
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Bromo-6-(chlorodiisopropylsilyl)phenyl tosylate as an efficient platform for intramolecular benzyne–diene [4 + 2] cycloaddition†
Arata
Nishii
a,
Hiroshi
Takikawa
 b and
Keisuke
Suzuki
*a
b and
Keisuke
Suzuki
*a
aDepartment of Chemistry, Tokyo Institute of Technology, 2-12-1 O-okayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: ksuzuki@chem.titech.ac.jp
bGraduate School of Pharmaceutical Sciences, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 6th March 2019
Abstract
An intramolecular benzyne–diene [4 + 2] cycloaddition with broad substrate scope has been realized by using a cleavable silicon tether, allowing access to various polycyclic structures. 2-Bromo-6-(chlorodiisopropylsilyl)phenyl tosylate serves as an efficient platform for (1) rapid attachment of various arynophiles to the benzyne precursor via a Si–O bond and (2) facile generation of benzyne via halogen–metal exchange with Ph3MgLi.
Introduction
Benzyne is a highly reactive species with a distorted formal triple bond.1,2 The frontier orbital is the unusually low-lying LUMO, that is the origin of four representative reactivities, (1) [4 + 2] cycloaddition, (2) [2 + 2] cycloaddition, (3) ene reaction, and (4) addition of a nucleophile. Among these, the [4 + 2] cycloaddition is useful for constructing benzo-fused six-membered rings embedded in various natural/unnatural products.2h–o However, two major problems have stymied further advances in synthetic applications. (1) Periselectivity: the [4 + 2] reactions, in particular of carbocyclic dienes or acyclic dienes, are often accompanied by competing [2 + 2] and ene reactions (Fig. 1a).3 (2) Regioselectivity: the formation and the ratio of two potential regioisomers depend on the steric and electronic effects of substituents A and B (Fig. 1b). Moreover, an effective frontier orbital interaction is essential for a successful [4 + 2] cycloaddition. Hence the diene component usually requires a high-lying HOMO and a constrained s-cis geometry, which are typically provided by a cyclic framework while limiting the scope of this reaction.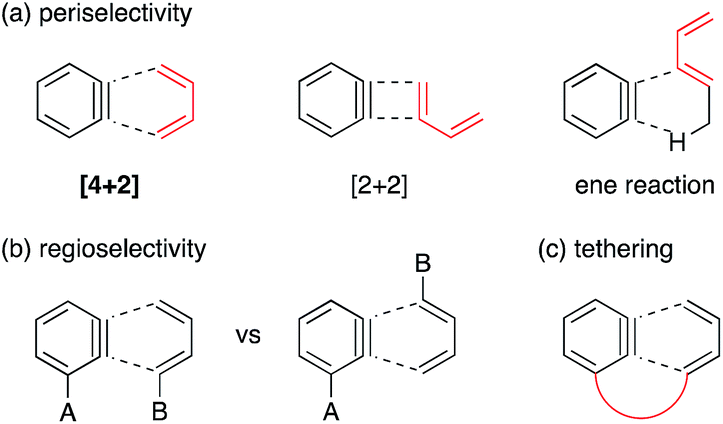 | ||
| Fig. 1 Peri- and regioselectivity of the benzyne reaction with dienes. | ||
One of the solutions to overcome these issues is to exploit the intramolecular reaction by tethering the benzyne progenitors to the dienes (Fig. 1c and Scheme 1).4 While furans are the most commonly used arynophiles for the intramolecular [4 + 2] cycloaddition as initially demonstrated by Wege,4a reactions with carbocyclic or acyclic dienes are rare. An early study by Buszek4c,d exemplifies the carbon tether for the benzyne–diene cycloaddition (Scheme 1a), which has later been investigated by Smith with the union of the anion relay chemistry (Scheme 1b).4g Danheiser reported an efficient intramolecular [4 + 2] cycloaddition of a benzyne with an acyclic diene by employing a nitrogen tether (Scheme 1c).4f Concerning a disposable linker,5 Martin pioneered the application of the silicon tether, connecting the benzyne precursor with the arynophile via a Si–C bond.4e However, furans were the only arynophiles used in the study and the method was elegantly exploited in the syntheses of various aryl C-glycoside natural products (Scheme 1d).6
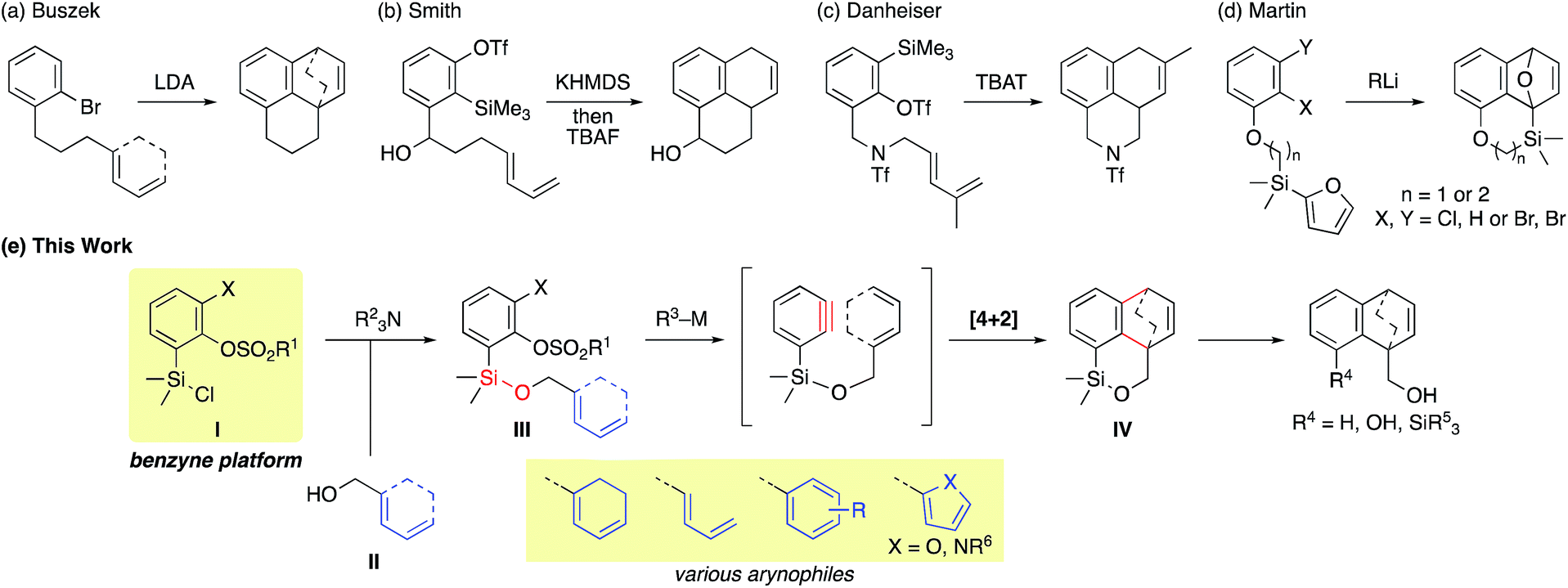 | ||
| Scheme 1 Overview of the reported intramolecular [4 + 2] cycloaddition of benzyne with dienes (top) and our strategy (bottom). | ||
In this context, we have focused our efforts on developing a viable strategy to further expand the utility of the intramolecular benzyne–diene [4 + 2] cycloaddition. Herein, we report a practical, robust implementation of this reaction by exploiting arylsilyl chloride I as a novel benzyne platform (Scheme 1e). Arylsilyl chloride I having a silicon atom directly attached to the benzyne precursor allows a facile assembly with the diene partner II having a pendant hydroxy group via a Si–O linkage under mild conditions to give cycloaddition precursor III. This strategy provides the following advantages: (1) the vicinal halogen–sulfonate functionalities of compound III allow facile benzyne generation initiated by a halogen–metal exchange4b,h,i,7 followed by the intramolecular cycloaddition. (2) The broad substrate scope resulting from various combinations of I and II gives useful cycloadduct IV that is amenable to numerous potential elaborations.
Results and discussion
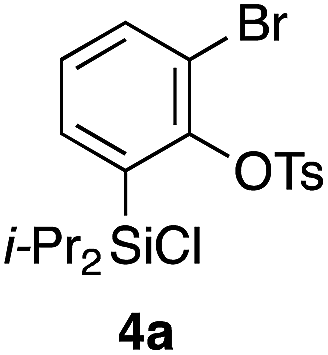
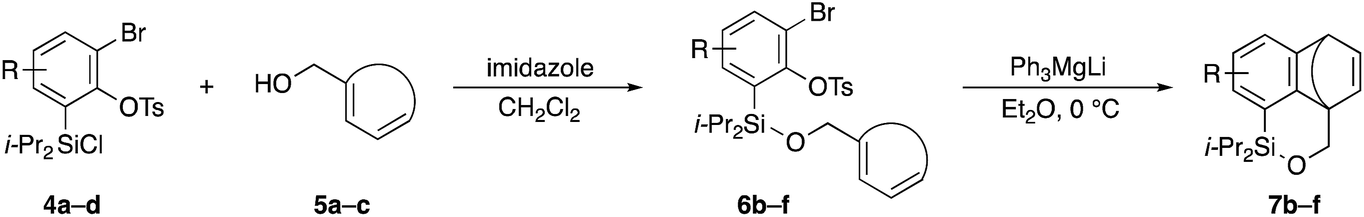
Scheme 2 shows the preparation of the model substrate 6a. 2,6-Dibromophenol (1a) was silylated with diisopropylsilyl chloride to give the corresponding silyl ether, which was treated with n-BuLi to induce a retro-Brook rearrangement8 to give o-silyl phenol 2a in 87% yield. Treatment of phenol 2a with TsCl (K2CO3, acetone, 0 °C → RT, 1.5 h) gave tosylate 3a in excellent yield,9 which reacted with trichloroisocyanuric acid (TCCA), giving silyl chloride 4a in quantitative yield.10 It turned out that 4a, once obtained as a white solid, was stable enough for storage over several months (at −15 °C).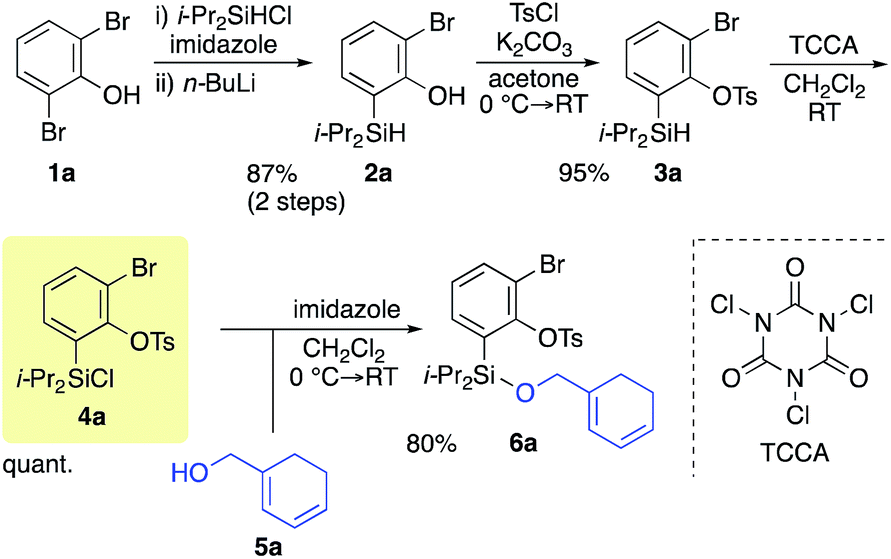 | ||
| Scheme 2 Preparation of the benzyne platform 4a and union with alcohol 5a. | ||
Silyl chloride 4a, thus obtained, served as a platform to combine various dienyl alcohols to give the cycloaddition precursors, as illustrated by the preparation of the model substrate 6a. In the presence of imidazole, 4a was combined with alcohol 5a having a cyclohexadienyl moiety to give silyl ether 6a in 80% yield.
Having 6a in hand, the intramolecular [4 + 2] cycloaddition was examined (Table 1). As the initial experiment, 6a was treated with n-BuLi (THF, 0 °C, 20 min), where the desired cycloadduct 7a was obtained in 53% yield (entry 1). The starting material 6a was recovered in 5% yield, and a side product 8 was obtained in ca. 8% yield, arising from the addition of a butyl anion to the benzyne followed by the protonation or Br–Li exchange reaction of the resulting aryl anion. Notably, no other byproducts, derived from potential side reactions such as the [2 + 2] cycloaddition and ene reaction, were detected, which is in sharp contrast to the corresponding intermolecular version.3,11 Concerning other organolithium reagents, s-BuLi gave lower yield of 7a (entry 2), while PhLi gave comparable results (entry 3). We turned our attention to organomagnesium reagents. The initial attempt with i-PrMgCl12 led to a slow reaction even at room temperature (entry 4). Pleasingly, Ph3MgLi improved the yield of 7a to 69% yield (entry 5).13,14 Among other solvents tested (entries 6–9), Et2O proved to be the solvent of choice, giving 7a in 87% yield (entry 6).
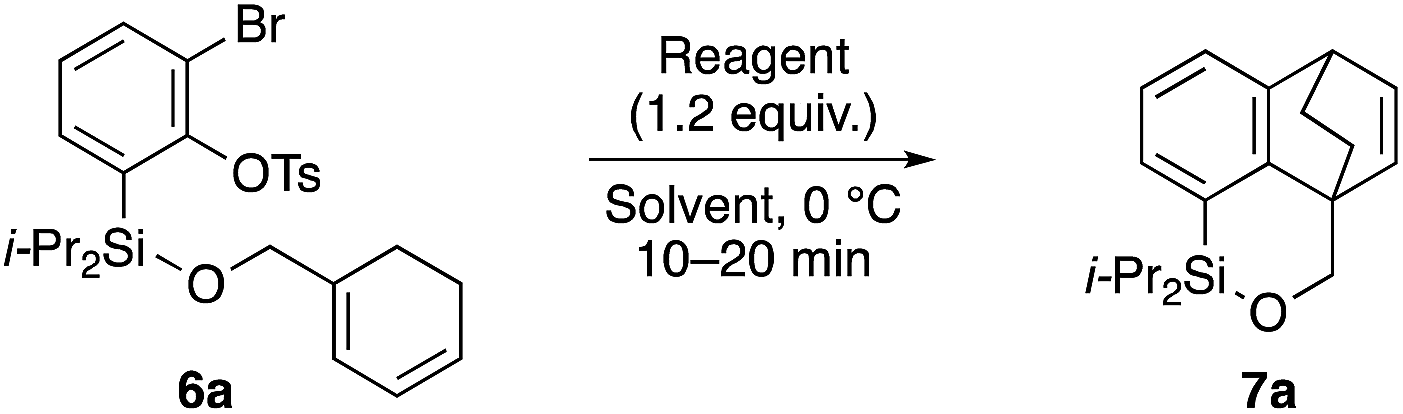
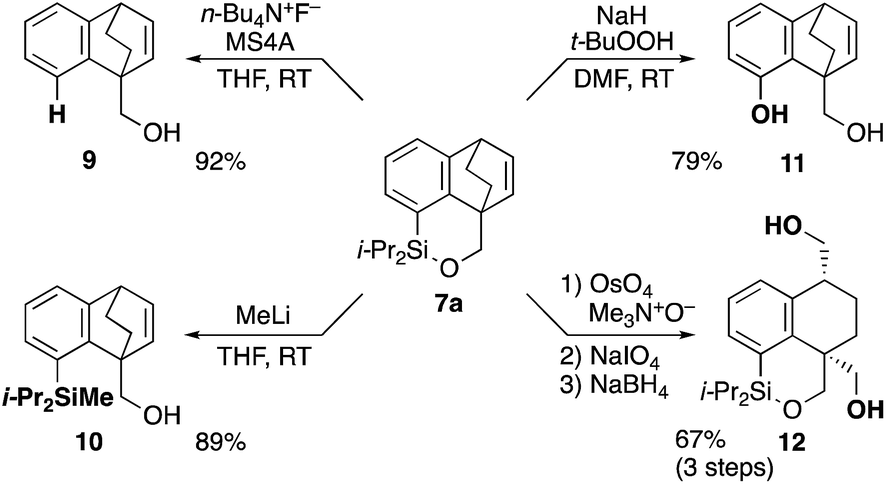
The synthetic utility of cycloadduct 7a was demonstrated by several orthogonal transformations (Scheme 3). The silyl group on 7a provided an opportunity for further elaboration of the aromatic ring. Indeed, by treating with tetrabutylammonium fluoride in the presence of 4 A molecular sieves, both the silicon–oxygen and silicon–carbon bonds in 7a were cleaved, giving alcohol 9 in 92% yield. By contrast, methyllithium cleaved only the silicon–oxygen bond to afford alcohol 10 in 89% yield. Tamao–Fleming oxidation15 of 7a by treatment with t-BuOOH and NaH gave phenol 11 in 79% yield. In addition, oxidative cleavage of the bridged double bond in 7a followed by reduction gave diol 12.
 | ||
| Scheme 3 Synthetic transformations. | ||
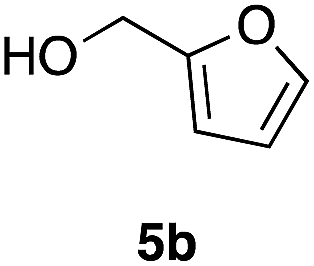
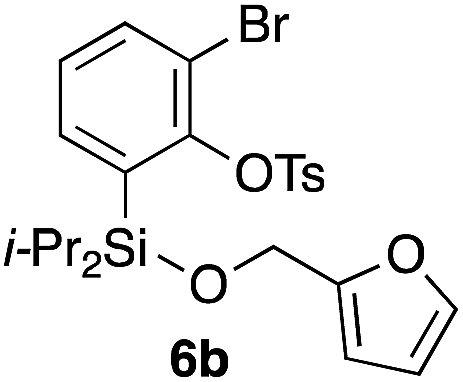
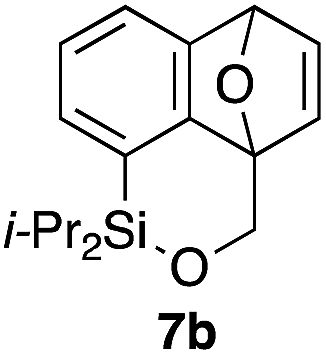
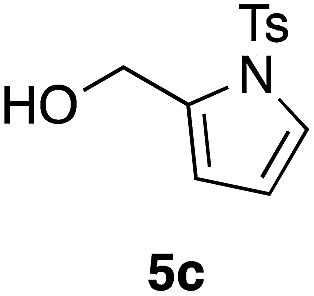
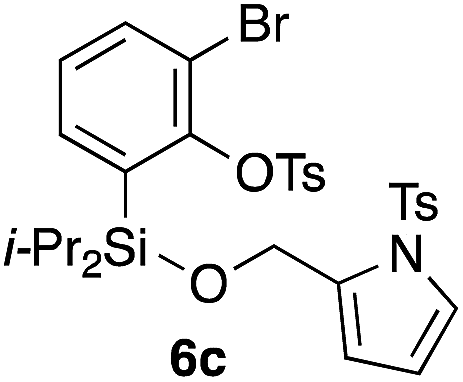
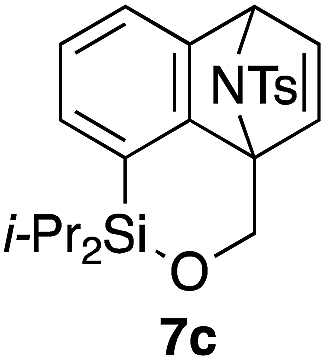
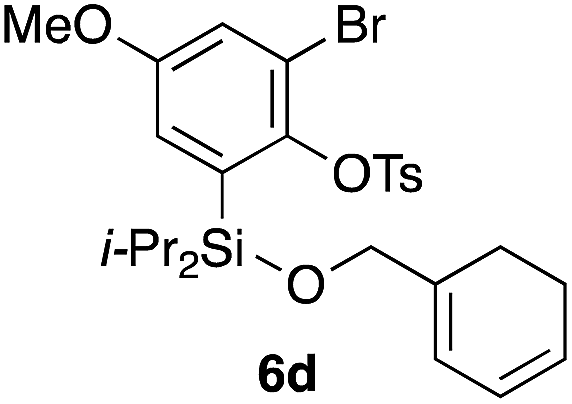
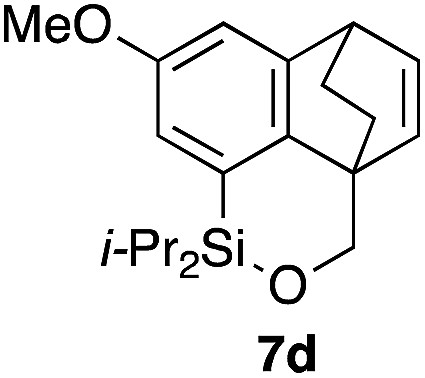
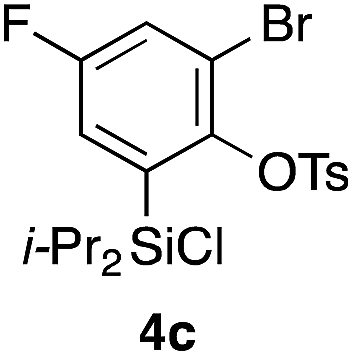
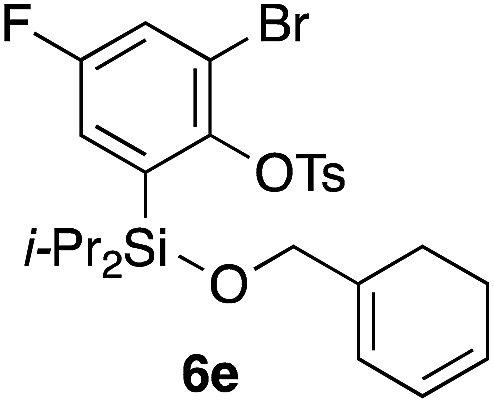
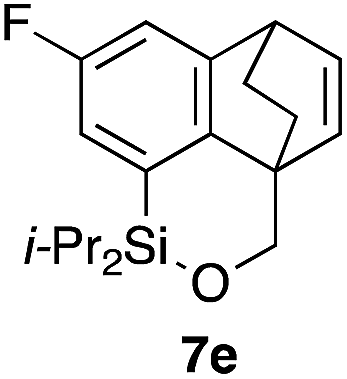
Table 2 shows the scope of this tethering strategy. The combination of 4a with furfuryl alcohol (5b) and pyrrolylmethanol 5c gave precursors 6b and 6c in high yields, which underwent the intramolecular [4 + 2] cycloaddition (Ph3MgLi, Et2O, 0 °C, 10 min) to give cycloadducts 7b and 7c in 90% and 84% yield, respectively (entries 1 and 2). Entries 3–5 show variation of the aryl moiety, using the precursors 6d–f having a methoxy or a fluoro substituent(s), prepared from chlorosilanes 4b–d and alcohol 5a. The reactions of 6d and 6e, bearing a methoxy and a fluoro group at the meta position to the respective silyl substituents, proceeded smoothly to give cycloadducts 7d and 7e both in high yield (entries 3 and 4). The precursor 6f, the ortho, para-dimethoxy derivative, also underwent a clean cycloaddition to give the desired product 7f in excellent yield (entry 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Silyl chloride 4 | Alcohol 5 | Precursor 6 | Yieldb (%) | Cycloadduct 7 | Yieldc (%) |
| a Conditions for the preparation of precursors 6b–f: see the ESI. Conditions of cycloadditions: precursor 6 (1.0 equiv.) and Ph3MgLi (1.2 equiv.) in Et2O (0.05 M) at 0 °C for 10 min. b Isolated yield. c Isolated yield from 6. d Overall yield for two steps from the corresponding arylhydrosilane: chlorination and O-silylation. | ||||||
| 1 |
|
|
|
82 |
|
90 |
| 2 | 4a |
|
|
81 |
|
84 |
| 3 |
|
|
|
83 |
|
78 |
| 4 |
|
5a |
|
82d |
|
82 |
| 5 |
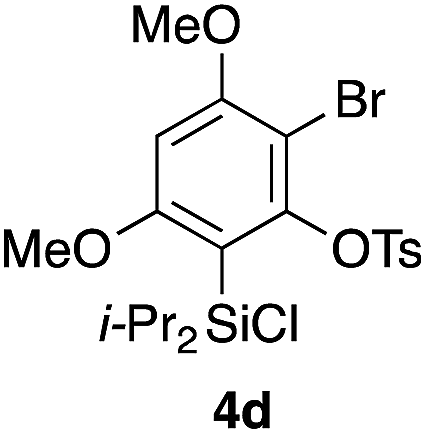
|
5a |
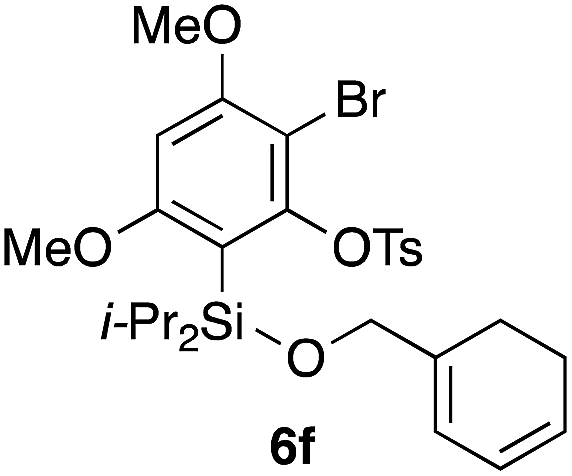
|
59d |
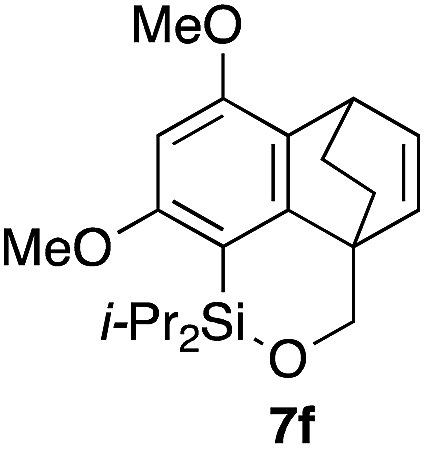
|
86 |
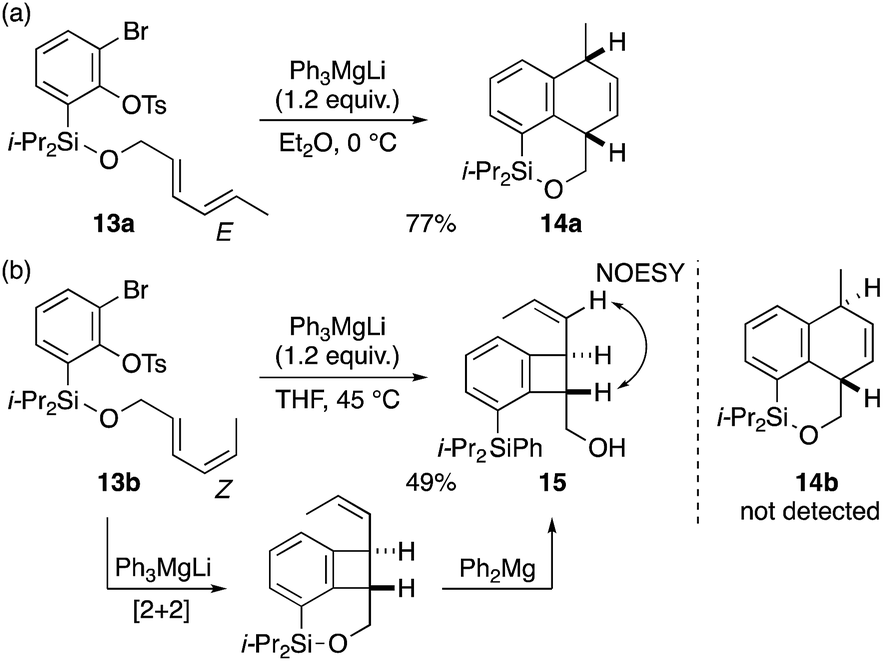
With excellent results obtained with cyclic dienyl arynophiles as discussed above, we further examined the applicability to the substrates with an acyclic diene, often less reactive than the cyclic counterpart,3c,f,4c,f,g,16 which gave interesting results (Scheme 4). When the E,E-diene substrate 13a was treated with Ph3MgLi at 0 °C (Et2O, 10 min), a clean [4 + 2] cycloaddition occurred to give dihydronaphthalene 14a in 77% yield (Scheme 4a). By contrast, the reaction of the E,Z-congener 13b (THF, 45 °C, 10 min) gave no [4 + 2] cycloadduct 14b, but instead, the [2 + 2] cycloadduct 15, albeit in 49% yield, which could be rationalized by a cycloaddition followed by Si–O bond scission by Ph2Mg:17 a remarkable finding given that, the regioselective [2 + 2] cycloaddition of an internal alkene in 1,3-dienes is less common (Scheme 4b).3c–g The relative trans configuration was verified by 1H–1H NOESY experiments.
 | ||
| Scheme 4 Reaction with acyclic dienes. | ||
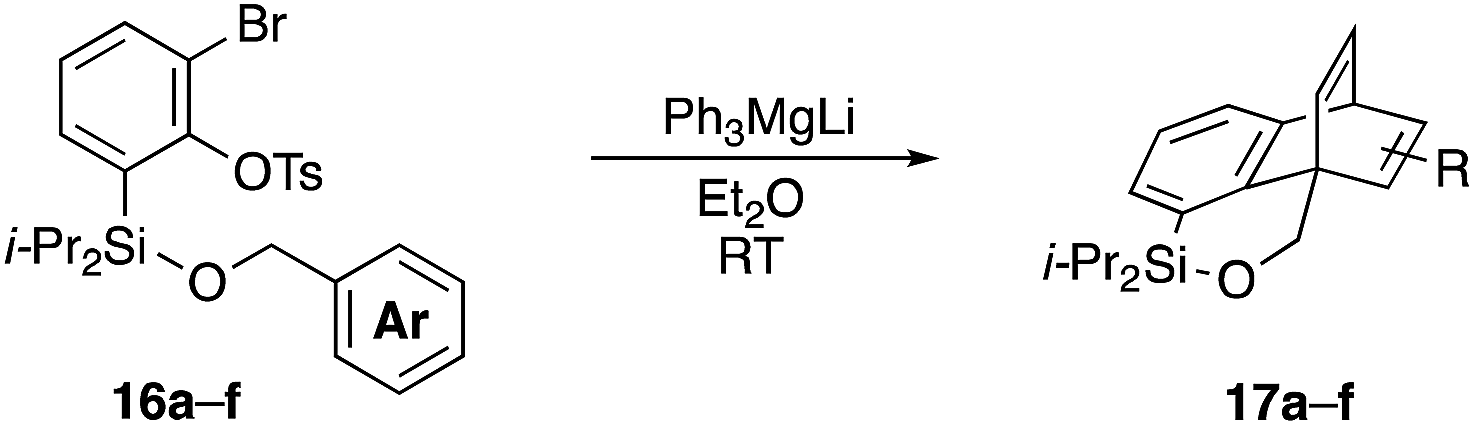
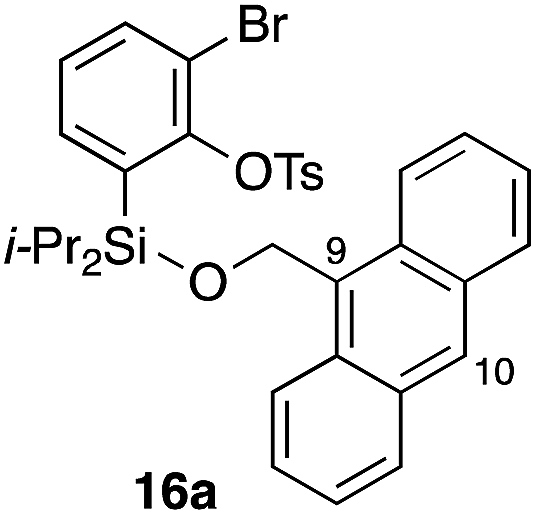
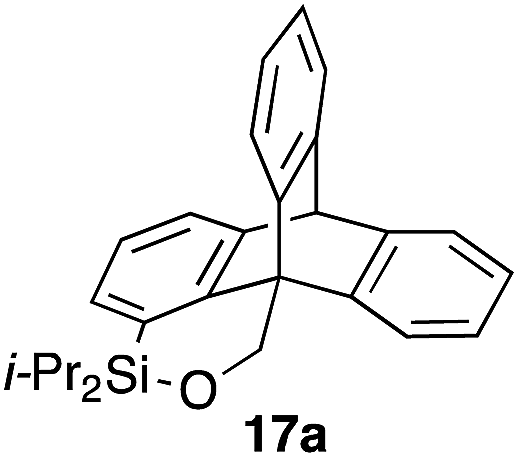
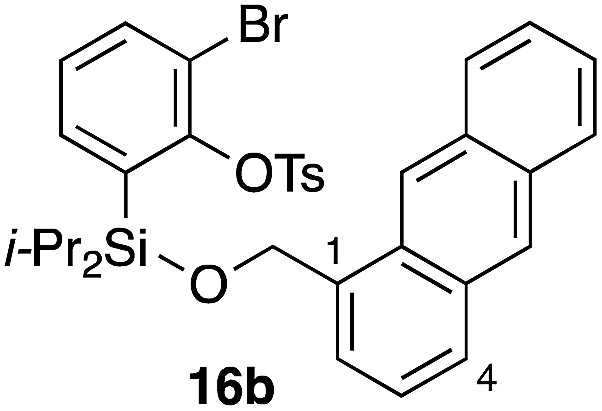
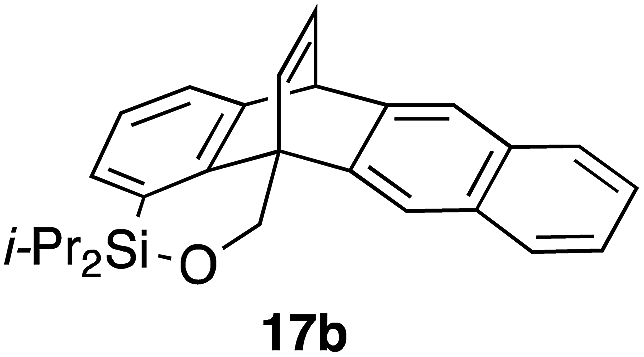
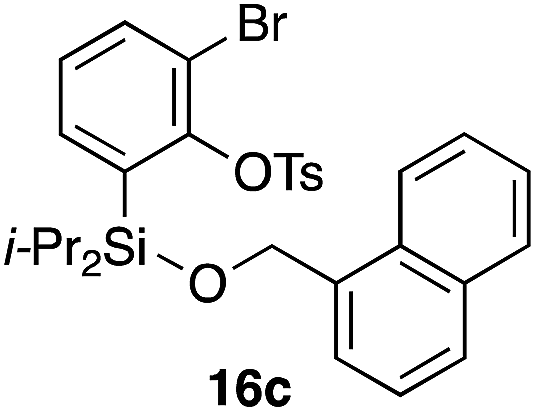
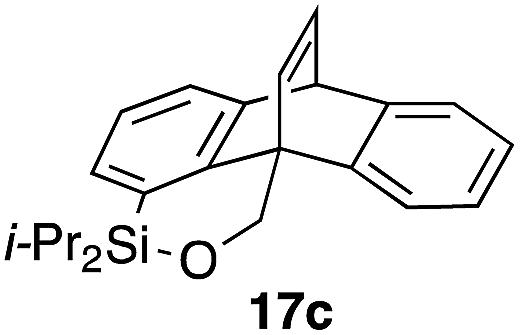
Furthermore, we addressed the applicability to the synthesis of barrelenes, an attractive class of bicyclic compounds.18 The question was whether or not the substrates having an internal arene moiety undergo the [4 + 2] cycloadditions with dearomatization, and the results are summarized in Table 3. Entries 1 and 2 show the cycloaddition reactions of bromoaryl tosylates 16a and 16b, whose anthracene moieties are tethered at the C9 and the C1 positions, respectively. Reaction of 16a gave the corresponding triptycene 17a in 64% yield (entry 1). By contrast, the precursor 16b underwent clean cycloaddition at the 1,4-position of the anthracene ring to afford naphthobarrelene 17b in 65% yield (entry 2). Note that here the C1 position of the anthracene served as the anchor for the tether, and therefore the benzyne reacted with the outer ring (1,4-position), despite the higher reactivity of the central ring (9,10-position).19 The [4 + 2] cycloaddition of naphthalene as a diene is more challenging than that of anthracene due to higher aromaticity.20 Nonetheless the reaction of precursor 16c was realized at an elevated temperature (40 °C), giving dibenzobarrelene 17c in 51% yield (entry 3).
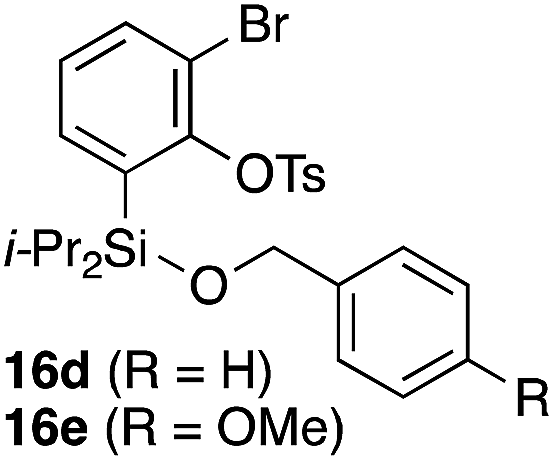
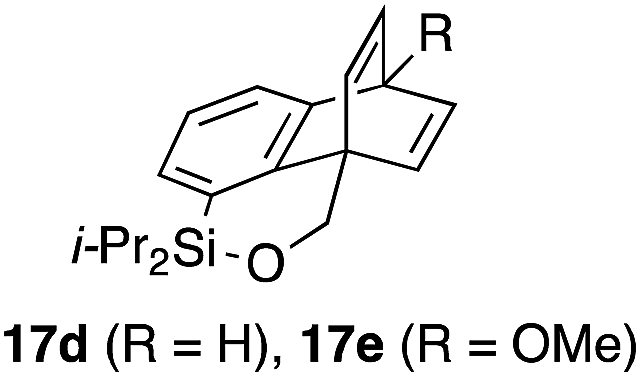
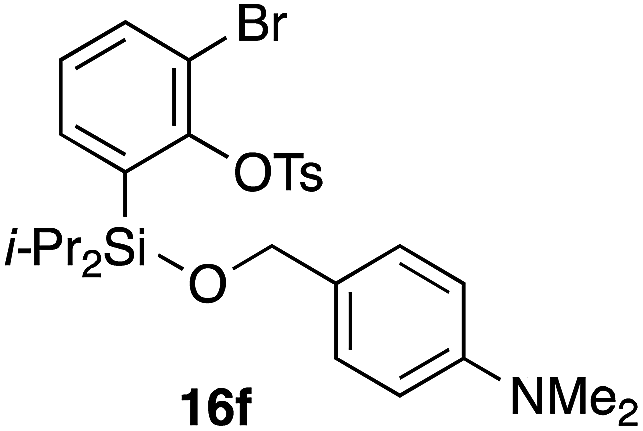
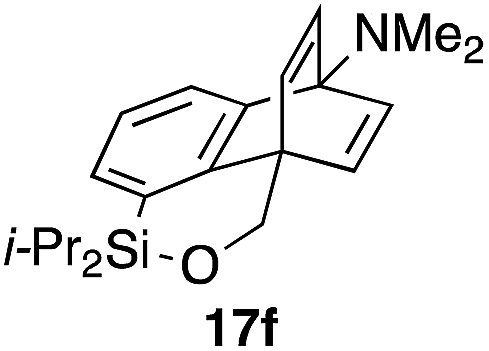
To explore the limits of this dearomatization, simple benzenes were examined as aromatic diene components (entries 4–6).21 The reaction of tosylate 16d with a simple benzene ring resulted in failure, giving no cycloadduct 17d (entry 4). With a hope of realizing the reaction by increasing the HOMO level of the benzene ring,21d,22 we examined substrates 16e and 16f, having a methoxy group and a dimethylamino group, respectively. It turned out that 16e gave no reaction (entry 5), whereas the dimethylamino group worked well for the cycloaddition to give benzobarrelene 17f in 44% yield (entry 6).
Conclusions
In summary, we have described a silicon-tether strategy for the intramolecular [4 + 2] cycloaddition of benzynes with dienes. 2-Bromo-6-(chlorodiisopropylsilyl)phenyl tosylate, a versatile benzyne platform, plays a key role in the facile assembly of various cycloaddition precursors by combining with dienes via a Si–O bond. This strategy would be applicable to various other potential arynophiles, and useful for complex polycyclic natural product syntheses. Further studies along these lines are in progress.Experimental
A typical procedure for the internal cycloaddition is described for the reaction of 6a with Ph3MgLi in Et2O (entry 6): To a solution of PhLi (0.67 M in cyclohexane and Et2O, 1.1 mL, 0.74 mmol) in Et2O (1.5 mL) was added PhMgBr (0.94 M in THF, 0.41 mL, 0.39 mmol) at 0 °C, and the mixture was stirred for 30 min. The resulting solution of Ph3MgLi was used in the following experiment. To a solution of bromoaryl tosylate 6a (165 mg, 0.300 mmol) in Et2O (6.0 mL) was added dropwise Ph3MgLi (vide supra) via cannula at 0 °C. After stirring for 10 min, the reaction was stopped by adding sat. NH4Cl aq., and the mixture was extracted with EtOAc (x3). The combined organic layer was washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was purified by preparative thin layer chromatography (silica gel, hexane, EtOAc = 98/2, x2) to afford cycloadduct 7a (77.8 mg, 87%) as a colorless oil.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Mark M. Maturi for careful proofreading. This work was supported by the Grant-in-Aid from Japan Society for the Promotion of Science (JSPS) (Grant No. JP16H06351 and JP18K06548).Notes and references
- R. W. Hoffmann, Dehydrobenzene and Cycloalkynes, Academic Press, New York, 1967 Search PubMed.
- For the selected recent reviews on aryne, see: (a) H. H. Wenk, M. Winkler and W. Sander, Angew. Chem., Int. Ed., 2003, 42, 502–528 CrossRef CAS PubMed; (b) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215–291 CrossRef CAS; (c) T. Kitamura, Aust. J. Chem., 2010, 63, 987–1001 CrossRef CAS; (d) C. Wentrup, Aust. J. Chem., 2010, 63, 979–986 CrossRef CAS; (e) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC; (f) S. Yoshida and T. Hosoya, Chem. Lett., 2015, 44, 1450–1460 CrossRef CAS; (g) J. Shi, Y. Li and Y. Li, Chem. Soc. Rev., 2017, 46, 1707–1719 RSC; (h) H. Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701–730 CrossRef CAS; (i) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS PubMed; (j) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 CrossRef CAS PubMed; (k) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 CAS; (l) A. E. Goetz, T. K. Shah and N. K. Garg, Chem. Commun., 2015, 51, 34–45 CAS; (m) J.-A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC; (n) T. Roy and A. T. Biju, Chem. Commun., 2018, 54, 2580–2594 CAS; (o) H. Takikawa, A. Nishii, T. Sakai and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030–8056 RSC.
- (a) H. E. Simmons, J. Am. Chem. Soc., 1961, 83, 1657–1664 CrossRef CAS; (b) R. Huisgen and R. Knorr, Tetrahedron Lett., 1963, 4, 1017–1021 CrossRef; (c) G. Wittig and H. Dürr, Justus Liebigs Ann. Chem., 1964, 672, 55–62 CrossRef CAS; (d) M. Jones Jr and R. H. Levin, J. Am. Chem. Soc., 1969, 91, 6411–6415 CrossRef; (e) P. Crews and J. Beard, J. Org. Chem., 1973, 38, 522–528 CrossRef CAS; (f) P. Crews and J. Beard, J. Org. Chem., 1973, 38, 529–532 CrossRef CAS; (g) E. E. Waali, J. Org. Chem., 1975, 40, 1355–1356 CrossRef CAS; (h) I. F. Eckhard, H. Heaney and B. A. Marples, J. Chem. Soc., 1969, 2098–2104 CAS; (i) D. Niu and T. R. Hoye, Nat. Chem., 2014, 6, 34–40 CrossRef CAS PubMed.
- (a) W. M. Best and D. Wege, Tetrahedron Lett., 1981, 22, 4877–4880 CrossRef CAS; (b) W. H. Darlington and J. Szmuszkovicz, Tetrahedron Lett., 1988, 29, 1883–1886 CAS; (c) K. R. Buszek, Tetrahedron Lett., 1995, 36, 9125–9128 CrossRef CAS; (d) K. R. Buszek and D. L. Bixby, Tetrahedron Lett., 1995, 36, 9129–9132 CrossRef CAS; (e) D. E. Kaelin Jr, S. M. Sparks, H. R. Plake and S. F. Martin, J. Am. Chem. Soc., 2003, 125, 12994–12995 CrossRef PubMed; (f) M. E. Hayes, H. Shinokubo and R. L. Danheiser, Org. Lett., 2005, 7, 3917–3920 CrossRef CAS PubMed; (g) A. B. Smith III and W.-S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6787–6792 CrossRef PubMed; (h) S. Yoshida, K. Shimizu, K. Uchida, Y. Hazama, K. Igawa, K. Tomooka and T. Hosoya, Chem.–Eur. J., 2017, 23, 15332–15335 CrossRef CAS PubMed; (i) S. Prévost, A. Dezaire and A. Escargueil, J. Org. Chem., 2018, 83, 4871–4881 CrossRef PubMed.
- (a) H. Nishiyama, T. Kitajima, M. Matsumoto and K. Itoh, J. Org. Chem., 1984, 49, 2298–2300 CrossRef CAS; (b) G. Stork and M. Kahn, J. Am. Chem. Soc., 1985, 107, 500–501 CrossRef CAS; (c) D. Craig and J. C. Reader, Tetrahedron Lett., 1990, 31, 6585–6588 CAS; (d) G. Stork, T. Y. Chan and G. A. Breault, J. Am. Chem. Soc., 1992, 114, 7578–7579 CAS ; for reviews, see:; (e) M. Bols and T. Skrydstrup, Chem. Rev., 1995, 95, 1253–1277 CrossRef CAS; (f) L. Fensterbank, M. Malacria and S. M. Sieburth, Synthesis, 1997, 813–854 CrossRef CAS; (g) D. R. Gauthier Jr, K. S. Zandi and K. J. Shea, Tetrahedron, 1998, 54, 2289–2338 CrossRef; (h) M. Parasram and V. Gevorgyan, Acc. Chem. Res., 2017, 50, 2038–2053 CrossRef CAS PubMed.
- (a) C.-L. Chen, S. M. Sparks and S. F. Martin, J. Am. Chem. Soc., 2006, 128, 13696–13697 CAS; (b) S. M. Sparks, C.-L. Chen and S. F. Martin, Tetrahedron, 2007, 63, 8619–8635 CrossRef CAS PubMed; (c) K. J. Procko, H. Li and S. F. Martin, Org. Lett., 2010, 12, 5632–5635 CrossRef CAS PubMed; (d) B. M. O'Keefe, D. M. Mans, D. E. Kaelin Jr and S. F. Martin, J. Am. Chem. Soc., 2010, 132, 15528–15530 CrossRef PubMed; (e) B. M. O'Keefe, D. M. Mans, D. E. Kaelin Jr and S. F. Martin, Tetrahedron, 2011, 67, 6524–6538 CrossRef PubMed.
- (a) W. Tochtermann, G. Stubenrauch, K. Reiff and U. Schumacher, Chem. Ber., 1974, 107, 3340–3352 CrossRef CAS; (b) T. Matsumoto, T. Hosoya, M. Katsuki and K. Suzuki, Tetrahedron Lett., 1991, 32, 6735–6736 CrossRef CAS.
- (a) J. L. Speier, J. Am. Chem. Soc., 1952, 74, 1003–1010 CrossRef CAS; (b) G. Simchen and J. Pfletschinger, Angew. Chem., Int. Ed., 1976, 15, 428–429 CrossRef; (c) D. Peña, A. Cobas, D. Pérez and E. Guitián, Synthesis, 2002, 1454–1458 Search PubMed.
- As a leaving group, we revealed that the tosyloxy group gave a better result than the triflyloxy group by an optimization process. For the difference of benzyne generation effected by the leaving group of halo-sulfonates, see: T. Hamura, T. Arisawa, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2006, 45, 6842–6844 CAS.
- (a) J. A. Tallarico, K. M. Depew, H. E. Pelish, N. J. Westwood, C. W. Lindsley, M. D. Shair, S. L. Schreiber and M. A. Foley, J. Comb. Chem., 2001, 3, 312–318 CrossRef CAS PubMed; (b) S. Varaprath and D. H. Stutts, J. Organomet. Chem., 2007, 692, 1892–1897 CrossRef CAS; (c) C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2009, 131, 10844–10845 CrossRef CAS; (d) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed.
- For comparison of the intra- and intermolecular reactions, the reaction of 2-bromophenyl tosylate with cyclohexadiene was attempted (n-BuLi, THF, 0 °C), giving no [4 + 2] cycloadduct.
- I. Sapountzis, W. Lin, M. Fischer and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 4364–4366 CrossRef CAS PubMed.
- (a) K. Kitagawa, A. Inoue, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2000, 39, 2481–2483 CrossRef CAS; (b) T. Higuchi, K. Ohmori and K. Suzuki, Chem. Lett., 2006, 35, 1006–1007 CrossRef CAS.
- Other magnesate complex, n-Bu2(i-Pr)MgLi, was not effective, and resulted in a low yield of 7a (28%).
- (a) K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694–1696 CrossRef CAS; (b) K. Tamao, T. Kakui, M. Akita, T. Iwahara, R. Kanatani, J. Yoshida and M. Kumada, Tetrahedron, 1983, 39, 983–990 CrossRef CAS; (c) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29–31 RSC.
- (a) R. R. Schmidt and R. Angerbauer, Angew. Chem., Int. Ed., 1979, 18, 304–305 CrossRef; (b) C. Dockendorff, S. Sahli, M. Olsen, L. Milhau and M. Lautens, J. Am. Chem. Soc., 2005, 127, 15028–15029 CrossRef CAS PubMed.
- A small amount (4%) of the cis isomer was observed, and other detectable products were the corresponding biphenylene, a homodimer of the in situ generated benzyne, and a phenyl adduct of the benzyne.
- (a) H. E. Zimmerman and G. L. Grunewald, J. Am. Chem. Soc., 1966, 88, 183–184 CrossRef CAS; (b) E. Ciganek, J. Am. Chem. Soc., 1966, 88, 2882–2883 CrossRef CAS; (c) H. E. Zimmerman, R. S. Givens and R. M. Pagni, J. Am. Chem. Soc., 1968, 90, 6096–6108 CAS ; for a review, see:; (d) H. E. Zimmerman and D. Armesto, Chem. Rev., 1996, 96, 3065–3112 CrossRef CAS PubMed.
- (a) B. H. Klanderman, J. Am. Chem. Soc., 1965, 87, 4649–4651 CrossRef CAS; (b) Y. Tsuchido, T. Ide, Y. Suzaki and K. Osakada, Bull. Chem. Soc. Jpn., 2015, 88, 821–823 CrossRef CAS.
- (a) R. G. Miller and M. Stiles, J. Am. Chem. Soc., 1963, 85, 1798–1800 CrossRef CAS; (b) P. W. Rabideau, Org. Prep. Proced. Int., 1986, 18, 113–116 CrossRef CAS.
- (a) D. T. Longone and J. A. Gladysz, Tetrahedron Lett., 1976, 4559–4562 CrossRef CAS; (b) W. J. Houlihan, Y. Uike and V. A. Parrino, J. Org. Chem., 1981, 46, 4515–4517 CrossRef CAS; (c) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–211 CrossRef CAS PubMed; (d) V. D. Pogula, T. Wang and T. R. Hoye, Org. Lett., 2015, 17, 856–859 CrossRef CAS PubMed.
- I. Tabushi, H. Yamada, Z. Yoshida and R. Oda, Bull. Chem. Soc. Jpn., 1977, 50, 285–290 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05518a |
| This journal is © The Royal Society of Chemistry 2019 |