
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipid-independent control of endothelial and neuronal TRPC3 channels by light†
Oleksandra
Tiapko‡
 a,
Niroj
Shrestha‡
a,
Sonja
Lindinger
b,
Gema
Guedes de la Cruz
c,
Annarita
Graziani
a,
Christiane
Klec
d,
Carmen
Butorac
b,
Wolfgang. F.
Graier
d,
Helmut
Kubista
e,
Marc
Freichel
f,
Lutz
Birnbaumer
gh,
Christoph
Romanin
b,
Toma
Glasnov
c and
Klaus
Groschner
*a
a,
Niroj
Shrestha‡
a,
Sonja
Lindinger
b,
Gema
Guedes de la Cruz
c,
Annarita
Graziani
a,
Christiane
Klec
d,
Carmen
Butorac
b,
Wolfgang. F.
Graier
d,
Helmut
Kubista
e,
Marc
Freichel
f,
Lutz
Birnbaumer
gh,
Christoph
Romanin
b,
Toma
Glasnov
c and
Klaus
Groschner
*a
aGottfried Schatz Research Center – Biophysics, Medical University of Graz, Neue Stiftingtalstraße 6/D/04, 8010 Graz, Austria. E-mail: klaus.groschner@medunigraz.at
bInstitute of Biophysics, University of Linz, Gruberstrasse 40/1, 4020 Linz, Austria
cInstitute of Chemistry, University of Graz, Heinrichstraße 28/I, 8010 Graz, Austria
dGottfried Schatz Research Center – Molecular Biology and Biochemistry, Medical University of Graz, Neue Stiftingtalstraße 6/6, 8010 Graz, Austria
eInstitute of Pharmacology, Medical University of Vienna, Währinger Straße 13A, 1090 Vienna, Austria
fPharmakologisches Institut, Universität Heidelberg, Im Neuenheimer Feld 366, D-69120 Heidelberg, Germany
gNeurobiology Laboratory, National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina 27709, USA
hInstitute of Biomedical Research (BIOMED), Catholique University of Argentina, Buenos Aires C1107AZZ, Argentina
First published on 15th January 2019
Abstract
Lipid-gated TRPC channels are highly expressed in cardiovascular and neuronal tissues. Exerting precise pharmacological control over their activity in native cells is expected to serve as a basis for the development of novel therapies. Here we report on a new photopharmacological tool that enables manipulation of TRPC3 channels by light, in a manner independent of lipid metabolism and with higher temporal precision than lipid photopharmacology. Using the azobenzene photoswitch moiety, we modified GSK1702934A to generate light-controlled TRPC agonists. We obtained one light-sensitive molecule (OptoBI-1) that allows us to exert efficient, light-mediated control over TRPC3 activity and the associated cellular Ca2+ signaling. OptoBI-1 enabled high-precision, temporal control of TRPC3-linked cell functions such as neuronal firing and endothelial Ca2+ transients. With these findings, we introduce a novel photopharmacological strategy to control native TRPC conductances.
Introduction
Non-selective cation channels of the TRPC family have been implicated in a variety of diseases although a comprehensive understanding of their specific role in the complex setting of organ physiopathology is lacking.1–3 TRPC channels are expressed throughout the human body with particular abundance in brain and cardiovascular tissues.2 Our current knowledge about the cell type-specific functions of TRPC molecules, their dependencies on temporal activity pattern and connections with down-stream signaling pathways is incomplete. This paucity of understanding is due to the difficulties encountered when attempting to precisely and specifically manipulate TRPC activity in native tissues. Hence, the development of photopharmacological strategies that target TRPC signaling pathways is needed to make advance to the field.Azobenzene photoswitches are both suitable and valuable for the high precision control of TRPC channels as has recently been demonstrated by reports on the spatial and temporal precision of TRPC activation achieved with photoconvertible diacylglycerols.4–6 Although exceptionally effective in terms of channel activation, this latter approach suffers from two inherent limitations, namely its general lack of selectivity and a certain temporal inaccuracy due to delay and frequency dependence of optical control based on cooperativity of lipid gating.5 The complexity of channel activation by lipid mediators impedes temporal precision of the manipulation of TRPC signaling with photolipids.
Over the past decade, an array of small molecules have been identified, which either inhibit or activate TRPC channels with variable degree of selectivity.7–10 Photoswitchable channel blocker do not appear perfectly suitable for efficient TRPC3 photopharmacology since the channels tend to inactivate or desensitize efficiently, and it is barely feasible to exert cyclic current control over cellular Ca2+ signaling by blocking and unblocking of a constitutively open TRPC3 pore. We therefore set out to generate a photoswitch based on the structural features of the recently characterized TRPC3 activator GSK1702934A (GSK).10 This molecule was found to activate native TRPC channel complexes with an apparently high degree of selectivity and reasonable potency. GSK acts independently of membrane lipid metabolism, and significant off-target effects on other ion conductances have not yet been detected.10 With this report, we provide proof of concept that a GSK-based azobenzene photoswitch enables efficient and temporally precise control over TRPC3 signaling.
Results and discussion
Optical control of TRPC3 signaling with photoswitchable diacylglycerol derivatives is generally feasible as shown at the whole-cell current level. Although, photoisomerization of lipid photoswitches into their active conformation is essentially fast, full activation of the channel requires multiple conformational steps, and is obtained with a certain delay during repetitive cycling. Inward currents through photolipid-activated TRPC3 were essentially small during the initial photoactivation cycle and increased with repetitive activation, as expected from the previously reported slow process of cooperative channel activation by the lipid photoswitch.5 To generate a photochromic activator that enables a higher degree of temporal precision and independence of lipid metabolism, we set out to develop an actuator based on GSK.Novel benzimidazole activators of TRPC3
As a first step, we synthetized a few selected GSK-related structures with potential agonist activity in biological activity tests (Scheme 1, Fig. 1). The newly synthesized compounds (Scheme 1 (1b–d)) were easily obtained in good to high yields by employing a synthetic procedure developed earlier9 and fully characterized by using analytical techniques (see ESI† for details). The obtained molecules, two different 1,3-dihydro-2H-benzo[d]imidazol-2-ones (BI-1, BI-2) and one 1,3-dihydro-2H-imidazo[4,5-b]pyridin-2-one derivative (PI) (Fig. 1a) were then initially compared with GSK in terms of their preserved activity at recombinant TRPC3 channels that were overexpressed in human embryonic kidney (HEK293) cells.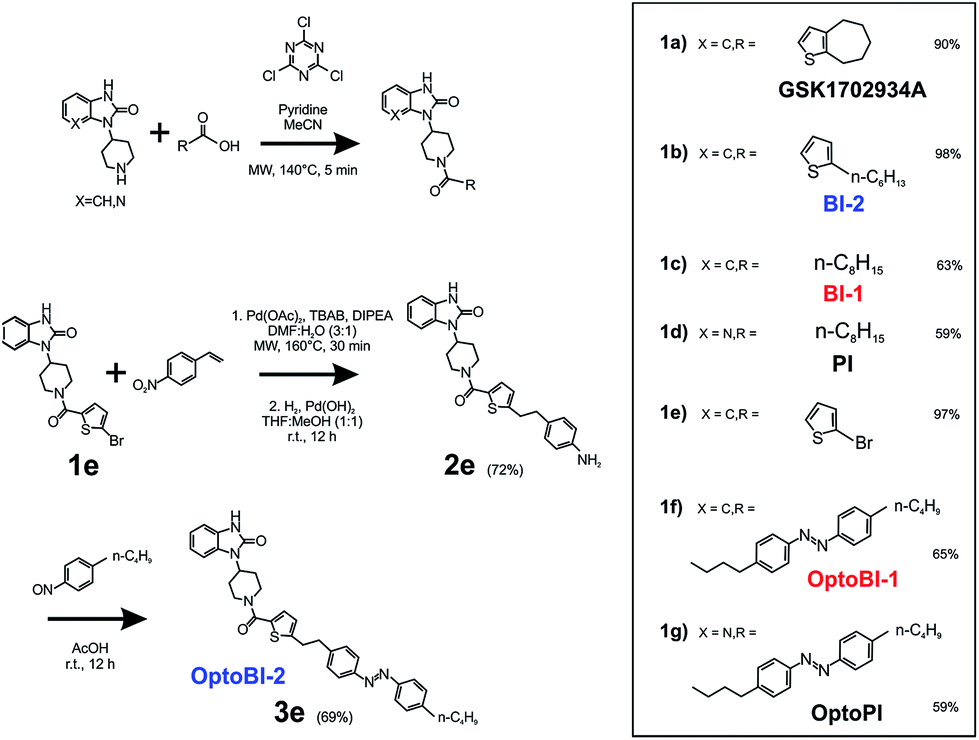 | ||
| Scheme 1 Synthesis of selected analogues of GSK1702934A (GSK), designated as BI-1, BI-2, PI, and the corresponding photoresponsive derivatives designated as OptoBI-1, OptoBI-2 and OptoPI. Yield is given in %. | ||
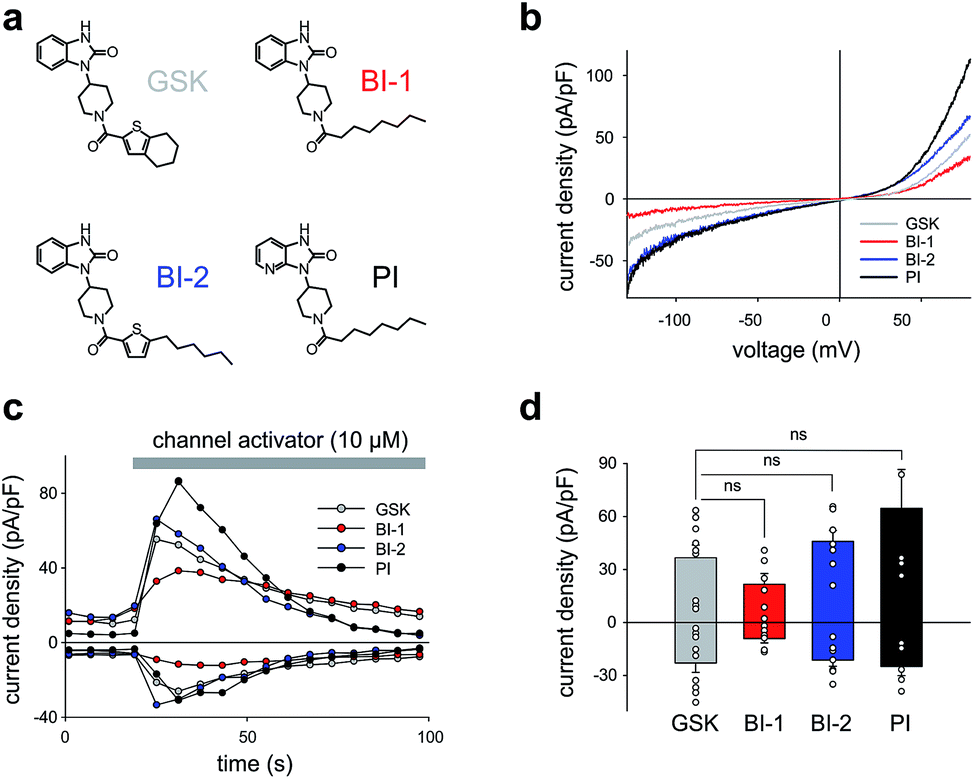 | ||
| Fig. 1 (a) Chemical structures of GSK1702934A (GSK) and GSK derivatives (BI-1, BI-2 and PI). (b) Current to voltage relations of the net conductances (Imax − Ibasal) induced by GSK, BI-1, BI-2 and PI (10 μM each) obtained in TRPC3-transfected HEK293 with voltage-ramp protocols. (c) Representative time courses of the TRPC3 conductances recorded at −90 mV and +70 mV during administration of channel activators (GSK, BI-1, BI-2 and PI as indicated) at 10 μM concentrations. (d) Current density of net, maximum responses obtained at −90 mV and +70 mV (mean ± SEM). Statistical significance was tested by two tailed t-test (normally distributed values) or Mann–Whitney tests (non-normally distributed values). ns = not significant (p > 0.05); N cells: GSK = 9; BI-1 = 6; BI-2 = 7; PI = 5. | ||
All three compounds activated recombinant TRPC3 channels in whole-cell voltage-clamp experiments. Fig. 1b illustrates the comparison of I–V relations among the peak conductances (ramp responses from −130 mV to +80 mV, 1 s) induced by GSK, BI-1, BI-2 and PI in HEK293 cells expressing a YFP-TRPC3 fusion construct. At a concentration of 10 μM, all compounds transiently induced currents that featured the double-rectifying I–V relation typical of TRPC3 conductances (Fig. 1b). Time-courses of the current activation are shown in Fig. 1c. The observed transient increase in conductance displayed a time course similar to that initiated by GSK, and peak current densities produced by BI-2 and PI were comparable to those evoked by GSK, whereas BI-1 induced slightly lower responses (Fig. 1d). In a previous study, we identified the aliphatic ring adjacent to the thiophene core in GSK1702934A as a determinant of agonist features. While reducing the ring size did not significantly modify activity, opening of the seven-membered aliphatic ring resulted in an increase of agonist efficacy as compared to GSK1702934A.9 Elimination of the thiophene core and introduction of an aliphatic chain instead, resulted in diminished agonist activity. These previous investigations also revealed that substitutions at the benzimidazole typically impair efficacy as a TRPC channel agonist. Unexpectedly, replacing the benzimidazole core with a pyridoimidazole led to an increase of agonist efficacy (PI, Fig. 1). As HEK293 cells essentially express low endogenous levels of TRPC proteins including TRPC3 while expressing other ion channel genes,11–13 the lack of effects observed in non-transfected HEK293 (ESI Fig. 1†) cells not only demonstrated that the activity of these compounds was strictly dependent on TRPC3 expression, but also indicated a certain degree of TRPC specificity. On the basis of these results, all three compounds were considered as suitable molecular scaffolds for the development of photoswitches to control of TRPC3.
Photochromic benzimidazoles for optical manipulation of TRPC3
In a next step, we generated three corresponding azobenzene derivatives (Scheme 1 and Fig. 2a), designated as OptoBI-1 (Scheme 1 (1f)), OptoBI-2 (Scheme 1 (3e)) and OptoPI (Scheme 1 (1g)), which feature efficient trans–cis–trans light-triggered photoisomerization (see ESI† for details). Based on the available knowledge about structure–activity relations as outlined above, we were prompted to generate photoswitchable OptoBI-1, OptoBI-2 and OptoPI by replacing the aliphatic chains for an photoswitchable diazo-moiety. Typically, these azobenzene-photoswitches reside in the thermodynamically more stable trans-conformation, while their irradiation with UV light (365 nm) initiates rapid isomerization into the cis-form. Triggering the opposite cis–trans transition can be rapidly and easily conducted by illuminating the photochromic ligand with visible light (blue, 430 nm).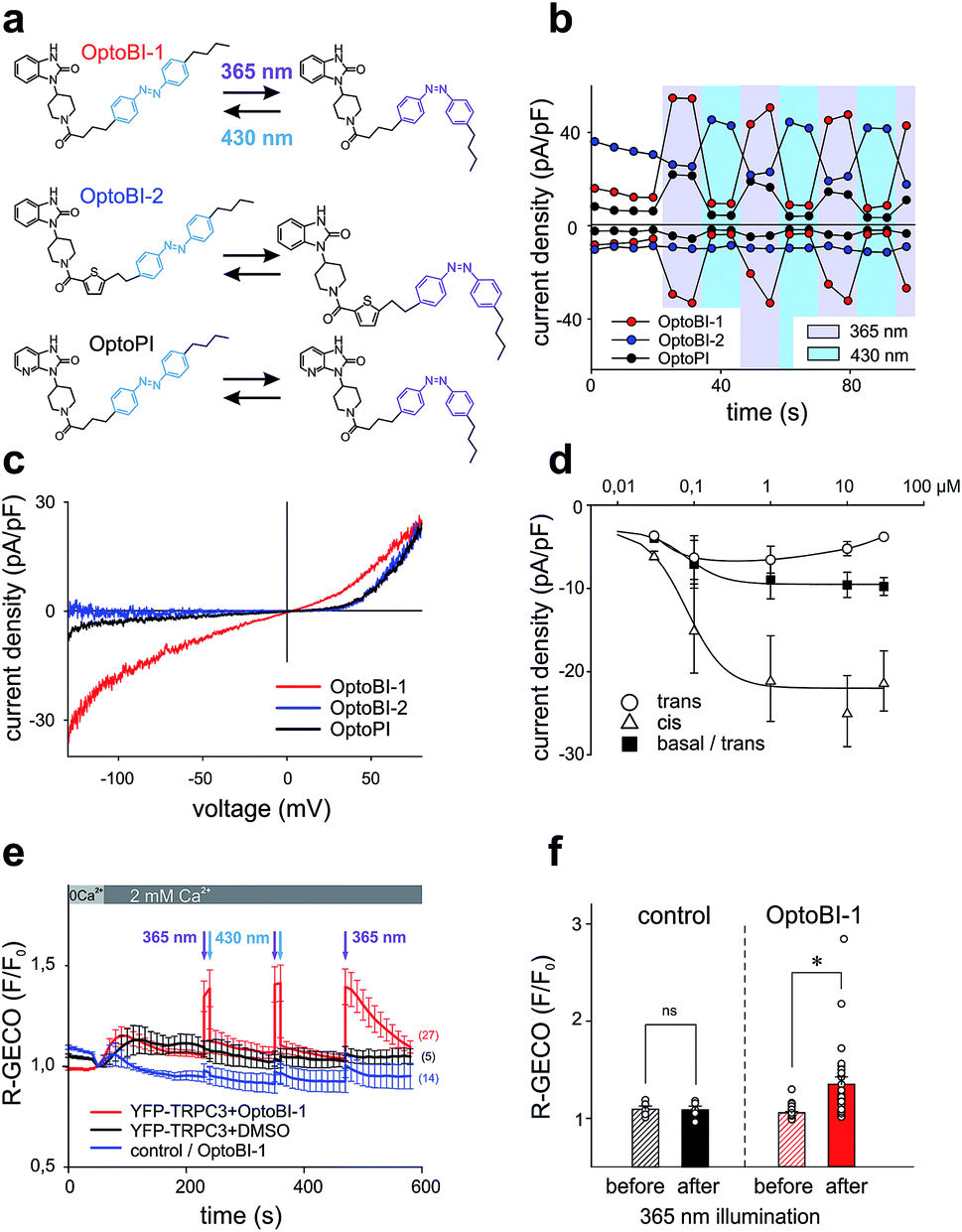 | ||
| Fig. 2 (a) Chemical structures of photoswitchable GSK derivatives: OptoBI-1, OptoBI-2, OptoPI. (b) Representative time courses of the TRPC3 conductances recorded at −90 mV and +70 mV during repetitive photoconversion of OptoBI-1, OptoBI-2 and OptoPI (10 μM). Cells were kept in dark prior to light application, following by cycling of UV (365 nm; violet) and blue light (430 nm; blue) illuminations, applied for 10 s each (indicated). (c) Representative net I–V relations (Imax − Ibasal) for OptoBI-1, OptoBI-2 and OptoPI-induced (10 μM) obtained in TRPC3-transfected HEK293 cells with voltage-ramp protocols. (d) Dependency of inward current densities (at −90 mV), induced by photoconversion of OptoBI-1 at increasing concentrations. Values (mean ± SEM) are given for currents before illumination (squares; basal/trans; N/cells = 8), during UV-induced cis isomerization (triangles; cis; N/cells = 8) and upon deactivation cycling by blue light (circles; trans; N/cells = 8). (e) Time courses of Ca2+-sensitive R-GECO fluorescence during OptoBI-1 (10 μM) photoconversion in TRPC3 plus R-GECO (red and black trace) and R-GECO only (blue trace) expressing in HEK293. Illumination (365 nm; 10 s and 430 nm; 30 s) performed before fluorescence recording, is indicated by arrows (N of cells: YFP-TRPC3 and OptoBI-1 = 27; YFP-TRPC3 and DMSO = 5; HEK293 WT and OptoBI-1 = 14). (f) Fluorescence levels before and after UV light-induced changes in cells co-transfected with TRPC3 and R-GECO in presence of DMSO (control) or cis-photoconversion of OptoBI-1 and OptoPI (10 μM). Mean ± SEM, ns = not significant (p > 0.05), *p < 0.05, statistical significance was tested by two tailed t-test; N = number of cells as indicated in (e); individual values are included (circles). | ||
The suitability of these photoswitches to control of TRPC3 was tested again in the HEK293 heterologous expression system by patch-clamp recordings. We characterized the activity of the GSK derivatives, OptoBI-1, OptoBI-2 and OptoPI (Fig. 2a; at 10 μM) on the basis of the conductance generated by recombinant TRPC3 during the cyclic photoisomerization. The compounds were present in the bath solution while measurements were taken and optical cycling was initiated after about 20 s of control recording (Fig. 2b). In contrast to OptoBI-1 and OptoPI, OptoBI-2 activated substantial constitutive TRPC3 activity in the dark-adapted trans conformation (before first illumination; Fig. 2b). Repetitive conformational cycling initiated by exposure to UV (365 nm) and blue light (430 nm) illumination rapidly changed the TRPC3 conductance in the presence of all three photoswitches. The current inactivation or desensitization that is typically observed with photoswitchable lipids or conventional agonist stimulation5 was barely detectable with these GSK derivatives using the short-term cyclic activation protocol. UV-induced cis isomerization activated the TRPC3 conductance in the presence of OptoBI-1 or OptoPI, but inhibited currents with OptoBI-2. Thus, all three photochromic modulators operated the channel between a basal and an activated state in a light dependent manner, OptoBI-2, however, displayed a reverse dependency on isomerization as compared to its congeners. Interestingly, the efficacy of current activation at 10 μM was highest in the presence of OptoBI-1, which originated from the relatively weak activator BI-1 (Fig. 1). Hence the incorporation of the azobenzene moiety did not disturb the activator efficacy of OptoBI-1 but reduced it for Opto-PI and thereby changed the order of efficacies in favor of OptoBI-1 as compared to OptoPI. Upon the photoisomerization of the derivatives, TRPC3 switched rapidly between a basal constitutive activity and a level of current density that is typically observed during maximum Gq-coupled receptor stimulation or direct lipid activation.5
Fig. 2c displays the peak current to voltage relations obtained for OptoBI-1 and OptoPI (at 365 nm) as well as for OptoBI-2 (at 430 nm) from voltage-ramp recordings. Double rectifying features were evident for the OptoBI-1 and OptoPI-induced conductance, while inward currents were essentially not observed with OptoBI-2. The largest inward currents were observed for OptoBI-1, indicating that profound physiological activity was occurring in terms of the control of TRPC3 Ca2+ signaling by this photoswitch (Fig. 2c). The maximum inward currents obtained in response to OptoBI-1 were −8.9 ± 2.2 pA pF−1 (Fig. 2c and d) and comparable to that of the parent compound BI-1 (−9.1 ± 2.4 pA pF−1, Fig. 1d). Specifically, the comparison of the inward current densities revealed that azobenzene modification substantially impaired the activator efficacy in Opto-PI, significantly reducing the maximum inward current densities from a level of −24.9 ± 5.2 pA pF−1, obtained with the parent compound PI (Fig. 1d), to −2.7 ± 1.0 pA pF−1 determined for Opto-PI (ESI Fig. 2†). The inward rectification of these currents was relatively weak as compared to that of the parent compound BI-2 (Fig. 1b) and the inward current densities were significantly smaller for OptoBI-2 (OptoBI-2 = −1.2 ± 0.6 pA pF−1vs. BI-2 = −21.3 ± 3.5 pA pF−1). These results identify OptoBI-1 as an effective TRPC3 actuator and a highly promising tool that can be used to exert control over TRPC3 signaling. This photochromic TRPC3 agonist was found able to activate also the closely related TRPC6 and TRPC7 isoforms, while lacking effects on TRPC4 and TRPC5 channels (ESI Fig. 3†). Of note, the switching amplitudes for OptoBI-1 in terms of fractional conversion into favorable conformations in the photo-stationary state in solution (0.4 mM) were >99% (UV, cis) and about 75% (blue light, trans; ESI Fig. 4†). In the dark, cis-OptoBI-1 converted slowly back to the trans form, which was first detectable by HPLC only after about 50 min at room temperature when dissolved (10 mM) in DMSO. To determine the relative potency and efficacy of the two isomers as a channel agonist we delineated their concentration–response relations as illustrated in Fig. 2d and ESI Fig. 5.† These experiments revealed that incubation of resting channels with trans-OptoBI-1 slightly enhanced basal activity that amounted to about doubling of the typical basal activity of TRPC3 at the highest concentration tested suggesting weak partial agonist activity of the trans isomer. Interestingly, when the trans conformation of OptoBI-1 is induced during cyclic photoisomerization, this conformational transition converts the channel effectively into its resting or closed state. Thereby, deactivation of TRPC signaling during optical cycling of OptoBI-1 is rapid and complete. So far, the binding site for benzimidazoles within the TRPC structure has not been identified, and it is unclear if and how these sites overlap with the recently identified lipid recognition/gating sites or sites that bind inhibitors.14,15 In view of the available knowledge on TRPC structure from CryoEM investigations14,16 it is tempting to speculate that the benzimidazoles, like other small molecule modulators intercalate between the S1–S4 voltage sensor like domain and the pore domain.
Next, we compared the effects of OptoBI-1, OptoBI-2 and OptoPI on Ca2+ signaling through recombinant TRPC3 channels by taking Ca2+ measurements in HEK293 cells. As this heterologous expression system allowed for the use of a genetically encoded Ca2+ sensor (R-GECO), which does not interfere with the azobenzene isomerization spectrum, we employed this reporter molecule to investigate the efficiency of optical control in terms of both activation and deactivation of the signaling pathway.
Ca2+ imaging in HEK293 cells co-expressing YFP-TRPC3 and R-GECO was performed along with cis–trans isomerization of the GSK photoswitches, triggered by exposure to 10 s illumination periods at 365 nm or 30 s periods at 430 nm. Fig. 2e illustrates the time course of the Ca2+-sensitive R-GECO fluorescence in experiments in which the initially extracellular Ca2+ was elevated from nominally free to 2 mM in the presence of OptoBI-1 (10 μM) in the dark. Cells exposed to either OptoBI-1 or solvent (DMSO, controls) displayed only small increases in fluorescence upon extracellular Ca2+ elevation in the dark (Fig. 2e). These results are consistent with a relatively low basal channel activity in the presence of trans-OptoBI-1. Notably, the modest partial agonist activity of the trans isoform, which resulted in about doubling of basal current levels (Fig. 2d) did not cause detectable basal Ca2+ entry in this experimental setting. UV illumination (365 nm) for a duration of 10 s triggered an elevation in the Ca2+-sensitive fluorescence signal, which was rapidly reverted to baseline levels upon illumination with blue light (430 nm; 30 s). The light-controlled initiation of intracellular Ca2+ signals in HEK293 cells was readily repeatable without desensitization when the active state (cis-form) of the photoswitch was restricted to relatively short intervals (10 s) as shown for two consecutive activation–deactivation cycles in Fig. 2e. When cells were exposed for prolonged periods to cis-OptoBI-1, as shown for the third activation cycle, slow desensitization/inactivation was suggested from the current decline observed within a period of about 1.5 min. The desensitization/inactivation process in TRPC3 is incompletely understood but most likely involves a current (Ca2+)-dependent component. This phenomenon is similarly observed when TRPC3 is activated by diacylglycerol photoswitches. Interestingly, activation of TRPC3 by lipid photoswitches such as OptoDArG15 features a slow component of activation that is not observed with OptoBI-1. Consequently, generation of TRPC3-mediated Ca2+ signals in HEK293 cells was faster with OptoBI-1 as compared to OptoDArG (ESI Fig. 6†). Fig. 2f provides a statistical summary of the changes observed in R-GECO fluorescence by photoisomerization of OptoBI-1. In contrast to OptoBI-1 both OptoBI-2 (not shown) and OptoPI (10 μM, ESI Fig. 7†) failed to induce significant alterations in cellular Ca2+ levels as expected from electrophysiological characterization (Fig. 2c).
Photochromic benzimidazoles allow optical control over native TRPC3
Our findings in the HEK293 expression system allowed us to identify OptoBI-1 as an efficient tool that can be used to exert optical control over TRPC3 signaling. This concept was further tested in two cell types that express endogenous TRPC channels and have been previously reported to feature TRPC3 as critical signaling elements in their physiopathology. As TRPC3 is prominently expressed in cardiovascular tissues and the brain2 we performed experiments in the cell line EA.hy926, which is derived from human umbilical vein endothelium, and in freshly isolated murine hippocampal neurons as test systems. In vascular endothelium TRPC3 has been shown to be essential for Ca2+ signaling processes,13,17,18 therefore we investigated whether OptoBI-1 could control TRPC3-mediated Ca2+ homeostasis in an endothelial cell background. The expression of endogenous TRPC3 channels in this cell preparation was also substantiated by experiments with GSK1702934A, which elicited oscillatory Ca2+ signals that were prevented by the TRPC channel blocker Pyr3 (10 μM; ESI Fig. 8†). As shown in Fig. 3a, native (non-transfected, wild type) EA.hy926 cells were shown to display profound Ca2+ transients as measured by Fluo-4 fluorescence (at 490 nm), in response to UV (365 nm, 15 s) illumination in the presence of OptoBI-1 (60 μM). This light-induced Ca2+ signal was detectable in about 80% of cells (ESI Fig. 9a†). The observation of divergent sensitivity of the endothelial cell population might reflect heterogeneity in terms of TRPC expression levels and/or endothelial phenotype as a determinant of TRPC function.19 The second cis OptoBI-1-induced Ca2+ transient displayed reduced amplitude, indicating that inactivation/desensitization of the Ca2+ entry pathway was taking place. The cells did not respond to the illumination protocol in the absence of the photoswitchable GSK-derivative, and responses were not observed in a nominally Ca2+-free solution, demonstrating that the OptoBI-1-induced response was exclusively due to Ca2+ entry (Fig. 3b). EA.hy926 cells with suppressed TRPC3 expression due to siRNA knock-down, displayed significantly suppressed responsiveness to cis-isomerization of OptoBI-1 (ESI Fig. 9a†) and the responses were eliminated in the presence of 10 μM Pyr3 (ESI Fig. 9c and d†). In turn, when TRPC3 channels were overexpressed in the endothelial cells, cis-isomerization elicited a significantly larger cellular Ca2+ response (Fig. 3b), which were observed in all cells tested (ESI Fig. 9a†), demonstrating the dependence of the light-controlled signaling process on TRPC3 expression.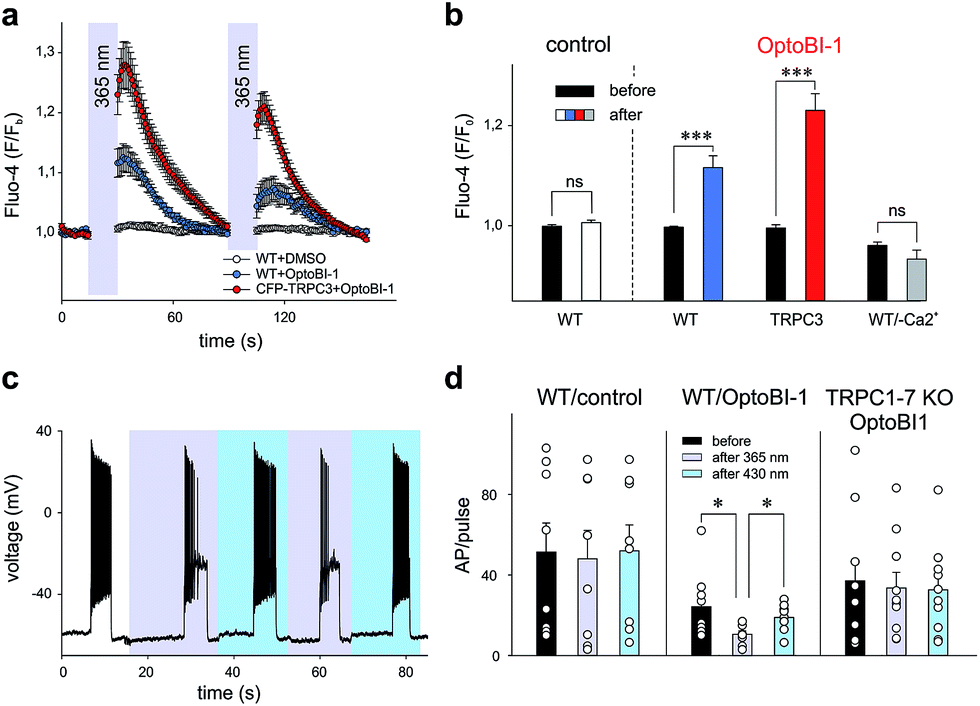 | ||
| Fig. 3 (a) Time course of Ca2+ sensitive Fluo-4 fluorescence in EA.hy926 non-transfected WT cells (control (DMSO) N cells used = 23; OptoBI-1, 60 μM, N = 23) or cells transfected with CFP-TRPC3 (OptoBI-1, 60 μM; N = 21) in 2 mM Ca2+ buffer upon cis-photoisomerization with UV light (365 nm; 15 s). (b) Change in bleaching-corrected Fluo-4 fluorescence evoked by cis-OptoBI-1 (60 μM) in EA.hy926 cells (WT, N = 23; CFP-TRPC3 transfected, N = 21 and WT in Ca2+-free, N = 10). Control cells (N = 23) were incubated with DMSO in 2 mM Ca2+ buffer; statistical significance was tested by two tailed t-test (normally distributed values) or Wilcoxon signed rank test (non-normally distributed values); ns = not significant (p > 0.05), ***p < 0.001. (c) Representative AP firing in primary hippocampal neurons (WT) induced by a short (5 s) current injection. The first current injection was performed in dark (trans conformation; control), following by cis isomerization with UV (365 nm) and subsequent reversal to trans conformation with blue (430 nm) light. (d) Mean AP count/pulse in dark, UV and blue light-stimulated in current-clamped WT controls (DMSO) and incubated with OptoBI-1 (20 μM) WT and TRPC1–7 KO neurons. Bar graphs show mean ± SEM from at least 3 different preparations; ns = not significant (p > 0.05), *p < 0.05; **p < 0.01; statistical significance was tested by two tailed t-test (normally distributed values) or Mann–Whitney tests (non-normally distributed values). Individual values are indicated (circles). | ||
In an additional test, we applied OptoBI-1 as a photopharmacological tool to control of neuronal firing. TRPC3 expression and TRPC3 activity levels have been reported to be inversely correlated with the firing of hippocampal neurons in mouse.20 Therefore, we utilized this cell type to test efficiency of our new photoswitch in intact neurons.
The electrical activity of cultured murine hippocampal neurons was monitored under whole-cell current clamp conditions, and firing was induced by repetitive depolarizing current injections (5 s, up to 100 pA) in the presence of OptoBI-1 (20 μM). The cyclic, light-induced photoisomerization of the activator was initiated after a control period to stabilize neuronal activity. UV light-induced cis-isomerization, (30 s) significantly reduced firing during the current injections (Fig. 3c and d). This light-induced suppression of neuronal activity was virtually instantaneous and could be rapidly reverted by trans-isomerization of OptoBI-1 (430 nm), albeit to a level slightly lower than that of the control. Subsequently, it was possible to perform the light-controlled cycling of neuronal firing between constant minimum and maximum levels for more than 5 times without observing apparent desensitization. The results of control experiments run in the absence of OptoBI-1 demonstrated that the alteration in neuronal activity was mediated by OptoBI-1 (ESI Fig. 10a†). The observed inhibition by cis-OptoBI-1, which activates TRPC channels is consistent with the reported inverse correlation of TRPC3 activity and hippocampal firing.20 The mechanism of this connection is still elusive, but may involve local Ca2+ signaling and activation of K+ conductances. The fact that a complete reversal of firing suppression was not achieved upon illumination with blue light (430 nm) may be explained by a high affinity interaction of the cis-isomer. This does not seem to be entirely eliminated by the photoconversion protocol. The dependency of OptoBI-1 effects on TRPC expression in this cell model was shown by the use of hippocampal neurons isolated from a mouse model that lacks all seven canonical TRPC isoforms (TRPC1–7 KO).21,22 The results indicated a tendency of trans-OptoBI-1 to reduce firing in WT neuron, which might be explained by its weak, partial agonist activity. Nonetheless, these differences were not statistically significant (Fig. 3d). Photoisomerization failed to induce a significant cycling of firing frequency in TRPC1–7 KO neurons (ESI Fig. 10b†), substantiating the conclusion that optical control was indeed achieved by interference with hippocampal TRPC activity.
Conclusions
Collectively our results obtained in experiments with vascular endothelial and neuronal cells confirm the value of OptoBI-1 as an effective photoswitch that can be utilized to control of TRPC3 activity. We have demonstrated that OptoBI-1 is suitable for the precise temporal manipulation of endogenously expressed TRPC-mediated signaling processes in human vascular endothelial cells and murine hippocampal neurons. This novel tool is expected to help researches gain a better understanding of TRPC physiopathology and to develop novel therapeutic strategies that target these ion channels in native tissues.Authors contributions
O. T., N. S., T. G. and K. G. wrote the manuscript; O. T., N. S., S. L., G. G. d. C., A. G., C. K., C. B., H. K. and T. G. performed experiments; W. F. G., M. F., C. R. and L. B. contributed essential tools and technical expertise; O. T., T. G. and K. G. designed the study.All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Medical University of Graz and Experiments were approved by the Animal Ethics Committee of Austrian Ministry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Michaela Janschitz and Patrik Wiedner for their excellent technical assistance. Oleksandra Tiapko, Niroj Shrestha and Christiane Klec are members of the PhD program (DK) “Metabolic and Cardiovascular Disease” (FWF W1226-B18 to KG and WFG). The work was supported by the FWF (P28243 to TG, P27263 and P26067 to CR, P28179 to HK) as well as the BMWFW HSRSM (PromOpt2.0 to CR and KG). Additional support came from the Intramural Research Program of the NIH (Project Z01-ES-101684 to LB) and from the Transregional Collaborative Research Centre (TR-SFB) 152 (MF), Collaborative Research Centre (SFB) 1118 (MF) and the DZHK (German Centre for Cardiovascular Research) (MF), the BMBF (German Ministry of Education and Research) (MF).Notes and references
- B. Nilius and V. Flockerzi, Handb. Exp. Pharmacol., 2014, 223, v–vi Search PubMed.
- V. Flockerzi and B. Nilius, Handb. Exp. Pharmacol., 2014, 222, 1–12 CrossRef CAS PubMed.
- P. Eder and K. Groschner, Channels, 2008, 2, 94–99 CrossRef PubMed.
- T. Leinders-Zufall, U. Storch, K. Bleymehl, M. M. Y. Schnitzler, J. A. Frank, D. B. Konrad, D. Trauner, T. Gudermann and F. Zufall, Cell Chem. Biol., 2017, 25, 1–13 Search PubMed.
- M. Lichtenegger, O. Tiapko, B. Svobodova, T. Stockner, T. N. Glasnov, W. Schreibmayer, D. Platzer, G. G. Cruz, S. Krenn, R. Schober, N. Shrestha, R. Schindl, C. Romanin and K. Groschner, Nat. Chem. Biol., 2018, 14, 1–17 CrossRef PubMed.
- O. Tiapko, B. Bacsa, G. G. de la Cruz, T. Glasnov and K. Groschner, Sci. China: Life Sci., 2016, 59, 802–810 CrossRef CAS PubMed.
- S. Kiyonaka, K. Kato, M. Nishida, K. Mio, T. Numaga, Y. Sawaguchi, T. Yoshida, M. Wakamori, E. Mori, T. Numata, M. Ishii, H. Takemoto, A. Ojida, K. Watanabe, A. Uemura, H. Kurose, T. Morii, T. Kobayashi, Y. Sato, C. Sato, I. Hamachi and Y. Mori, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5400–5405 CrossRef CAS.
- D. G. Washburn, D. A. Holt, J. Dodson, J. J. McAtee, L. R. Terrell, L. Barton, S. Manns, A. Waszkiewicz, C. Pritchard, D. J. Gillie, D. M. Morrow, E. A. Davenport, I. M. Lozinskaya, J. Guss, J. B. Basilla, L. K. Negron, M. Klein, R. N. Willette, R. E. Fries, T. C. Jensen, X. Xu, C. G. Schnackenberg and J. P. Marino Jr, Bioorg. Med. Chem. Lett., 2013, 23, 4979–4984 CrossRef CAS PubMed.
- G. Guedes de la Cruz, B. Svobodova, M. Lichtenegger, O. Tiapko, K. Groschner and T. Glasnov, Synlett, 2017, 28, 695–700 CAS.
- B. Doleschal, U. Primessnig, G. Wolkart, S. Wolf, M. Schernthaner, M. Lichtenegger, T. N. Glasnov, C. O. Kappe, B. Mayer, G. Antoons, F. Heinzel, M. Poteser and K. Groschner, Cardiovasc. Res., 2015, 106, 163–173 CrossRef CAS.
- O. Moran, M. Nizzari and F. Conti, FEBS Lett., 2000, 473, 132–134 CrossRef CAS PubMed.
- M.-Y. Amarouch, N. Syam and H. Abriel, Neurosci. Lett., 2013, 541, 105–110 CrossRef CAS PubMed.
- K. Groschner, S. Hingel, B. Lintschinger, M. Balzer, C. Romanin, X. Zhu and W. Schreibmayer, FEBS Lett., 1998, 437, 101–106 CrossRef CAS.
- C. Fan, W. Choi, W. Sun, J. Du and W. Lu, eLife, 2018, 7, e36852 CrossRef PubMed.
- M. Lichtenegger, O. Tiapko, B. Svobodova, T. Stockner, T. N. Glasnov, W. Schreibmayer, D. Platzer, G. G. Cruz, S. Krenn, R. Schober, N. Shrestha, R. Schindl, C. Romanin and K. Groschner, Nat. Chem. Biol., 2018, 14, 1–9 CrossRef.
- Q. Tang, W. Guo, L. Zheng, J.-X. Wu, M. Liu, X. Zhou, X. Zhang and L. Chen, Cell Res., 2018, 27, 1–10 Search PubMed.
- M. Kamouchi, S. Philipp, V. Flockerzi, U. Wissenbach, A. Mamin, L. Raeymaekers, J. Eggermont, G. Droogmans and B. Nilius, J. Physiol., 1999, 2, 345–358 CrossRef.
- M. Poteser, A. Graziani, C. Rosker, P. Eder, I. Derler, H. Kahr, M. X. Zhu, C. Romanin and K. Groschner, J. Biol. Chem., 2006, 281, 13588–13595 CrossRef CAS PubMed.
- A. Graziani, M. Poteser, W.-M. Heupel, H. Schleifer, M. Krenn, D. Drenckhahn, C. Romanin, W. Baumgartner and K. Groschner, J. Biol. Chem., 2010, 285, 4213–4223 CrossRef CAS.
- S. M. Neuner, L. A. Wilmott, K. A. Hope, B. Hoffmann, J. A. Chong, J. Abramowitz, L. Birnbaumer, K. M. O'Connell, A. K. Tryba, A. S. Greene, C. Savio Chan and C. C. Kaczorowski, Behav. Brain Res., 2015, 281, 69–77 CrossRef CAS.
- L. Birnbaumer, J. Mol. Med., 2015, 93, 941–953 CrossRef CAS.
- A. Lutas, L. Birnbaumer and G. Yellen, J. Neurosci, 2014, 34, 16336–16347 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05536j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |