
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective [1,3] O-to-C rearrangement: dearomatization of alkyl 2-allyloxy/benzyloxy-1/3-naphthoates catalyzed by a chiral π–Cu(II) complex†
Lu
Yao
 and
Kazuaki
Ishihara
*
and
Kazuaki
Ishihara
*
Graduate School of Engineering, Nagoya University, B2-3(611) Furo-cho, Chikusa, Nagoya 464-8603, Japan. E-mail: ishihara@cc.nagoya-u.ac.jp
First published on 10th January 2019
Abstract
An unprecedented catalytic asymmetric [1,3] O-to-C rearrangement of alkyl 2-allyloxy/benzyloxy-1/3-naphthoates was realized under the catalysis of a chiral π–Cu(II) complex (1–10 mol%). This dearomatization strategy provides facile access to highly functionalized β-naphthalenone derivatives bearing an all-carbon quaternary stereogenic center in high yield with excellent enantioselectivity. The π–cation interaction between the aromatic substituent of the ligand and the Cu(II) center was proved by X-ray diffraction analysis and shown to be crucial for enantioselective control. Further preliminary mechanistic studies suggest that this intramolecular reaction proceeds through a contact ion pair intermediate.
1. Introduction
O-to-C rearrangements are among the most powerful C–C bond forming strategies in organic synthesis. One such well-known reaction is Claisen rearrangement,1 which has been shown to transfer stereochemistry from a cleaved C–O bond to a formed C–C bond via a concerted [3,3] sigmatropic pathway. In contrast, non-concerted [m,n] rearrangement such as [1,3] rearrangement has gained less attention. This lack of research is probably due to high activation barriers, since suprafacial [1,3]-sigmatropic rearrangement is an orbital symmetry-forbidden process, and typically requires harsh reaction conditions such as high temperature to proceed through radical intermediate.2 To overcome this limitation, tremendous effort has been devoted to the activation of a substrate by various catalytic systems (transition metal catalysis,3 nucleophilic catalysis4 and Lewis acid catalysis5). Among these systems, Lewis acid catalysis has been most intensively studied. Mechanistic studies have shown that, in general, the Lewis acid-promoted reaction proceeds with heterolytic cleavage of the O–C bond of the vinyl ether, in which the ion pair intermediates (the carbocation and the enolate counter anion) would be generated, and the recombination of the resulting intermediates would lead to the formation of the desired product. This mechanism suggests that an appropriate, well-designed substrate that can generate a stable ion pair, along with a carefully selected Lewis acid, might lead to successful [1,3] O-to-C rearrangement. Although significant effort has been made in this area, there is still room for the improvement of the current method. To the best of our knowledge, catalytic enantioselective [1,3] rearrangement has remained elusive to date.4Our group recently developed a group of small-molecule Cu(II) catalysts based on intramolecular π–cation interactions and has demonstrated their efficiency in several enantioselective cycloaddition reactions of α,β-unsaturated carboxamides.6 As part of our ongoing interest in extending the application of these chiral π–Cu(II) catalysts, we started to consider its capability in the catalysis of [1,3] rearrangement reactions. In 2002 and 2003, Gansäuer demonstrated that, in the presence of catalytic amount of Cu(OTf)2, the vinyl ether could lead to [1,3] and [3,3] products in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 1a).7 In the year 2005, Rovis reported the regioselective [1,3] rearrangement of allyl vinyl ether catalyzed by aluminum and copper Lewis acids.8 They mentioned that the rearrangement of trisubstituted alkenes would preferentially produce the [1,3] adduct due to steric congestion for normal [3,3] rearrangement. Thus, as shown in Scheme 1b, the [1,3] product could be obtained in moderate to good yields with up to 95:5 regioselectivity. Inspired by these pioneering studies, we envisioned that catalytic enantioselective [1,3] O-to-C rearrangement might be realized with suitable functionalized substrates by using our catalytic system. Here we report the first enantioselective [1,3] O-to-C rearrangement reaction of alkyl 2-allyloxy/benzyloxy-1-naphthoates 1 and methyl 1-substituted 2-allyloxy-3-naphthoates 3 catalyzed by the chiral π–Cu(II) complex (Scheme 1c).
:1 ratio (Scheme 1a).7 In the year 2005, Rovis reported the regioselective [1,3] rearrangement of allyl vinyl ether catalyzed by aluminum and copper Lewis acids.8 They mentioned that the rearrangement of trisubstituted alkenes would preferentially produce the [1,3] adduct due to steric congestion for normal [3,3] rearrangement. Thus, as shown in Scheme 1b, the [1,3] product could be obtained in moderate to good yields with up to 95:5 regioselectivity. Inspired by these pioneering studies, we envisioned that catalytic enantioselective [1,3] O-to-C rearrangement might be realized with suitable functionalized substrates by using our catalytic system. Here we report the first enantioselective [1,3] O-to-C rearrangement reaction of alkyl 2-allyloxy/benzyloxy-1-naphthoates 1 and methyl 1-substituted 2-allyloxy-3-naphthoates 3 catalyzed by the chiral π–Cu(II) complex (Scheme 1c).
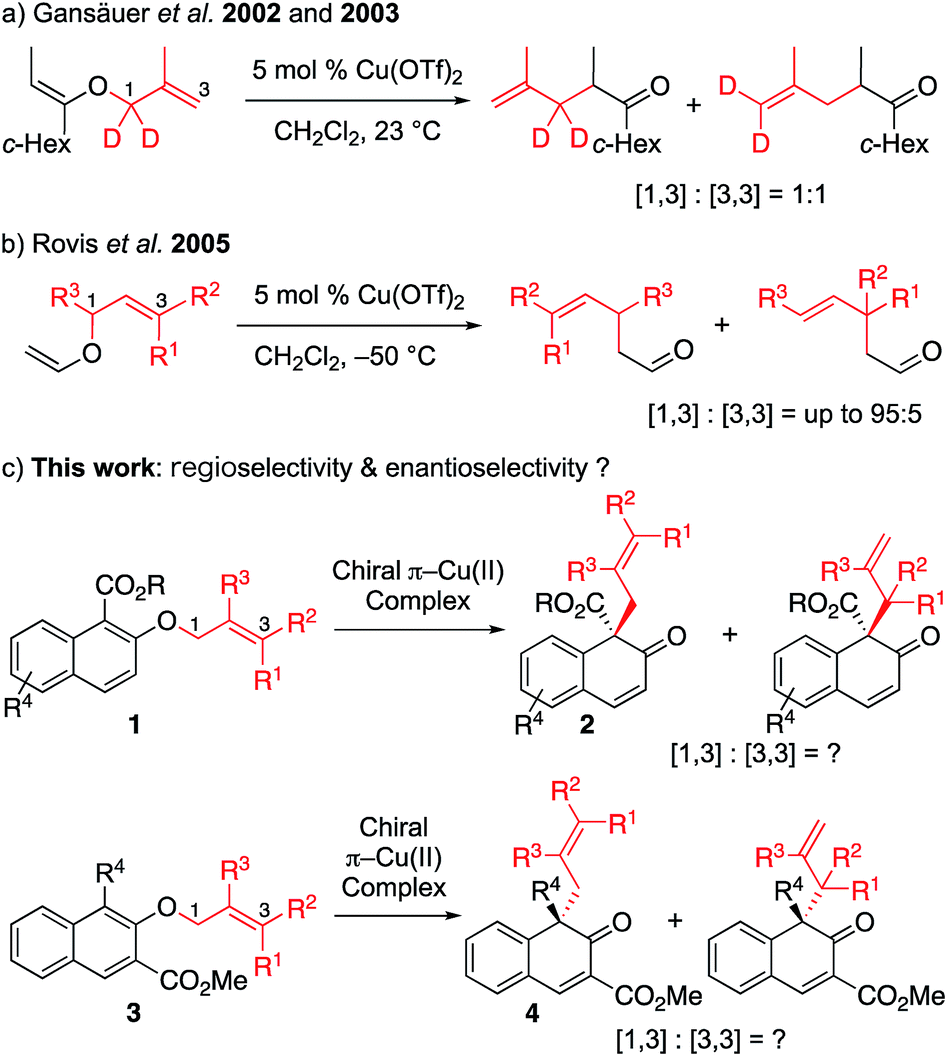 | ||
| Scheme 1 Cu(II)-catalyzed [1,3] rearrangement reactions of allyl vinyl ethers or allyl naphthyl ether: (a) Gansäuer et al. 2002 and 2003. (b) Rovis et al. 2005. (c) This work: regioselectivity & enantioselectivity? | ||
2. Results and discussion
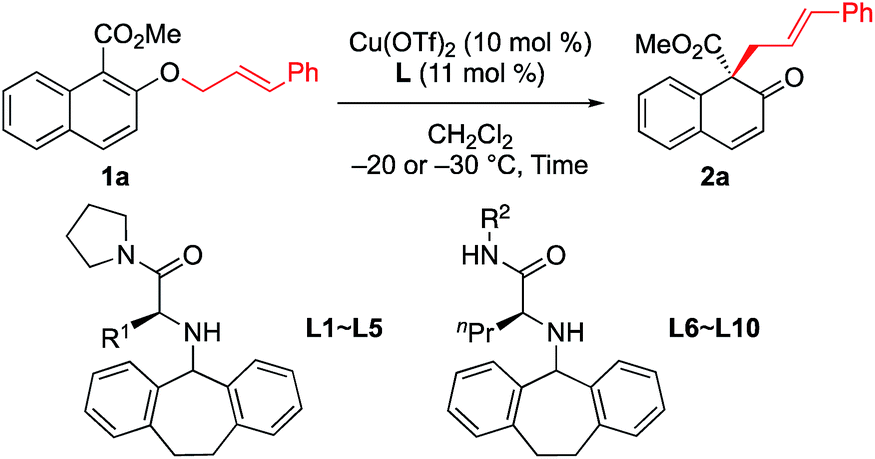
First, we sought to identify appropriate substrates. Considering that naphthols are readily available starting materials that can be used to access functionalized chiral naphthalenones, we decided to investigate the feasibility of naphthol-derived vinyl ethers as potential substrates.9 Recently, You10 and Zhong11 reported the catalytic asymmetric allylic dearomatization (CADA) of naphthol derivatives.12 We anticipate that the same product would be generated if allyl naphthyl ethers undergo [1,3] O-to-C rearrangement under catalysis by the Cu(II) complex (Scheme 1c). If this works, our strategy could avoid the need to use precious metals such as palladium or iridium.After a brief screening of substituents on both naphthalene rings and the allylic chain, we were very pleased to find that 1a could undergo [1,3] O-to-C rearrangement smoothly under catalysis of Cu(OTf)2, affording the dearomatization product 2a with an all-carbon quaternary stereogenic center in low yield. However, no [3,3] rearrangement product was observed during the process.13 This result encouraged us to perform further ligand screening.14 Initial optimization showed that the N-dibenzosuberyl substituted group is crucial for enantioselective control, which is consistent with our original intention to introduce π–cation interaction into the catalyst design. Hence, we decided to keep this moiety intact while evaluating other substituted groups on the chiral ligand (Table 1). The R1 group that originated from l-amino acids had noticeable effects on the enantioselectivity; the results of L1 to L5 indicate that the steric hindrance of this substituent should neither be too large nor too small (entries 1–5). L-Norvaline-derived ligand L4 (R1 = propyl) gave the desired product 2a in 85% NMR yield with 89% ee (entry 4). The amide moiety of the ligand was also examined. The structure of the amide moiety strongly influenced the reaction rate. For example, tertiary amide- and bulky secondary amide-substituted ligands, such as L4, L9 and L10, were not so effective for promoting the reaction (entries 4, 9 and 10). In comparison, the reaction was complete within only 6 hours when less-bulky secondary amide-substituted ligands, such as L6–L8, were used at – 20 °C (entries 6–8). Finally, we found that the desired [1,3] O-to-C rearrangement product 2a could be obtained in 90% isolated yield with excellent enantioselectivity (91% ee) at −30 °C using 10 mol% L-norvaline-derived chiral π–Cu(II) complex as a catalyst in dichloromethane15 (entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Time (h) | 2a | ||
| Conv.b (%) | Yieldb (%) | Eec (%) | |||
| a Unless otherwise noted, all reactions were carried out with 0.15 mmol of 1a in dichloromethane (0.75 mL) at −20 °C. b Yield of 2a based on 1H NMR. Small amount of methyl 3-cinnamyl-2-hydroxy-naphthoate (2a′) was included in crude products. c Determined by HPLC analysis. d Isolated yield of 2a. e The reaction was carried out at −40 °C. | |||||
| 1 | L1 (R1 = Me) | 14 | 96 | 74 | 73 |
| 2 | L2 (R1 = Et) | 14 | 96 | 77 | 86 |
| 3 | L3 (R1 = iPr) | 14 | 72 | 55 | 77 |
| 4 | L4 (R1 = Pr) | 14 | 94 | 85 | 89 |
| 5 | L5 (R1 = Bu) | 14 | 93 | 72 | 83 |
| 6 | L6 (R2 = Et) | 6 | 99 | 83 | 85 |
| 7 | L7 (R2 = Pr) | 6 | 98 | 80 | 90 |
| 8 | L8 (R2 = Bu) | 6 | 99 | 82 | 90 |
| 9 | L9 (R2 = CH2tBu) | 12 | 98 | 80 | 90 |
| 10 | L10 (R2 = iPr) | 60 | 86 | 68 | 78 |
| 11e | L8 (R2 = Bu) | 24 | 99 | 90d | 91 |
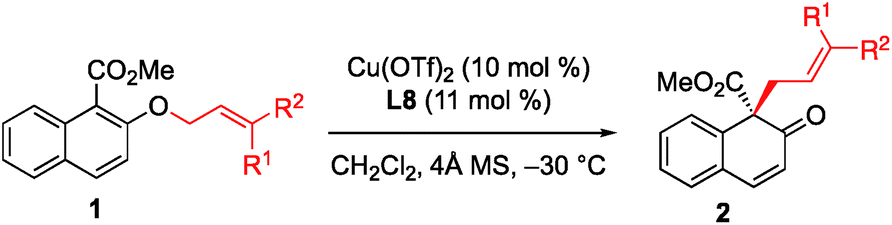
With the optimized conditions in hand, we started to evaluate the generality of this [1,3] rearrangement reaction. First, we assessed the substituent effect on the allyl moiety. As revealed in Table 2, cinnamyl groups bearing electron-rich (entries 2–4), electron-neutral (entry 1), and electron-deficient (entries 5 and 6) substituents were all well-accommodated and gave the corresponding products 2 in good to high yield with excellent enantioselectivity. Additionally, naphthyl-substituted substrates were also tolerated in this reaction, and the desired products were obtained in excellent yield and with high enantioselectivity (entries 7 and 8). We were very pleased to find that the rearrangement of heteroaryl-substituted substrates was also successful, and gave the corresponding β-naphthalenones in moderate to good yield with high ee values (entries 9 and 10). Disubstituted substrates were compatible with this rearrangement protocol. The desired products were obtained in high yield with high enantioselectivity when 1k and 1l were used in this reaction (entries 11 and 12), while prenyl naphthyl ether (1m) gave the desired product 2m in only 58% yield with 68% ee (entry 13). These results indicate that at least one aryl group is needed to achieve both high yield and high enantioselectivity.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (R1, R2) | Time (h) | 2 | ||
| Yieldb (%) | Eec (%) | ||||
| a All reactions were carried out with 0.15 mmol of 1 in dichloromethane (0.75 mL) at −30 °C. b Isolated yield. c Determined by HPLC analysis. d The reaction was carried out at −60 °C. e The reaction was carried out at −40 °C. | |||||
| 1 | 1a (H, Ph) | 24 | 2a | 90 | 91 |
| 2 | 1b (H, o-MeC6H4) | 16 | 2b | 90 | 95 |
| 3 | 1c (H, m-MeC6H4) | 20 | 2c | 91 | 92 |
| 4 | 1d (H, p-MeC6H4) | 10 | 2d | 87 | 90 |
| 5 | 1e (H, p-BrC6H4) | 60 | 2e | 76 | 91 |
| 6 | 1f (H, p-ClC6H4) | 60 | 2f | 78 | 92 |
| 7 | 1g (H, 1-naphthyl) | 10 | 2g | 95 | 92 |
| 8 | 1h (H, 2-naphthyl) | 10 | 2h | 86 | 92 |
| 9d | 1i (H, 3-furyl) | 14 | 2i | 63 | 96 |
| 10 | 1j (H, 3-thienyl) | 1 | 2j | 85 | 94 |
| 11e | 1k (Me, Ph) | 10 | 2k | 92 | 94 |
| 12e | 1l (R1 = R2: p-ClC6H4) | 36 | 2l | 89 | 90 |
| 13 | 1m (Me, Me) | 24 | 2m | 58 | 68 |
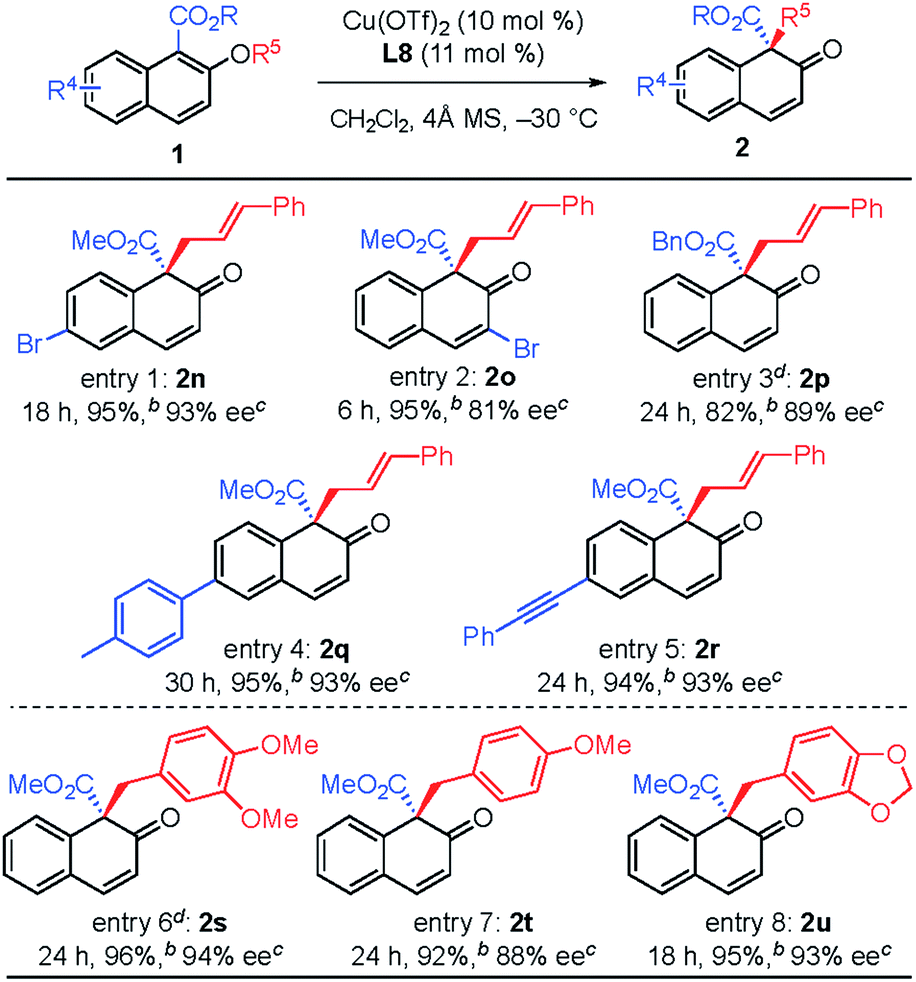
The substituent effect on the naphthalene ring was also investigated (Table 3). Various substituents were well-tolerated at the 3- and 6-positions on the naphthol framework. Rearrangement of allyl naphthyl ethers containing bromo (1n and 1o), para-methylphenyl (1q) and phenylethynyl (1r) gave the corresponding products 2 in excellent yields and with high enantioselectivities. Single-crystal X-ray analysis of compound 2l allowed us to establish the stereogenic center has an S configuration (see the ESI†). The investigation of the substrate scope revealed that substrates with an electron-donating substituent on R2 or an electron-withdrawing substituent on the naphthalene ring that is capable of stabilizing the allylic cation or the counter anion would undergo rearrangement smoothly and give the products 2 in high yield (entries 1–5). In addition, we found that the reactions of benzyl naphthyl ethers (1s, 1t and 1u) with a benzyl group bearing strong electron-donating substituents proceeded smoothly to give the corresponding products 2 in high yield with enantioselectivity (entries 6–8).
To further demonstrate the utility of this strategy, a more challenging substrate, methyl (E)-2-[(2-methyl-3-phenylallyl)oxy]-1-naphthoate (1v), was used. To our delight, the rearrangement reaction proceeded smoothly to give 2v in 71% yield with 88% ee (Scheme 2a). Furthermore, the rearrangement of methyl 1-substituted 2-[3-(3-thienyl)allyloxy]-3-naphthoates 3a and 3b gave the desired products 4a and 4b with 92% ee and 90% ee, respectively (Scheme 2b). The rearrangement reaction of 1a was conducted on a gram scale, and 2a was obtained in 87% yield with 92% ee when only 5 mol% catalyst was used. Notably, when the amount of catalyst was reduced to 1 mol%, the ee value was maintained while only moderate yield was observed (Scheme 2c). Treatment of 2a with NaBH4 and cerium(III) chloride heptahydrate in MeOH at −78 °C for 2 h gave the corresponding allyl alcohol 5 in 88% yield and 96:4 diastereoselectivity with the retention of enantiomeric purity (Scheme 2d).
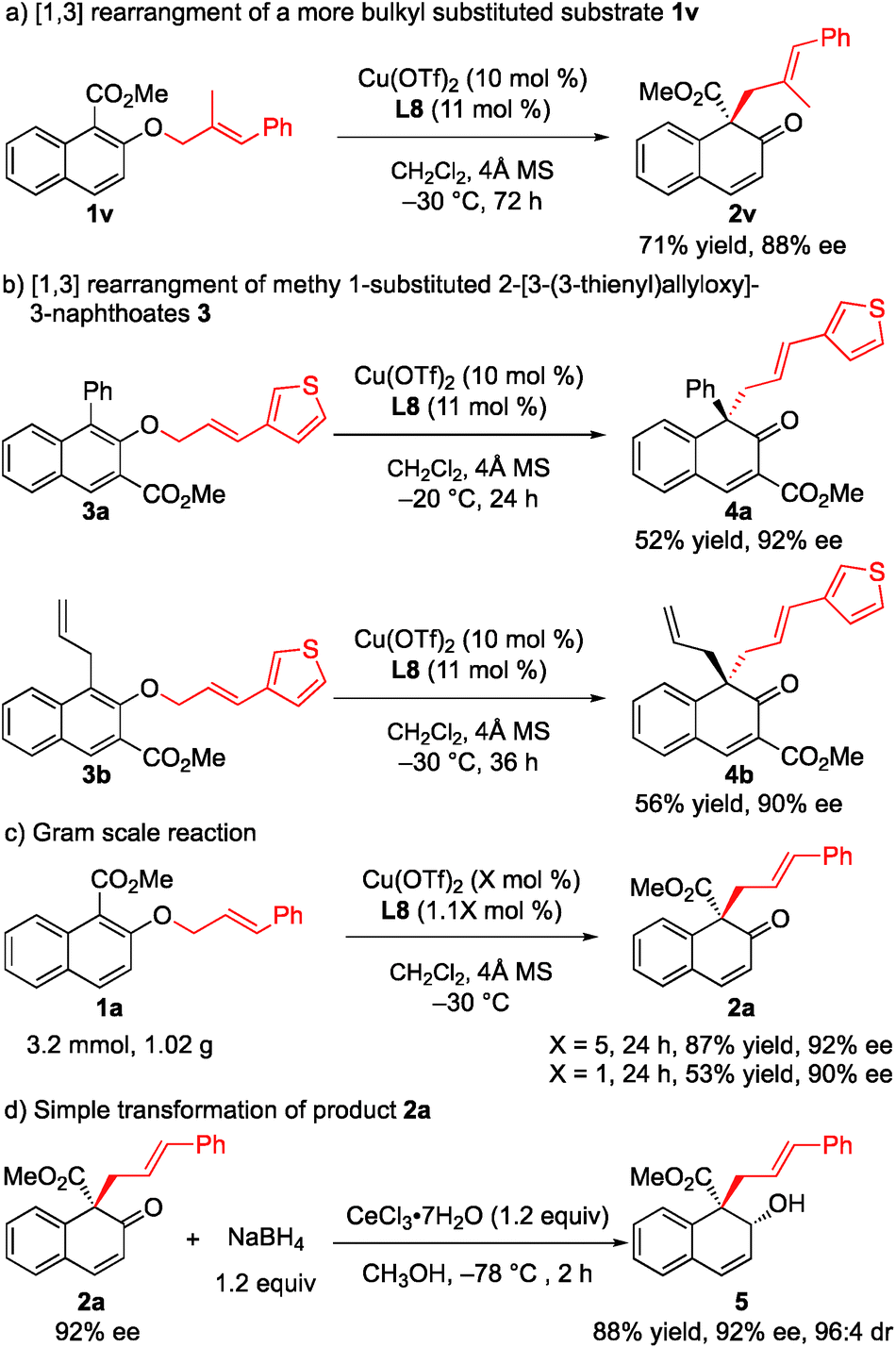 | ||
| Scheme 2 Further synthetic study of the rearrangement reaction: (a) [1,3] rearrangement of a more bulky substituted substrate 1v. (b) [1,3] rearrangement of methyl 1-substituted 2-[3-(3-thienyl)allyloxy]-3-naphthoates 3. (c) Gram scale reaction. (d) Simple transformation of product 2a. | ||
As observed in the above reaction, the N-5-dibenzosuberyl-substituted group of the ligand proved to be crucial in regulating asymmetric induction. To get a clear understanding of the exact role that the N-5-dibenzosuberyl-substituted group plays in this reaction, we tried to cultivate a single crystal of the copper complex. To our delight, we successfully obtained a single crystal of copper and the ligand in a 1:2 ratio. X-ray crystallographic analysis indicated that the close contact between the Cu(II) center and the benzene ring of the ligand should be a result of π–cation interaction,16 which we believe is responsible for the asymmetric induction (Fig. 1a).
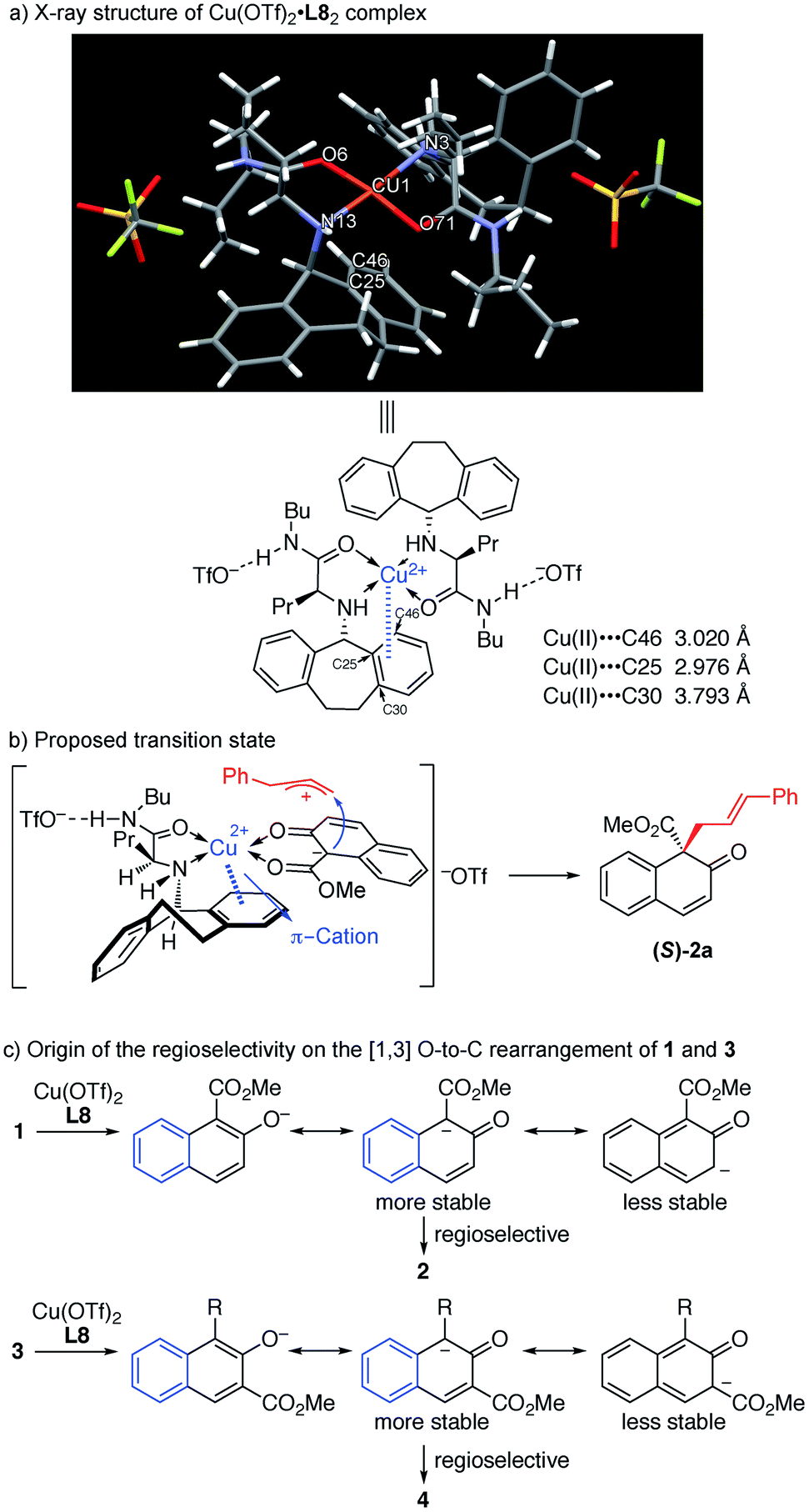 | ||
| Fig. 1 (a) X-ray structure of Cu(OTf)2·L8 complex. (b) Proposed transition state. (c) Origin of the regioselectivity on the [1,3] O-to-C rearrangement of 1 and 3. | ||
In order to gain insight into the reaction mechanism, a crossover reaction was carried out using substrates 1v and 1p. As shown in Scheme 3, products 2v and 2p were obtained cleanly while no crossover products were detected. This indicates that the rearrangement is an intramolecular reaction and should involve a tight ion pair intermediate. The regioselectivity in the rearrangement of 1 and 3 can be understood by the resonance stability of naphthoxy anions (Fig. 1c).
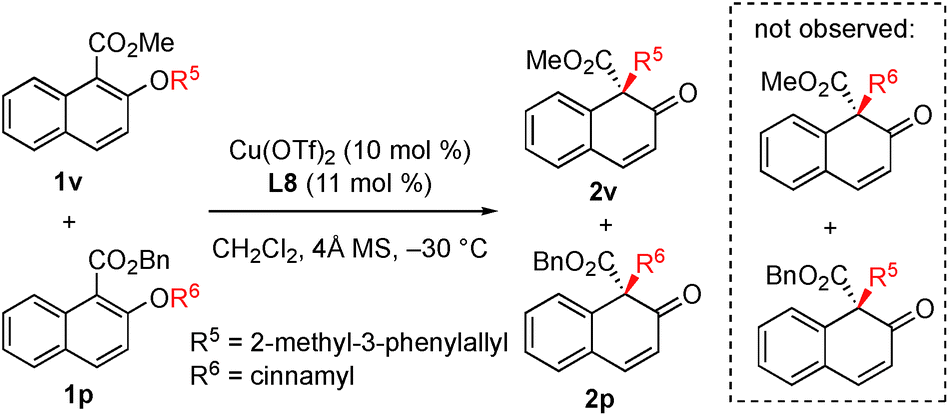 | ||
| Scheme 3 Crossover experiments. | ||
In the light of these observations, we proposed the following transition state for the π–Cu(II) complex-catalyzed [1,3] O-to-C rearrangement of allyl naphthyl ethers (Fig. 1b). The square-planar Cu(II) center is coordinated with the N and O atoms from ligand L8 and the two O atoms from substrate 1a in trans chelation to avoid steric hindrance between the N-5-dibenzosuberyl group of L8 and the cinnamyl group of 1a.15 Simultaneously, substrate 1a undergoes heterolytic C–O cleavage to form a reactive tight ion pair intermediate between the allyl cation and the counter enolate anion. The π–cation interactions between the Cu center and the N-5-dibenzosuberyl group of L8 lead to the folding of the N-5-dibenzosuberyl group, which perfectly shields the re face of the naphthalenolate. The enolate then uses its si face to approach the allyl cation, affording the rearrangement product 2a in S configuration.
3. Conclusions
In conclusion, we reported the successful development of enantioselective [1,3] O-to-C rearrangement of methyl 2-allyloxy/benzyloxy-1/3-naphthoates using a chiral π–Cu(II) catalyst (1–10 mol%) for the first time. This method is practical and scalable (1 g scale). X-ray crystallographic analysis of the Cu(II) complex provided solid evidence for the existence of π–cation interaction, which was shown to be crucial for inducing enantioselectivity. Based on the experimental results and mechanistic studies, we proposed a possible transition state involving a tight ion pair intermediate. We believe that this catalytic system may be useful for inducing asymmetry in other types of orbital symmetry-forbidden [m,n] rearrangement reactions. Studies along these lines are underway.4. Methods
The ESI† contains details of X-ray crystallographic analysis of the π–Cu(II) complex, experiments, product analyses and spectra of all characterized compounds.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Numbers JP15H05755 and JP15H05810 for Precisely Designed Catalysts with Customized Scaffolding, and the cutting-edge, international research units, Nagoya University. We thank Drs Manabu Hatano, Takahiro Horibe, Yanhui Lu and Yan Zhou for X-ray diffraction analysis.Notes and references
- (a) R. P. Lutz, Chem. Rev., 1984, 84, 205–247 CrossRef CAS; (b) H. Ito and T. Taguchi, Chem. Soc. Rev., 1999, 28, 43–50 RSC; (c) M. Hiersemann and L. Abraham, Eur. J. Org. Chem., 2002, 1461–1471 CrossRef CAS; (d) A. M. Martín Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef PubMed; (e) K. C. Majumdar, S. Alam and B. Chattopadhyay, Tetrahedron, 2007, 64, 597–643 CrossRef; (f) J. Rehbein and M. Hiersemann, Synthesis, 2013, 45, 1121–1159 CrossRef CAS; (g) T. C. A. F. Rodrigues, W. A. Silva and A. H. L. Machado, Curr. Org. Synth., 2015, 12, 795–805 CrossRef CAS.
- (a) W. M. Lauer and M. A. Spielman, J. Am. Chem. Soc., 1933, 55, 4923–4930 CrossRef; (b) K. B. Wiberg, T. M. Shryne and R. R. Kintner, J. Am. Chem. Soc., 1957, 79, 3160–3164 CrossRef CAS; (c) K. B. Wiberg, R. Roy Kintner and E. L. Motell, J. Am. Chem. Soc., 1963, 85, 450–454 CrossRef CAS.
- (a) P. A. Grieco, J. D. Clark and C. T. Jagoe, J. Am. Chem. Soc., 1991, 113, 5488–5489 CrossRef CAS; (b) J. C. R. Brioche, T. A. Barker, D. J. Whatrup, M. D. Barker and J. P. A. Harrity, Org. Lett., 2010, 12, 4832–4835 CrossRef CAS PubMed; (c) S. Hou, X. Li and J. Xu, J. Org. Chem., 2012, 77, 10856–10869 CrossRef CAS PubMed; (d) C. N. Kona, M. N. Patil and C. V. Ramana, Org. Chem. Front., 2016, 3, 453–456 RSC; (e) I. Nakamura, M. Owada, T. Jo and M. Terada, Org. Lett., 2017, 19, 3059–3062 CrossRef CAS PubMed; (f) Z. Jiantao, L. Zhehui, C. Lianfen, J. Huanfeng and Z. Shifa, Chem.–Eur. J., 2018, 24, 6927–6931 CrossRef PubMed.
- (a) J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532–11533 CrossRef CAS; (b) S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368–13369 CrossRef CAS PubMed; (c) S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va and E. Vedejs, J. Am. Chem. Soc., 2006, 128, 925–934 CrossRef CAS PubMed.
- (a) S. J. Meek and J. P. A. Harrity, Tetrahedron, 2007, 63, 3081–3092 CrossRef CAS; (b) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240–254 RSC.
- (a) K. Ishihara and M. Fushimi, Org. Lett., 2006, 8, 1921–1924 CrossRef CAS PubMed; (b) K. Ishihara, M. Fushimi and M. Akakura, Acc. Chem. Res., 2007, 40, 1049–1055 CrossRef CAS PubMed; (c) K. Ishihara and M. Fushimi, J. Am. Chem. Soc., 2008, 130, 7532–7533 CrossRef CAS PubMed; (d) A. Sakakura, M. Hori, M. Fushimi and K. Ishihara, J. Am. Chem. Soc., 2010, 132, 15550–15552 CrossRef CAS PubMed; (e) A. Sakakura and K. Ishihara, Chem. Soc. Rev., 2011, 40, 163–172 RSC; (f) M. Hori, A. Sakakura and K. Ishihara, J. Am. Chem. Soc., 2014, 136, 13198–13201 CrossRef CAS PubMed.
- (a) A. Gansauer, D. Fielenbach and C. Stock, Adv. Synth. Catal., 2002, 344, 845–848 CrossRef; (b) A. Gansäuer, D. Fielenbach, C. Stock and D. Geich-Gimbel, Adv. Synth. Catal., 2003, 345, 1017–1030 CrossRef.
- C. G. Nasveschuk and T. Rovis, Org. Lett., 2005, 7, 2173–2176 CrossRef CAS PubMed.
- (a) T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2012, 134, 20017–20020 CrossRef CAS PubMed; (b) J. Nan, J. Liu, H. Zheng, Z. Zuo, L. Hou, H. Hu, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 2356–2360 CrossRef CAS PubMed; (c) S.-G. Wang, X.-J. Liu, Q.-C. Zhao, C. Zheng, S.-B. Wang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 14929–14932 CrossRef CAS PubMed; (d) L. Zhou, X.-Q. Li, H. Yang, J.-J. Wang, B.-B. Gou and J. Chen, Chem.–Eur. J., 2017, 23, 5381–5385 CrossRef PubMed; (e) Q. Guo, M. Wang, H. Liu, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 4747–4751 CrossRef CAS PubMed.
- (a) C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2013, 52, 10056–10059 CrossRef CAS PubMed; (b) C.-X. Zhuo and S.-L. You, Adv. Synth. Catal., 2014, 356, 2020–2028 CrossRef CAS; (c) H. F. Tu, C. Zheng, R. Q. Xu, X. J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 3237–3241 CrossRef CAS PubMed.
- D. Shen, Q. Chen, P. Yan, X. Zeng and G. Zhong, Angew. Chem., Int. Ed., 2017, 56, 3242–3246 CrossRef CAS PubMed.
- For reviews on CADA, see: (a) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed; (b) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570–1580 RSC.
- See Scheme S1 in the ESI† for details of the substrate screening.
- See Table S1 in the ESI† for details of the ligand screening.
- See Table S2 in the ESI† for details of the solvent effect.
- D. Van der Helm and C. E. Tatsch, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 2307–2312 CrossRef CAS . In the X-ray structure of Cu(II)·L8 complex in Fig. 1a, there are two close approaches between the Cu(II) and carbon atoms of the N-5-dibenzosuberyl group [C(25): 2.976 Å and C(46): 3.020 Å] which are believed to constitute the π–cation interactions between the Cu(II) and the benzene ring of the N-5-dibenzosuberyl group. The distance between the benzene plane of the N-5-dibenzosuberyl group and Cu(II) is 2.692 Å. In addition, the basal plane [O(6), N(3), O(71), N(13)] of the square pyramid is tetrahedrally distorted; the Cu(II) is displayed 0.064 Å away from this plane towards the top of the pyramid. This distortion is likely related to the π–cation interactions between Cu(II) and the N-5-dibenzosuberyl group.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1865509 and 1865510. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05601c |
| This journal is © The Royal Society of Chemistry 2019 |