electrophilic carbon atom of the acylphosphine functional group in the rate limiting step of the reaction.
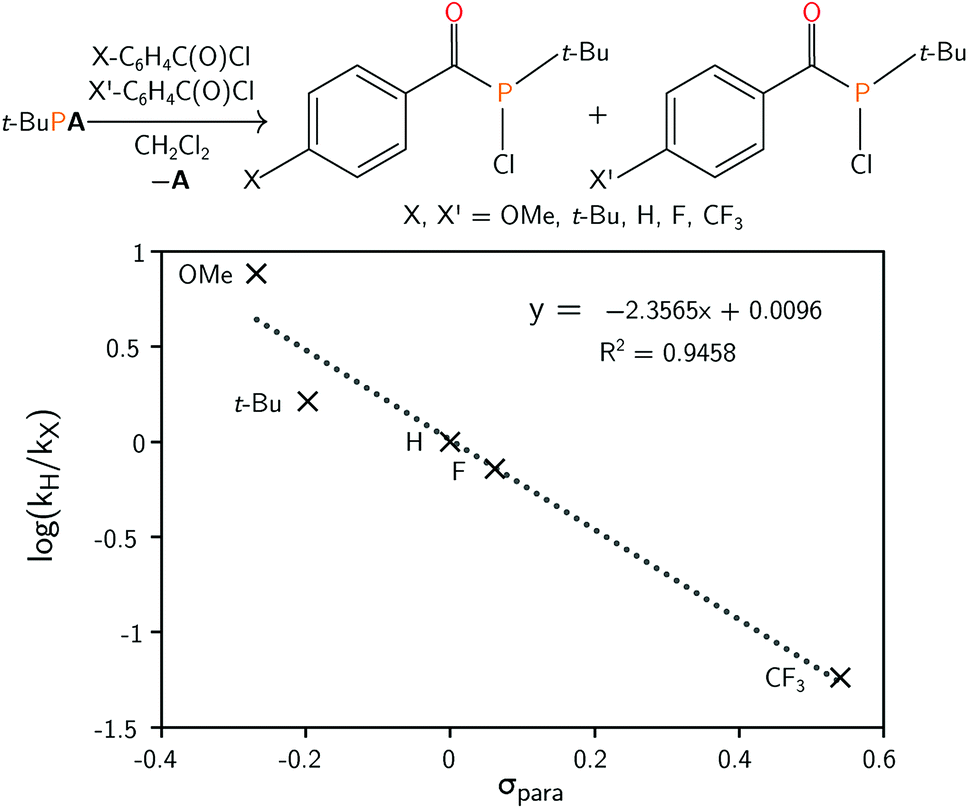 | ||
| Fig. 2 (Top) Preparation of acyl(chloro)phosphines starting from t-BuPA and para-substituted benzoyl chlorides. (Bottom) Hammett plot for the reaction, obtained by competition experiments. | ||
Quantum chemical calculations were employed to study the formation of 1. These were performed using ORCA 4.0.0.246 at the B3LYP/Def2-TZVP level of theory and the results are summarised in Fig. 3. According to our calculations, initial nucleophilic attack of t-BuPA on benzoyl chloride proceeds with an activation barrier of +17.9 kcal mol−1TS1, in which the phosphorus atom occupies a tetrahedral environment, leads to an intermediate comprising a phosphonium and chloride ion-pair, I1. The distance between the phosphorus and chlorine atoms is 2.55 Å which is significantly longer than the sum of the single-bond covalent radii, 2.10 Å.38 However, the phosphorus center features a trigonal bipyramidal structure instead of the ideal tetragonal coordination sphere of a 4-coordinate phosphonium species, suggestive of a strong interaction between the phosphonium centre and chloride. A second transition state in the reaction pathway TS2 was located, which corresponds to formation of a P–Cl bond and concomitant loss of anthracene to give 1. TS2 had a relative Gibbs free energy of +15.6 kcal mol−1, which is lower than the Gibbs free energy for TS1. The loss of anthracene in TS2 is highly concerted and appears to proceed by a cheletropic extrusion pathway. This type of formal reductive elimination from λ-5 phosphoranes to give a phosphorus(III) product has previously been described in the literature.47–50 Finally, we investigated the possibility of a phosphenium chloride salt intermediate (I2, see ESI†), which would arise from loss of anthracene from cation 3 and is the formal reverse of the McCormack reaction.49–51 We found the relative Gibbs free energy of intermediate I2 to be 65.0 kcal mol−1 which is significantly higher than either TS1 of TS2, ostensibly ruling it out as an intermediate in the reaction mechanism (see ESI†).
 | ||
| Fig. 3 Calculated lowest energy pathway for the reaction of t-BuPA with benzoyl chloride, at the B3LYP/Def2-TZVP level of theory (see ESI† for full computational details). ΔG is shown in orange and ΔH is shown in blue. Hydrogen atoms are omitted for clarity. | ||
The higher Gibbs free energy of the first transition state compared to the second is consistent with experiment, where a phosphonium intermediate is not observed when benzoyl chloride is used as the acylating reagent. Thus it is indicated that, when switching from chloride to triflate, there is a change in the rate determining step from TS1 to TS2, which are in proximity (17.9 and 15.6 kcal mol−1, respectively) as determined by DFT calculations.
Efforts to further derivatise 1 were undertaken by reduction of the P–Cl bond. Treatment of compound 1 with 2 equivalents of freshly-prepared sodium naphthalenide52 led to the formation of a new species with concomitant formation of naphthalene and sodium chloride (Scheme 2). Addition of 15-crown-5 to the reaction mixture led to the isolation of [Na(15-crown-5)]4 as a crystalline orange powder in 49% yield. The connectivity of anion 4 as [OC(Ph)Pt-Bu]− was confirmed by a single crystal X-ray diffraction experiment (Fig. 4). In the solid state, the P–C and C–O bond lengths are 1.756(2) and 1.271(2) Å, respectively. The short C–P bond and long C–O bond are consistent with delocalization of electron density from a phosphide lone pair into the π*(CO) orbital.
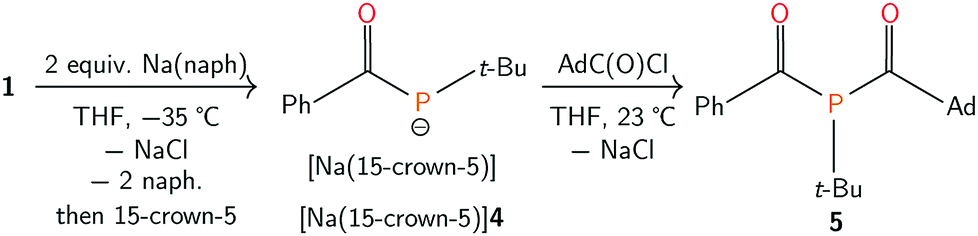 | ||
| Scheme 2 Synthesis of [Na(15-crown-5)]4 and 5. | ||
 | ||
| Fig. 4 Molecular structure of [Na(15-crown-5)]4 with ellipsoids set at the 50% probability level and hydrogen atoms omitted for clarity. | ||
To quantify the degree of electron delocalization over the acylphosphide functional group, the electronic structure was evaluated using natural bond orbital (NBO) methods.53 Natural resonance theory (NRT) analysis, implemented within the NBO package, showed that the primary resonance structure (4a, Fig. 5) had a contribution of 39%. A second resonance structure, containing a carbon-phosphorus double bond and carbon-oxygen single bond (4b, Fig. 5), closely followed with a contribution of 33%, while the remaining resonance structures each had less than 2% weight. The C–O and C–P natural bond orders were remarkably similar at 1.51 and 1.49, respectively. The high degree of delocalization of the phosphide lone pair into the carbonyl unit seems to be a result of the anionic charge and of having two lone pairs at P, one of which is higher in energy and has more p-character and is thus more prone to delocalization, since for neutral acylphosphine-containing molecules, minimal delocalization has been observed.54,55
 | ||
| Fig. 5 Leading resonance structures of anion 4, as determined by Natural Resonance Theory (NRT), implemented within the Natural Bond Orbital (NBO) package. | ||
The combination of [Na(15-crown-5)]4 and AdC(O)Cl (Ad = 1-adamantyl) in tetrahydrofuran generated 5, which was isolated as a crystalline yellow solid in 55% yield (Scheme 2). In the 31P{1H} NMR spectrum of 5, a singlet resonance is observed at δ 37 ppm. Two diagnostic doublet resonances are observed in the downfield region of the 13C{1H} NMR spectrum at δ 225 (JPC = 50 Hz) and 214 ppm (JPC = 46 Hz), which are assigned to the two acyl carbon atoms. The IR spectrum of 5 shows two CO absorption peaks at 1665 and 1629 cm−1. These spectroscopic data inform our formulation of 5 as OC(Ph)P(C(O)Ad)t-Bu. Single crystals of 5 were grown from a concentrated pentane solution and analyzed by an X-ray diffraction study, which featured 5 as a single (R)-enantiomer (see ESI†) in the chiral space group P21 (Fig. 6).
 | ||
| Fig. 6 Molecular structure of (R)-5 with ellipsoids set at the 50% probability level and hydrogen atoms omitted for clarity. P1–C1, C1–O1, P1–C2 and C2–O2 bond lengths are 1.209(4), 1.902(3), 1.212(4) and 1.888(3) Å, respectively. | ||
The formation of 5 from [Na(15-crown-5)]4 provides an approach for preparing dissymmetric bis(acyl)phosphines.56 Interestingly, a previous report of a (W(CO)5) P-protected acylphosphide anion describes C–O instead of C–P bond formation upon treatment with an acyl chloride, to give an ester-containing phosphaalkene.57
In summary, we have described a novel reaction of t-BuPA with acyl chlorides to give acyl(chloro)phosphine-containing products, which were not accessible previously.36,37 Experimental mechanistic studies agree well with computational insights, and clearly demonstrate the presence of an intermediate acylphosphonium species. The acyl(chloro)phosphine functional group can be reduced to the corresponding acylphosphide anion, which reacts further with an acyl chloride to provide access to a dissymmetric bis(acyl)phosphine. Other synthetic pathways to related acylphosphine derivatives are the subject of on-going investigations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge computational support from the Massachusetts Green High-Performance Computing Center. We thank the National Science Foundation (NSF-CHE-1664799) for financial support of this research. D. W. S. is grateful for the award of an NSERC Canada Research Chair and a visiting Einstein Professorship at TU Berlin (2016–2019). K. M. S. acknowledges the support of an NSERC PGS-D award and the University of Toronto Department of Chemistry for a Special Opportunity Graduate Travel Fellowship. Dr Eric Arndt of Formlabs is thanked for stimulating discussion. Dr Peter Müller is gratefully thanked for his assistance with crystallographic studies.Notes and references
- L. Gonsalvi and M. Peruzzini, Angew. Chem., Int. Ed., 2012, 51, 7895–7897 CrossRef CAS PubMed.
- K. Dietliker, A compilation of photoinitiators: commercially available for UV today, SITA Technology Ltd, Edinburgh, London, 2002 Search PubMed.
- K. Dietliker, T. Jung, J. Benkhoff, H. Kura, A. Matsumoto, H. Oka, D. Hristova, G. Gescheidt and G. Rist, Macromol. Symp., 2004, 217, 77–98 CrossRef CAS.
- E. Schrader, Doctoral thesis, ETH Zürich, 2018.
- P. Lechtken, I. Buethe, M. Jacobi and W. Trimborn, US4298738A, 1981.
- P. Lechtken, B. Bronstert, G. Hoffmann, R. Vyvial and J. Lynch, US4292152A, 1981.
- P. Lechtken, I. Buethe and A. Hesse, US4324744A, 1982.
- A. Henne, A. Hesse, M. Jacobi, G. Schornick, R. Vyvial and K. Holoch, US4447520A, 1984.
- C.-T. Huang, P. D. Hammesfahr and S. R. Jefferies, US5367002A, 1994.
- H. Grützmacher, T. Ott and K. Dietliker, WO2011003772A1, 2011.
- N. Karim, T. D. Jones, K. M. Lewandowski, B. D. Craig, S. B. Mitra and J. Yang, EP2073782B1, 2011.
- S. Stelzig, J. Klee, A. Facher and C. Weber, EP2497454A1, 2012.
- A. A. Guimont, C. Li, X. Zhou, H. S. Bui and J.-T. Simonnet, US9636293B2, 2017.
- X. Zhou, C. Li, H. S. Bui and J.-T. Simonnet, US9801803B2, 2017.
- G. Becker, Z. Anorg. Allg. Chem., 1981, 480, 38–48 CrossRef CAS.
- G. Becker, M. Rössler and W. Uhl, Z. Anorg. Allg. Chem., 1981, 473, 7–19 CrossRef CAS.
- G. Becker, Z. Anorg. Allg. Chem., 1981, 480, 21–37 CrossRef CAS.
- S. A. Kosarev and S. J. Collier, Tris(trimethylsilyl)phosphine, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, 2011 Search PubMed.
- D. S. Bohle, G. R. Clark, C. E. F. Rickard and W. Roper, J. Organomet. Chem., 1988, 353, 355–381 CrossRef.
- D. S. Bohle, G. R. Clark, C. E. F. Rickard and W. R. Roper, J. Organomet. Chem., 1990, 393, 243–285 CrossRef.
- J.-J. Brunet, A. Capperucci, R. Chauvin and B. Donnadieu, J. Organomet. Chem., 1997, 533, 79–81 CrossRef CAS.
- J.-J. Brunet, R. Chauvin, B. Donnadieu and E. Thepaut, J. Organomet. Chem., 1999, 579, 198–205 CrossRef CAS.
- A. Antiñolo, S. Gacía-Yuste, A. Otero, R. Reguillo-Carmona and M. T. Tercero-Morales, C. R. Chim., 2010, 13, 929–934 CrossRef.
- C. L. Liotta, M. L. McLaughlin and B. A. O'Brien, Tetrahedron Lett., 1984, 25, 1249–1252 CrossRef CAS.
- C. L. Liotta, M. L. McLaughlin, D. G. Van Derveer and B. A. O'Brien, Tetrahedron Lett., 1984, 25, 1665–1668 CrossRef CAS.
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS.
- B. A. Trofimov, S. N. Arbuzova and N. K. Gusarova, Russ. Chem. Rev., 1999, 68, 215–228 CrossRef CAS.
- J. Wang, G. Siqueira, G. Müller, D. Rentsch, A. Huch, P. Tingaut, J. Levalois-Grützmacher and H. Grützmacher, Chem. Commun., 2016, 52, 2823–2826 RSC.
- A. Huber, A. Kuschel, T. Ott, G. Santiso-Quinones, D. Stein, J. Bräuer, R. Kissner, F. Krumeich, H. Schönberg, J. Levalois-Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2012, 51, 4648–4652 CrossRef CAS PubMed.
- A. Eibel, M. Schmallegger, M. Zalibera, A. Huber, Y. Bürkl, H. Grützmacher and G. Gescheidt, Eur. J. Inorg. Chem., 2017, 2017, 2469–2478 CrossRef CAS.
- K. X. Bhattacharyya, S. Dreyfuss, N. Saffon-Merceron and N. Mézailles, Chem. Commun., 2016, 52, 5179–5182 RSC.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- J.-P. Wolf and G. Hug, US20050245768A1, 2005.
- A. Velian and C. C. Cummins, J. Am. Chem. Soc., 2012, 134, 13978–13981 CrossRef CAS PubMed.
- W. J. Transue, A. Velian, M. Nava, C. García-Iriepa, M. Temprado and C. C. Cummins, J. Am. Chem. Soc., 2017, 139, 10822–10831 CrossRef CAS PubMed.
- Y. A. Veits, M. V. Filippov, A. A. Borisenko and V. L. Foss, Zh. Obshch. Khim., 1991, 61, 108–113 Search PubMed.
- Y. A. Veits, E. G. Neganova, M. V. Filippov, A. A. Borisenko and V. L. Foss, Zh. Obshch. Khim., 1991, 61, 114–118 Search PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- S. Gowrisankar, H. Neumann, A. Spannenberg and M. Beller, Organometallics, 2014, 33, 94–99 CrossRef CAS.
- K. M. Szkop, A. R. Jupp and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 12751–12755 CrossRef CAS PubMed.
- L. Brown and M. Koreeda, J. Org. Chem., 1984, 49, 3875–3880 CrossRef CAS.
- A. Jayaraman and B. T. Sterenberg, Organometallics, 2016, 35, 2367–2377 CrossRef CAS.
- A. Jayaraman, S. Nilewar, T. V. Jacob and B. T. Sterenberg, ACS Omega, 2017, 2, 7849–7861 CrossRef CAS.
- S. Nilewar, A. Jayaraman and B. T. Sterenberg, Organometallics, 2018, 37, 4699–4710 CrossRef CAS.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science, Sausalito, CA, 2006 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- D. B. Denney, D. Z. Denney, C. D. Hall and K. L. Marsi, J. Am. Chem. Soc., 1972, 94, 245–249 CrossRef CAS.
- K. A. Mesch and L. D. Quin, Tetrahedron Lett., 1980, 21, 4791–4794 CrossRef CAS.
- T. Kee, in Phosphorus-Carbon Heterocyclic Chemistry, ed. F. Mathey, Elsevier Science Ltd, Oxford, 2001, pp. 195–218 Search PubMed.
- K. D. Reichl and A. T. Radosevich, Chem. Commun., 2014, 50, 9302–9305 RSC.
- A. H. Cowley, R. A. Kemp, J. G. Lasch, N. C. Norman and C. A. Stewart, J. Am. Chem. Soc., 1983, 105, 7444–7445 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- F. Weinhold and C. R. Landis, Valency and bonding: a natural bond orbital donor-acceptor perspective, Cambridge University Press, Cambridge, UK; New York, 2005 Search PubMed.
- A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed.
- M. B. Geeson, A. R. Jupp, J. E. McGrady and J. M. Goicoechea, Chem. Commun., 2014, 50, 12281–12284 RSC.
- R. H. Sommerlade, S. Boulmaâz, J.-P. Wolf, J. Geier, H. Grützmacher, M. Scherer, H. Schönberg, D. Stein, P. Murer and S. Burkhardt, WO2005014605A1, 2005.
- V. Nesterov, L. Duan, G. Schnakenburg and R. Streubel, Eur. J. Inorg. Chem., 2011, 2011, 567–572 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1882890–1882892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05657a |
| This journal is © The Royal Society of Chemistry 2019 |