
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiacetyl as a “traceless” visible light photosensitizer in metal-free cross-dehydrogenative coupling reactions†
Chia-Yu
Huang‡
 ,
Jianbin
Li‡
,
Wenbo
Liu
and
Chao-Jun
Li
*
,
Jianbin
Li‡
,
Wenbo
Liu
and
Chao-Jun
Li
*
Department of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. W., Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 8th April 2019
Abstract
Minisci alkylation is of prime importance for its applicability in functionalizing diverse heteroarenes, which are core structures in many bioactive compounds. In alkyl radical generation processes, precious metal catalysts, high temperatures and excessive oxidants are generally involved, which lead to sustainability and safety concerns. Herein we report a new strategy using diacetyl (2,3-butanedione) as an abundant, visible light-sensitive and “traceless” hydrogen atom abstractor to achieve metal-free cross-dehydrogenative Minisci alkylation under mild conditions. Mechanistic studies supported hydrogen atom transfer (HAT) between an activated C(sp3)–H substrate and diacetyl. Moreover, with the assistance of di-tert-butyl peroxide (DTBP), the scope of the reaction could be extended to strong aliphatic C–H bonds via diacetyl-mediated energy transfer. The robustness of this strategy was demonstrated by functionalizing complex molecules such as quinine, fasudil, nicotine, menthol and alanine derivatives.
Introduction
Heteroarenes are ubiquitous skeletons for pharmaceutical and agricultural uses, as well as small molecule studies. Therefore, functionalization of these molecules especially during the late stage to create diversity is a long-lasting topic.1 Minisci alkylation is a powerful reaction to construct a C(sp2)–C(sp3) bond between electron-deficient heteroarenes and electron-rich alkyl radicals, and is complementary to Friedel–Crafts alkylation involving electron-rich arenes. Over the past few decades, many efforts have been devoted to the development of novel Minisci alkylations, predominantly focusing on alkyl radical generation in more efficient and greener ways. Although alkyl radicals could be generated through carbon–heteroatom bond cleavage of alkyl iodides,2 boronic acids3 and others,4 or carbon–carbon bond cleavage of aldehydes,5 acid derivatives,6 and oxime esters,7 pre-synthesis of alkyl radical precursors is required. Alternatively, direct alkyl radical generation via hydrogen atom abstraction from alkanes is the most desirable because alkyl structures are naturally abundant, and formal removal of H2 through cross-dehydrogenative coupling (CDC) between alkanes and arenes gives the greatest atom and step economy (Fig. 1).8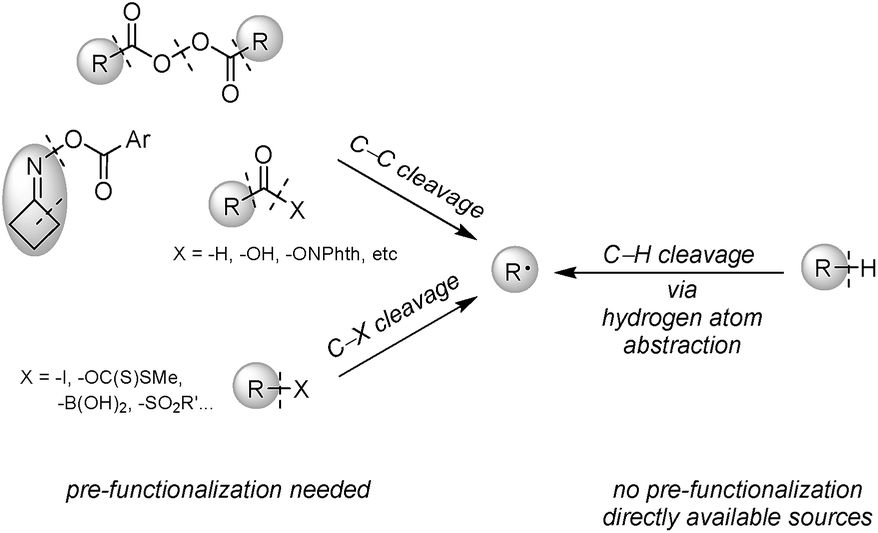 | ||
| Fig. 1 Alkyl radical generation through C–C, C–X and C–H bond cleavage. | ||
The key to cross-dehydrogenative alkylation is the activation of inert C(sp3)–H bonds, and oxidative C–H activation is a straightforward way to generate carbon radicals via hydrogen atom abstraction;9 thereby an efficient hydrogen atom abstractor is a requisite for the reaction design. Most of the strategies used oxygen-centered radicals, routinely generated from peroxides or persulfates, to perform hydrogen atom abstraction from alkanes. Traditional methods relied on thermo-cleavage of O–O bonds; however, demands of high reaction temperatures might deteriorate the reaction selectivity and functional group tolerance. Also, heating explosive peroxides could cause safety concerns and limit the reaction scale.10 To solve these issues, visible light-mediated CDC with an iridium photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 at room temperature was introduced by MacMillan.11 Alternatively, Ryu's group employed solar light to activate a decatungstate photocatalyst (TBADT) as a hydrogen atom abstractor, with persulfate salt being added to regenerate the catalyst.12 Other than oxy radical-based oxidants, hydrogen atom abstractors such as nitrogen-centered radical cations from N-hydroxysuccinimide (NHS) and Selectfluor,13 thiyl radicals from thiols,14 iodine/azide-centered radicals from phenyliodine bis(trifluoroacetate) (PIFA)/sodium azide,15 and chlorine radicals generated from dichloromethane (DCM),16 were also feasible under mild reaction conditions.
Although numerous facile synthetic strategies have been developed, they are far from being ideal. It would be more desirable to conduct a CDC reaction under milder conditions without using costly photocatalysts and special light sources, while avoiding concomitant production of excess waste salts and harmful byproducts. Herein we wish to report a new approach to cross-dehydrogenative Minisci-type alkylation enabled by visible light using triplet state ketones as the hydrogen atom abstractor.
Research design
Triplet state ketones and peroxides/persulfates display similar redox reactivities to a certain degree. For example, Baran has designed a series of sulfinate salts for C–H functionalization of heteroarenes mediated by peroxides.4b,c Later, our group reported light-induced C(sp2)–H trifluoromethylation with NaSO2CF3 enabled by excited acetone or diacetyl (2,3-butanedione) as transient single-electron oxidants (Fig. 2a).17 Inspired by persulfate-based bimolecular homolytic substitution (SH2) reactions of organoboranes,18 we demonstrated that alkyl/aryl trifluoroborates could be converted to alkyl/aryl radicals through SH2 by diacetyl under visible light irradiation (Fig. 2b).19 Based on these studies, we hypothesized that triplet state ketones could substitute peroxides/persulfates in other carbon-centered radical generation processes, such as hydrogen atom transfer. Indeed, ketones have been used in some hydrogen atom transfer processes;20 however, they were rarely applied in CDC reactions.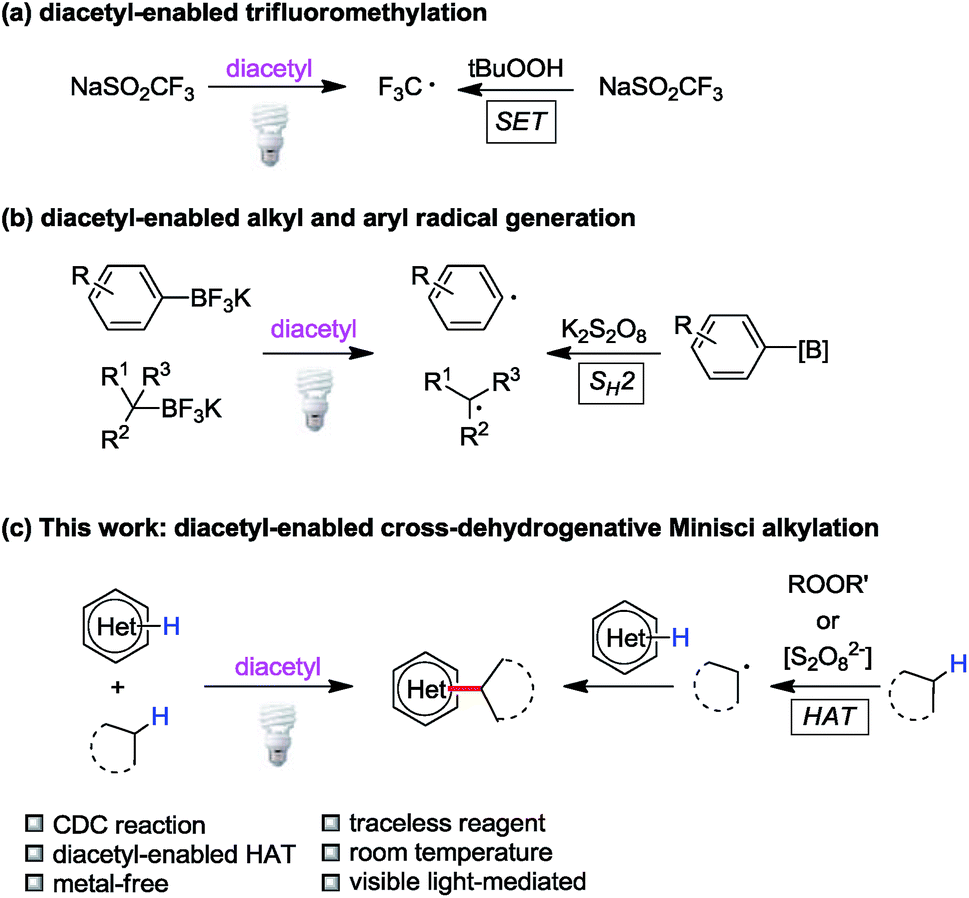 | ||
| Fig. 2 Diacetyl as a peroxide/persulfate surrogate. | ||
As a stoichiometric amount of the oxidant is required for the dehydrogenation process, the removal of excessive oxidant and its byproducts needs tedious and costly laboratory processes. Diacetyl is the smallest visible light sensitive ketone (380–460 nm), which can be a traceless photosensitizer due to its high volatility and solubility in water (200 g L−1 at 20 °C). The reduced product of diacetyl, acetoin, is miscible with water (1 kg L−1 at 20 °C). Considering this, byproducts derived from diacetyl could be removed by aqueous workup and reduced pressure. Furthermore, diacetyl is readily available and non-toxic. These advantages make diacetyl an ideal candidate for the cross-dehydrogenative system.
Combining the literature reports and our previous experience, we hypothesized that diacetyl could serve as a hydrogen atom abstractor in cross-dehydrogenative Minisci alkylation due to its diradical character in the excited state (Fig. 2c). We envisioned that the success of this reaction would be a milestone in the exploration of ketone-enabled Minisci reactions and advance the development of greener cross-coupling reactions.
Results and discussion
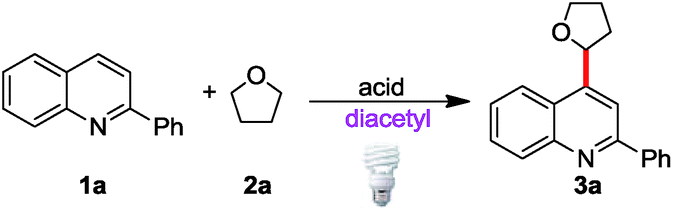
In the preliminary studies, 2-phenylquinoline (1a) was selected as the radical acceptor due to its high reactivity. At the beginning, a few representative compounds (tetrahydrofuran (THF), cyclohexane and toluene) were selected as the alkyl radical sources to test our hypothesis. To our delight, all of the corresponding alkylated products were detected by GC-MS under visible light irradiation using a 40 W compact fluorescent lamp (CFL) as the light source (see ESI Table S1†). Then we chose THF (2a) as the model substrate to perform our optimization as it produced the highest yield along with its ease of use in spectral analysis. As given in Table 1, trifluoroacetic acid (TFA) was essential for the transformation while acetic (AcOH) or triflic acids (TfOH) were not effective proton sources (Table 1, entries 2 and 3). Different loadings of THF and diacetyl were investigated; the combination of 0.2 mL of THF and 0.2 mL of diacetyl gave the highest yield (entry 4). The reaction was completed by prolonging the reaction time to 36 h (entry 7). In this reaction, a blue LED gave inferior yield (entry 9). Finally, the control experiments showed that the light, diacetyl and an inert atmosphere are all important for the reaction (entries 8, 10 and 11). As expected, most of the byproducts of this reaction could be simply removed by aqueous workup and reduced pressure without chromatographic purification (see ESI Fig. S2†).|
|
|||||
|---|---|---|---|---|---|
| Entrya | 2a (mL) | Diacetyl (mL) | Acid | 1a (%) | 3a (%) |
| a All reactions were conducted with 0.1 mmol of 1a and 2a, and 2 equiv of acid and diacetyl, and a 40 W CFL at room temperature under argon for 20 h. b The yield was determined by 1H NMR using mesitylene as the internal standard. Isolated yield in parentheses. c The reaction was run for 36 h. d In air. e The reaction was irradiated using a blue LED. f The reaction was heated to 70 °C in the dark. | |||||
| 1 | 0.4 | 0.2 | TFA | 26 | 71 |
| 2 | 0.4 | 0.2 | AcOH | >99 | — |
| 3 | 0.4 | 0.2 | TfOH | >99 | — |
| 4 | 0.2 | 0.2 | TFA | 10 | 88 |
| 5 | 0.2 | 0.1 | TFA | 43 | 55 |
| 6 | 0.1 | 0.1 | TFA | 40 | 53 |
| 7c | 0.2 | 0.2 | TFA | — | 90 (86) |
| 8d | 0.2 | 0.2 | TFA | 20 | 64 |
| 9c,e | 0.2 | 0.2 | TFA | — | 52 |
| 10 | 0.4 | — | TFA | >99 | — |
| 11f | 0.2 | 0.2 | TFA | >99 | — |
With the optimized conditions in hand, we tested the generality of this method (Scheme 1). The reaction tolerated diverse functional groups such as halides (3b–3d, 3l), cyano (3e), acetyl (3g), methoxy (3i and 3l) and alkyl groups (3k–3m). Most of the substrates provided moderate to excellent yields of the corresponding products (50–83%), and no significant reactivity bias between C2- and C4-alkylations (3a and 3n, and 3m and 3r) was observed. Substrates with a steric effect (3o) or a strong electron-donating group (3i), which might obstruct radical addition, could still provide moderate yields (34–42%). It is notable that 3-substituted quinoline selectively gave the C4-alkylated product 3o, and C4-alkylated quinoline 3m could be obtained as the major product when unsubstituted quinoline was used. This showed that the C4 position is the more active position in our method. Other heterocycles such as isoquinolines (3s and 3t), pyridines (3u and 3v) and pyrazine (3w) could be functionalized smoothly by this protocol, although the reaction failed with heterocycles such as benzothiazole, benzoxazole and quinoxaline and gave poor yields (see ESI Scheme S1†). To our delight, pharmaceutically valuable molecules such as quinine (3ad) and fasudil (3ae), or complex menthol and L-alanine derivatives (3af and 3ag) could be functionalized with this method. To show the potential applications in industrial and pharmaceutical uses, we demonstrated a gram-scale reaction with 2-phenylquinoline (1a), in which 89% yield of the product (3a) could be obtained (for details see the ESI†).
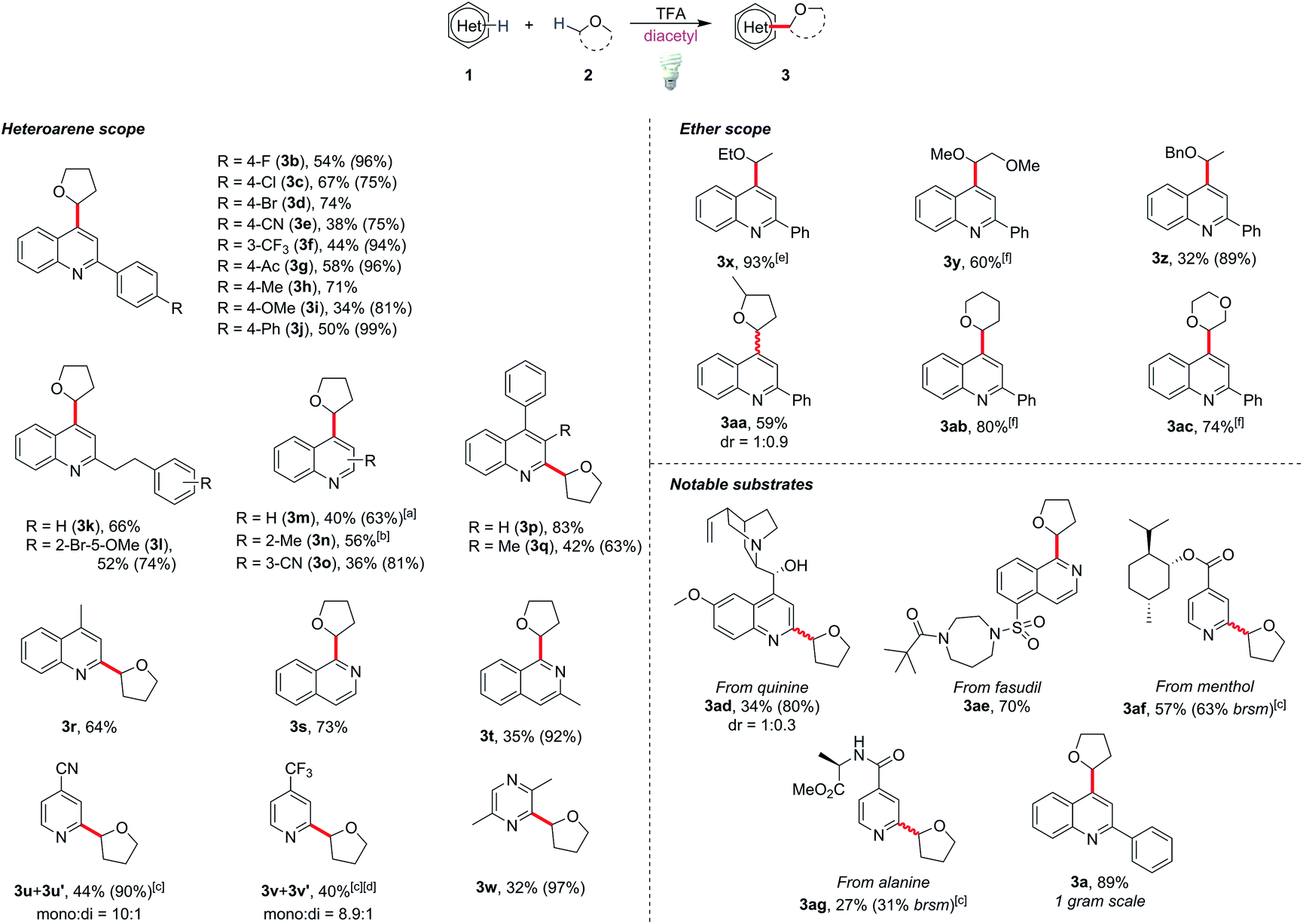 | ||
| Scheme 1 Scope of Minisci alkylation with ethers. All reactions were conducted under optimized conditions for 48 h unless otherwise specified, and the yields were isolated ones. Yields in parentheses were based on recovered starting materials. [a] 0.1 mL of diacetyl and 0.2 mL of MeCN was added. [b] The reaction was run for 36 h. [c] 20 equiv of TFA was used. [d] Yield is from 1H NMR using mesitylene as the internal standard. [e] 0.3 mL of ether was used. [f] 2 equiv of DTBP was added to the reaction, and the reactions were run for 20 h. | ||
To consolidate our hypothesis that diacetyl is a hydrogen atom abstractor, mechanistic studies were conducted. When 2 equiv of radical inhibitor such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT) was added to the model reaction, the reaction was significantly suppressed, implying that radical formation is involved (Scheme 2a). A prominent primary isotope effect was observed in the KIE experiments (kH/kD = 4), indicating that the α-C–H homocleavage of THF is the rate-determining step in this reaction.21 Furthermore, hydrogen atom abstraction is quite selective as no proton/deuterium exchange was observed from the reaction with THF-d8 (Scheme 2b and ESI Fig. S4†). When cyclopropyl phenyl ethylene (6) was tested, we were able to isolate an appreciable amount of the alkylated adduct 7, confirming the formation of the THF radical (Scheme 2c). To rule out the acyl radical-involved HAT pathway,22 we replaced diacetyl with benzil (8) and 75% alkylated quinoline 3a was obtained from the model substrates 1a and 2a, while only a trace amount of benzaldehyde (11) was observed from the crude NMR spectra. In contrast, vicinal diol 9 and benzoin (10) were isolated from the reaction as byproducts, indicating direct HAT from THF to the excited benzil (Scheme 2d). Although we could not completely exclude the possibility that an acyl radical took part in HAT, these results strongly suggested that diketones do play an indispensable role in hydrogen atom abstraction to generate alkyl radicals.
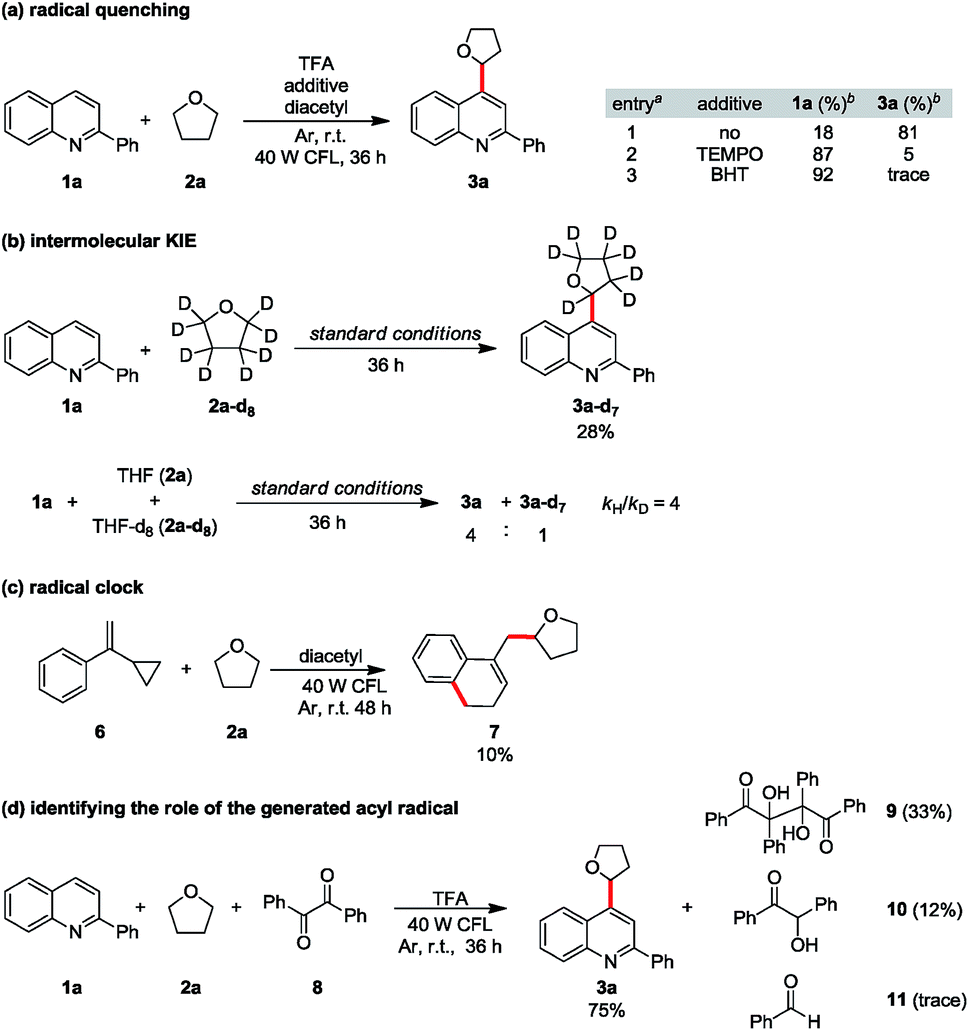 | ||
| Scheme 2 Mechanistic studies: (a) radical quenching, (b) intermolecular KIE studies, (c) radical clock, and (d) identifying the role of the generated acyl radical. | ||
For the coupling of 2-phenylquinoline with other ether derivatives (Scheme 1), diethyl ether gave an excellent yield of the alkylated product 3x. Reaction with 2-methyl tetrahydrofuran also gave moderate yield of the desired product 3aa where the reaction occurred selectively on the less hindered α-carbon. Surprisingly, some ethers such as 1,2-dimethoxyethane (DME), tetrahydropyran (THP) and dioxane yielded only 10 to 20% of the corresponding products. According to the literature,23 the bond dissociation energy of these ethers is slightly higher, roughly 2 to 4 kcal mol−1, than that of THF. We postulated that unlike traditionally persistent oxyradicals, the transient diketone diradical is a milder and more selective oxidant toward C–H species, which resulted in a low efficiency in oxidizing stronger C–H bonds. To overcome this obstacle, a stronger oxidant is necessary. It is known that triplet state photosensitizers can serve as energy transfer agents to facilitate alkene isomerization,24 cycloaddition,25 metallic reductive elimination26 and homolytic bond cleavage.27 Theoretically, the triplet state energy of diacetyl, about 55 to 57 kcal mol−1,28 would be enough to induce the cleavage of weak peroxide O–O bonds (BDE is generally lower than 50 kcal mol−129). Considering that di-tert-butyl peroxide (DTBP) has a low O–O bond-dissociation energy (38 kcal mol−1) and the byproduct tert-butanol is easily removable, 2 equiv of DTBP was chosen to be added to the reaction system. The reactions proceeded smoothly at room temperature and reached completion after 20 h to give good yields of the products 3y, 3ab and 3ac.
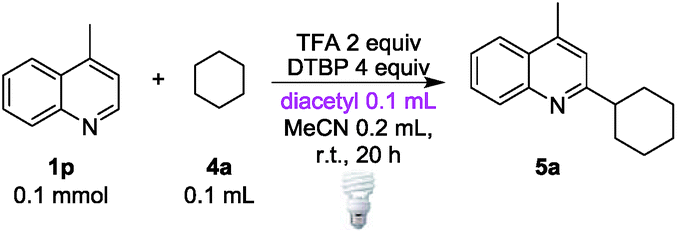
When diacetyl was used to cleave the strong C(sp3)–H bond of cyclohexane (4a), it was not surprising that only a trace amount of the cyclohexyl adduct 5a was observed (Table 2, entry 2). Further optimizations of the protocol were performed by adding DTBP as the oxidant, and 84% of 5a could be obtained with MeCN as the cosolvent (for detailed optimizations see ESI Table S2†). In order to eliminate the possibility of thermal cleavage of peroxides at room temperature, a reaction was conducted at 4 °C, and the same yield could be obtained by prolonging the reaction time to 24 h (entry 3). In contrast, no reaction took place in the absence of light, even when the reaction was heated to 70 °C (entry 4). These control experiments supported energy transfer-induced peroxide cleavage rather than thermal-induced peroxide cleavage. The efficiency of this energy transfer at low temperature also provided a synthetic option for thermosensitive compounds. Further analysis of the crude components from the reaction also supported the energy transfer rather than radical substitution process between diacetyl and DTBP (see ESI Schemes S5 and S6†).
The scope of this modified protocol was then examined. Various heterocycles including phenanthridine (5e), isoquinoline (5f), quinazoline (5g), pyrimidine (5h) and benzothiazole (5l) could be functionalized under mild conditions and gave moderate yields (40–50%) of the corresponding products (Scheme 3). Valuable substrates, for example, quinine (5s), nicotine (5t), and menthol or L-alanine derivatives (5u and 5v), could be functionalized albeit with lower yields (27–37%). The method is also useful to modify heterocyclic ligands such as 4,4′-di-tert-butyl-2,2′-dipyridyl, yielding 34% dialkylated product 5w. Diverse alkylated products could be obtained by replacing cyclohexane with other alkyl substrates such as cyclopentane and cyclooctane in excellent yields (89–97%) of heterocycle derivatives (5m and 5n). Reactions with bridged hydrocarbon norbornane gave 84% alkylated product 5p, while those with cyclopentanone, 7-oxabicyclo[2.2.1]heptane and toluene gave only poor to moderate yields of the corresponding products 5o, 5q and 5r.
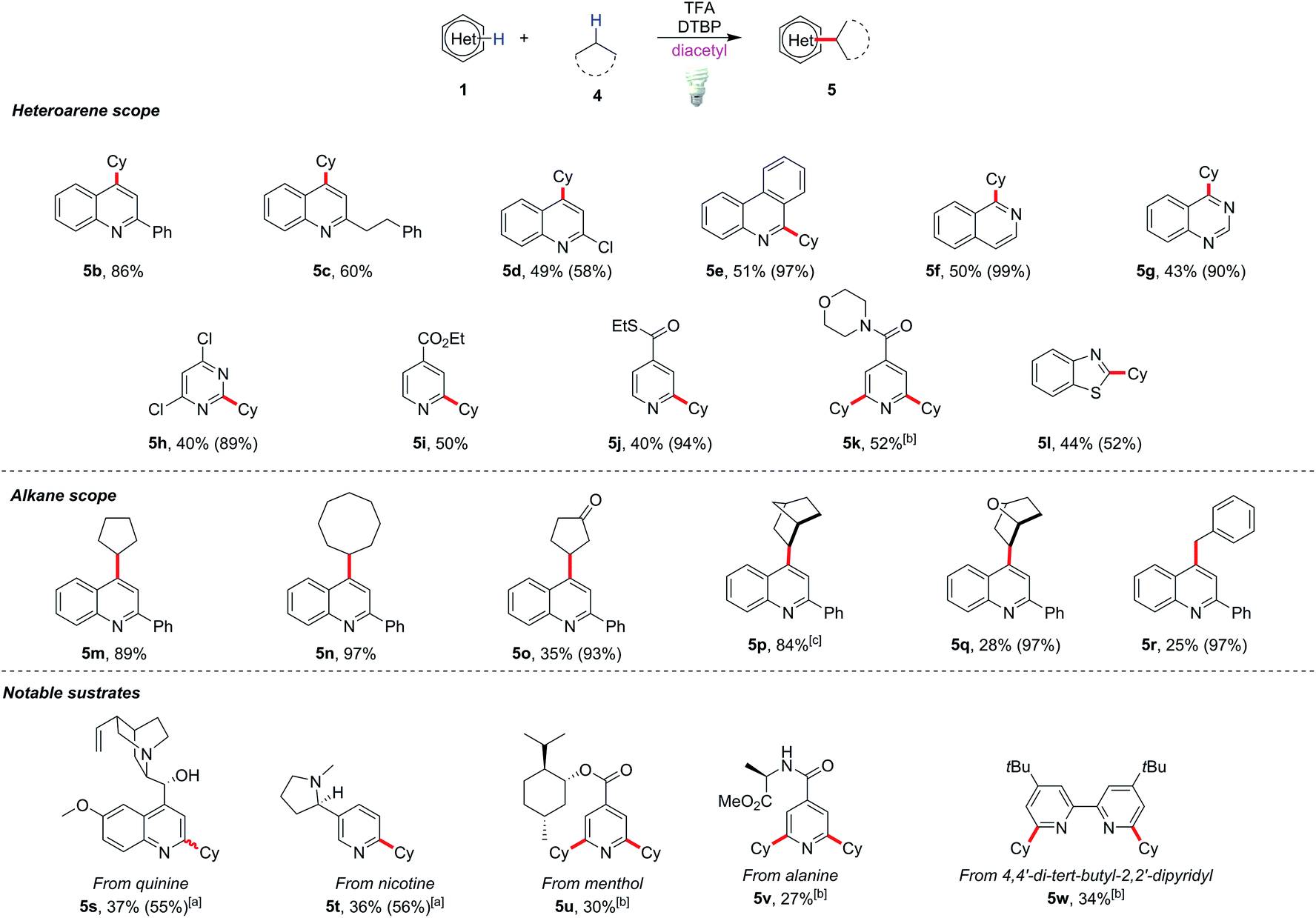 | ||
| Scheme 3 Scope of Minisci alkylation with unactivated C(sp3)−H species. All reactions were conducted under optimized conditions unless otherwise specified, and the yields were isolated ones. Yields in parentheses were based on recovered starting materials. [a] 3 equiv of TFA was used. [b] The reactions were conducted with 0.2 mL of alkane, 8 equiv of DTBP, 0.2 mL of diacetyl, and 0.4 mL of MeCN. [c] 5 equiv of alkane was used. | ||
Based on previous literature and the mechanistic investigations presented above, a plausible mechanism for the diacetyl-enabled CDC reaction was proposed as follows: the excited ketone 12 acts as a hydrogen atom abstractor to generate the alkyl radical 13 to couple with protonated heteroarene 1-H+ (Scheme 4, right). The protonated ketyl radical 15 might undergo tautomerization to form an enol radical 16 and perform a second hydrogen atom abstraction to rearomatize the alkyl adduct 14, which gives the product 3-H+. When a peroxide (DTBP) was added to enhance the HAT, the role of the triplet ketone is switched to that as the energy transfer agent (Scheme 4, left). By energy transfer from the excited ketone 12 to a peroxide 18, the excited peroxide 19 cleaves into 2 equiv of the oxy radical 20 to abstract a hydrogen atom from the alkane 2/4 and the alkyl adduct 14, respectively, to give a protonated heteroarene 3-H+/5-H+ as the product.
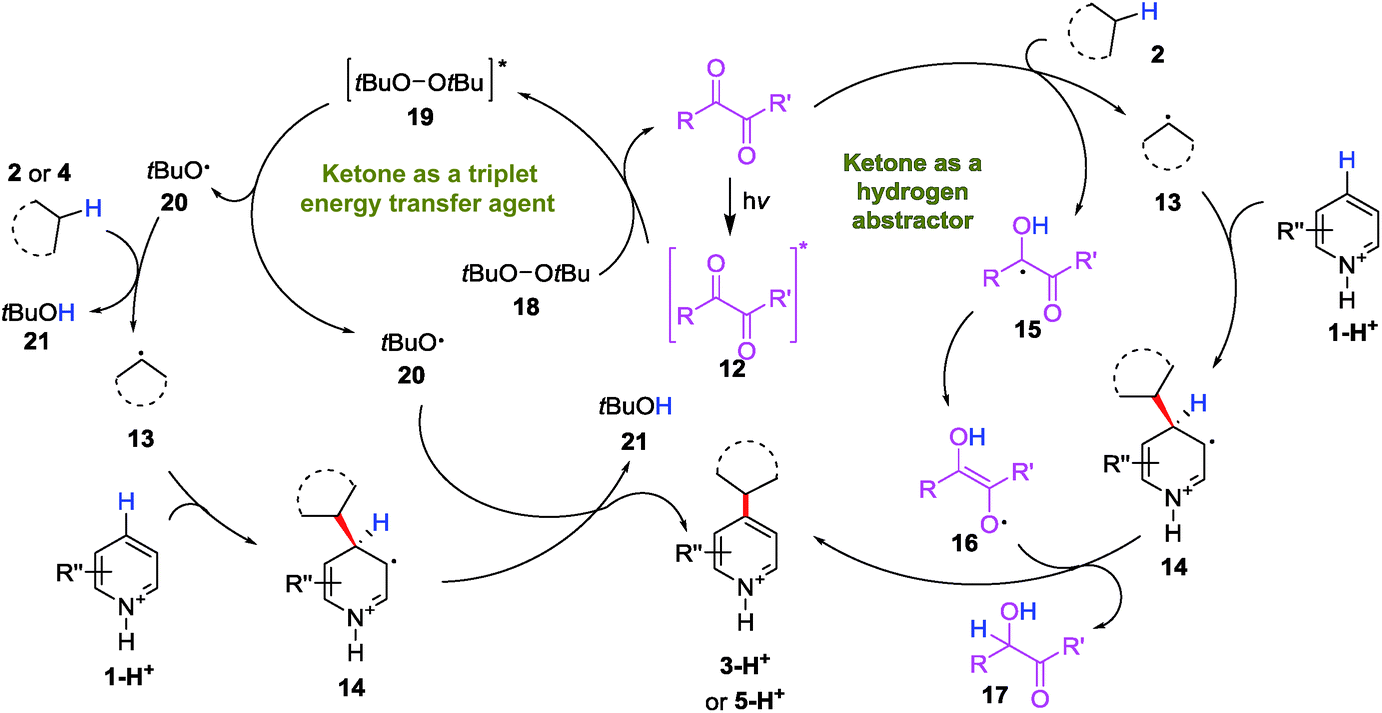 | ||
| Scheme 4 Proposed ketone-enabled CDC reaction mechanism. | ||
Conclusions
In summary, the first ketone-enabled cross-dehydrogenative Minisci alkylation using diacetyl as a traceless and sustainable photosensitive reagent has been developed. This approach utilized triplet diacetyl as either a hydrogen atom abstractor or an energy transfer agent for the coupling of heterocycles and alkanes under mild conditions. This strategy allowed functionalization of a wide range of substrates bearing copious functional groups. Control experiments and mechanistic studies suggest the involvement of a radical process. Further studies will allow us to understand the hydrogen atom abstraction ability of diacetyl and other potential traceless diketone photosensitizers.Experimental section
Detailed experimental procedures are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.-J. L.), the Canada Foundation for Innovation, the FQRNT Center in Green Chemistry and Catalysis, the Natural Sciences and Engineering Research Council of Canada, and McGill University for supporting our research. C.-Y. H. and J. L. are grateful for the discussion with Z. Q., Dr L. L., C.-C. L., and H. K. (McGill University).Notes and references
- (a) J. Wu, Y. Liu, C. Lu and Q. Shen, Chem. Sci., 2016, 7, 3757–3762 RSC; (b) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS PubMed.
- F. Fontana, F. Minisci and E. Vismara, Tetrahedron Lett., 1988, 29, 1975–1978 CrossRef CAS.
- (a) G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC; (b) L. Zhang and Z.-Q. Liu, Org. Lett., 2017, 19, 6594–6597 CrossRef CAS.
- (a) F. Coppa, F. Fontana, F. Minisci, G. Pianese, P. Tortoreto and L. Zhao, Tetrahedron Lett., 1992, 5, 687–690 CrossRef; (b) Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 14411–14415 CrossRef CAS PubMed; (c) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed; (d) P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef CAS PubMed.
- R.-J. Tang, L. Kang and L. Yang, Adv. Synth. Catal., 2015, 357, 2055–2060 CrossRef CAS.
- (a) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS; (b) R. Xia, M.-S. Xie, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2014, 16, 444–447 CrossRef CAS PubMed; (c) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS; (d) W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem.–Eur. J., 2017, 23, 2537–2541 CrossRef CAS PubMed.
- (a) B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 12727–12731 CrossRef CAS PubMed; (b) X.-Y. Yu, J.-R. Chen, P.-Z. Wang, M.-N. Yang, D. Liang and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738–743 CrossRef CAS PubMed; (c) L. Yang, P. Gao, X.-H. Duan, Y.-R. Gu and L.-N. Guo, Org. Lett., 2018, 20, 1034–1037 CrossRef CAS PubMed.
- (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (b) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed; (c) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- For selected oxidative C–H activations, see (a) D. Liu, C. Liu, H. Li and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 4453–4456 CrossRef CAS; (b) D. Liu, C. Liu, H. Li and A. Lei, Chem. Commun., 2014, 50, 3623–3626 RSC; (c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed; (d) L. Zhang, H. Yi, J. Wang and A. Lei, J. Org. Chem., 2017, 82, 10704–10709 CrossRef CAS PubMed; (e) X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS; Y. Zhang and C.-J. Li, Eur. J. Org. Chem., 2007, 2007, 4654–4657 Search PubMed; G. Deng, L. Zhao and C.-J. Li, Angew. Chem., Int. Ed., 2008, 47, 6278–6282 Search PubMed; G. Deng, K. Ueda, S. Yanagisawa, K. Itami and C.-J. Li, Chem.–Eur. J., 2009, 15, 333–337 Search PubMed; X. Guo and C.-J. Li, Org. Lett., 2011, 13, 4977–4979 Search PubMed.
- (a) C. Giordano, F. Minisci, E. Vismara and S. Levi, J. Org. Chem., 1986, 51, 536–537 CrossRef CAS; (b) G. Deng and C.-J. Li, Org. Lett., 2009, 11, 1171–1174 CrossRef CAS PubMed; (c) Z. Xie, Y. Cai, H. Hu, C. Lin, J. Jiang, Z. Chen, L. Wang and Y. Pan, Org. Lett., 2013, 15, 4600–4603 CrossRef CAS PubMed; (d) X. Li, H.-Y. Wang and Z.-J. Shi, New J. Chem., 2013, 37, 1704–1706 RSC; (e) N. Okugawa, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2015, 2015, 4973–4981 CrossRef CAS; (f) A. Correa, B. Fiser and E. Gómez-Bengoa, Chem. Commun., 2015, 51, 13365–13368 RSC; (g) B. A. Mir, A. Banerjee, S. K. Santra, S. Rajamanickam and B. K. Patel, Adv. Synth. Catal., 2016, 358, 3471–3476 CrossRef CAS; (h) J. Yuan, J. Fu, J. Yin, Z. Dong, Y. Xiao, P. Mao and L. Qu, Org. Chem. Front., 2018, 5, 2820–2828 RSC.
- (a) J. Jin and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS ; for another iridium photocatalyst-based CDC, see ; (b) J. Dong, Q. Xia, X. Lv, C. Yan, H. Song, Y. Liu and Q. Wang, Org. Lett., 2018, 20, 5661–5665 CrossRef CAS PubMed.
- M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335–2338 RSC.
- (a) S. Liu, A. Liu, Y. Zhang and W. Wang, Chem. Sci., 2017, 8, 4044–4050 RSC . During the preparation of our manuscript, a related study was published:; (b) L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019 DOI:10.1038/s41467-019-08413-9.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS.
- A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
- W. Liu, X. Yang, Z.-Z. Zhou and C.-J. Li, Chem, 2017, 2, 688–702 CAS.
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed.
- (a) I. B. Sieple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef PubMed; (b) J. W. Lockner, D. D. Dixon, R. Risgaard and P. S. Baran, Org. Lett., 2011, 13, 5628–5631 CrossRef CAS PubMed.
- (a) P. J. Krusic and J. K. Kochi, J. Am. Chem. Soc., 1969, 91, 3942–3944 CrossRef CAS; (b) A. G. Davies and B. P. Roberts, Nature, 1971, 229, 221–223 CrossRef CAS; (c) A. G. Davies and B. P. Roberts, Acc. Chem. Res., 1972, 5, 387–392 CrossRef CAS; (d) C. Carra and J. C. Scaiano, Eur. J. Org. Chem., 2008, 2008, 4454–4459 CrossRef; (e) W. Liu, P. Liu, L. Lv and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 13499–13503 CrossRef CAS.
- (a) C. Walling and M. J. Gibian, J. Am. Chem. Soc., 1965, 87, 3361–3364 CrossRef CAS; (b) H. A. J. Carless and S. Mwesigye-Kibende, J. Chem. Soc. Chem. Commun., 1987, 1673–1674 RSC; (c) K.-i. Wada, M. Yamamoto and J.-i. Setsune, Tetrahedron Lett., 1999, 40, 2773–2776 CrossRef CAS; (d) A. M. Cardarelli, M. Fagnoni, M. Mella and A. Albini, J. Org. Chem., 2001, 66, 7320–7327 CrossRef CAS; (e) S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928–5931 CrossRef CAS PubMed; (f) J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed; (g) T. Hoshikawa, S. Kamijo and M. Inoue, Org. Biomol. Chem., 2013, 11, 164–169 RSC; (h) A. Lipp, G. Lahm and T. Opatz, J. Org. Chem., 2016, 81, 4890–4897 CrossRef CAS PubMed; (i) S. Kamijo, G. Takao, K. Kamijo, T. Tsuno, K. Ishiguro and T. Murafuji, Org. Lett., 2016, 18, 4912–4915 CrossRef CAS.
- Y. Zhu and Y. Wei, Chem. Sci., 2014, 5, 2379–2382 RSC.
- L. Zhang, X. Si, Y. Yang, M. Zimmer, S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 1823–1827 CrossRef CAS.
- (a) E. T. Denisov, I. B. Afanas'ev, Oxidation and Antioxidants in Organic Chemistry and Biology, Taylor & Francis Group, 2005, pp. 281−298 CrossRef; (b) H. Matsubara, S. Suzuki and S. Hirano, Org. Biomol. Chem., 2015, 13, 4686–4692 RSC.
- (a) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275–5278 CrossRef CAS PubMed; (b) J. B. Metternich and R. Gilmour, Synlett, 2016, 27, 2541–2552 CrossRef CAS.
- (a) P. Sharma, B. Lygo, W. Lewis and J. E. Moses, J. Am. Chem. Soc., 2009, 131, 5966–5972 CrossRef CAS PubMed; (b) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed; (c) J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807–9810 CrossRef CAS PubMed; (d) N. Münster, N. A. Parker, L. van Dijk, R. S. Paton and M. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 9468–9472 CrossRef PubMed.
- (a) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed; (b) E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS; (c) Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS.
- (a) E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793–797 CrossRef CAS PubMed; (b) E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed; (c) M. Teders, C. Henkel, L. Anhäuser, F. Strieth-Kalthoff, A. Gómez-Suárez, R. Kleinmans, A. Kahnt, A. Rentmeister, D. Guldi and F. Glorius, Nat. Chem., 2018, 10, 981–988 CrossRef CAS PubMed.
- (a) M. W. Schmidt and E. K. C. Lee, J. Am. Chem. Soc., 1970, 92, 3579–3586 CrossRef CAS; (b) A. W. Jackson and A. J. Yarwood, Can. J. Chem., 1972, 50, 1331–1337 CrossRef CAS.
- (a) S. L. Khursan, D. A. Mikhailov, A. A. Gusmanov and I. M. Borisov, Russ. J. Phys. Chem. A, 2001, 75, 724–732 Search PubMed; (b) F. Agapito, B. J. C. Cabral and J. A. M. Simões, J. Mol. Struct. THEOCHEM, 2005, 729, 223–227 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05631e |
| ‡ C.-Y. Huang and J. Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |