
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide-based capsules with chirality-controlled functionalized interiors – rational design and amplification from dynamic combinatorial libraries†
Hanna
Jędrzejewska
 and
Agnieszka
Szumna
*
and
Agnieszka
Szumna
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: agnieszka.szumna@icho.edu.pl
First published on 20th March 2019
Abstract
Peptides are commonly perceived as inapplicable components for construction of porous structures. Due to their flexibility the design is difficult and shape persistence of such putative structures is diminished. Notwithstanding these limitations, the advantages of peptides as building blocks are numerous: they are functional and functionalizable, widely available, diverse and biocompatible. We aimed at the construction of discrete porous structures that exploit the inherent functionality of peptides by an approach that is inspired by nature: structural pockets are defined by the backbones of peptides while functionality is introduced by their side chains. In this work peptide ribbons were preorganized on a macrocyclic scaffold using azapeptide–aldehyde reactions. The resulting cavitands with semicarbazone linkers arrange the peptide backbones at positions that are suitable for self-assembly of dimeric capsules by formation of binding motifs that resemble eight-stranded β-barrels. Self-assembly properties and inside/outside positions of the side chains depend crucially on the chirality of peptides. By rational optimization of successive generations of capsules we have found that azapeptides containing three amino acids in a (L, D, D) sequence give well-defined dimeric capsules with side chains inside their cavities. Taking advantage of the reversibility of the reaction of semicarbazone formation we have also employed the dynamic covalent chemistry (DCC) for a combinatorial discovery of capsules that could not be rationally designed. Indeed, the results show that stable capsules with side chains positioned internally can be obtained even for shorter sequences but only for combination peptides of (L, L) and (D, L) chirality. The hybrid (L, L)(D, L) capsule is amplified directly from a reaction mixture containing two different peptides. All capsules gain substantial ordering upon self-assembly, which is manifested by a two orders of magnitude increase of the intensity of CD spectra of capsules compared with non-assembled analogs. Temperature-dependent CD measurements indicate that the capsules remain stable over the entire temperature range tested (20–100 °C). Circular dichroism coupled with TD DFT calculations, DOSY measurements and X-ray crystallography allow for elucidation of the structures in the solid state and in solution and guide their iterative evolution for the current goals.
Introduction
Proteins, with their variety of biological functions and diversity of structural motifs, constitute an unlimited source of inspiration for scientists in many fields. Unsurpassed models of high binding affinity, selectivity and catalytic efficiency are found among proteins' recognition sites and enzymatic pockets. Such effectiveness comes from the presence of precisely organized confined spaces in proteins' interiors: shape-persistent cavities with well-defined positions of functional groups. Artificial porous materials and molecules that contain cavities also aim at mimicking these structural features for similar purposes (storage, separation and catalysis). The current state-of-the-art techniques in this field allow for the synthesis of a vast variety of porous materials either in the form of discrete capsules1 or as infinite crystalline frameworks (MOFs,2 COFs3 or SOFs4). However, decoration of the cavities with functional groups is still a tall order. Among porous solid materials metal–organic frameworks (MOFs) with functional organic sites,5 MOFs mimicking natural enzymes,6 peptide functionalized MOFs7 and peptide-based porous materials have been reported.8,9 Seminal examples of discrete porous molecules – capsules – that form directional interactions with encapsulated guests were presented by the group of Ballester,10,11 and by our group.12 In these examples functional groups constitute integral parts of the capsules' walls, and therefore they are highly conserved and cannot be modified without affecting the main structural framework.In the current study we aim at developing a more general approach towards functionalization of cavities using nature-inspired building blocks – peptides. Peptides are commonly perceived as inapplicable components for the construction of porous structures due to their flexibility and poorly defined non-covalent interactions. These features reduce shape persistence of the putative porous products and make their design difficult. We have previously shown that, despite flexibility and low predictability, short peptides can be used to form discrete porous structures provided that they are properly preorganized by positioning at semi-rigid scaffolds and stabilized by the formation of β-sheet-like binding motifs.13,14 In this work we develop this concept towards internal functionalization of the cavities. We propose construction of capsules in which structural frameworks are formed by peptides' backbones while the functionality is provided by their side chains, positions of which are determined by chirality. This strategy opens up the possibilities of diverse functionalization using a rich pool of amino acids while retaining the integrity of the main structural framework. Such an approach is closely analogous to the design exploited by nature, which constructs enzymatic/recognition sites using amino acids' side chains, while the shapes of the binding pockets are determined by their backbones. However, our approach also introduces new elements: abiotic macrocyclic fragments (required for preorganization) and peptides of unnatural chirality15 (for internal functionalization). The final structural results of this combination are only vaguely predictable based on structural motifs present in natural proteins. Therefore, in order to allow for both the rational design and an element of serendipity we employ two approaches: (a) a strategy of rational iterative improvement and complexity increase in successive generations of capsules and (b) the dynamic covalent chemistry approach (DCC) to potentially amplify the capsules overlooked by rational design. Our results show that the combination of both approaches produces a unique set of peptide-based porous capsules with functionalized interiors, of which only some could be rationally designed.
Rational design
We have previously reported the synthesis and self-assembly of cavitands composed of peptides of different lengths, sequences and chiralities, which were attached to macrocyclic scaffolds (resorcin[4]arenes) using either methylene bridges16, imines13 or acylhydrazone linkers.14 These semi-open cavitands form non-covalent dimers by hydrogen bonding arranged in β-barrel type binding motifs that involve peptides' backbones, while their side chains are potentially available for other functions. Molecular modelling indicates that imine-based and acylhydrazone-based capsules have enough space inside the cavities to accommodate some side chains; however, all of our experimental attempts to synthesize such capsules have failed resulting in disintegration of the dimers or hydrolysis of the dynamic covalent bonds. We hypothesize that steric crowding due to an acute internal curvature or inappropriate stereochemistry is the limiting factor. Indeed the internal diameters of such artificial capsules are in the range 13.1–14.7 Å (measured between opposite Cα carbons), while for eight-stranded β-barrels the internal diameter is at least 16 Å (Fig. 1a). Therefore, in the current work we designed capsules with increased diameters by employing semi-rigid semicarbazone linkers (Fig. 1a). The semicarbazone linkers can be synthesized by reaction of azapeptides with aromatic aldehydes. In the predominant extended conformation (all amides trans) the linker extends the internal diameter of the putative capsule up to ca. 17 Å (Fig. 1a–c) which should be sufficient to accommodate side chains. In order to control the positions of side chains the chirality of peptides was modulated. In homochiral peptide ribbons the side chains of subsequent amino acids point alternatively to the opposite surfaces of the ribbon. In heterochiral peptide ribbons all side chains are positioned at the same surface of the ribbon, but they point towards slightly different directions (Fig. 1b). Upon self-assembly of cavitands chirality is expected to control the position of the side chains.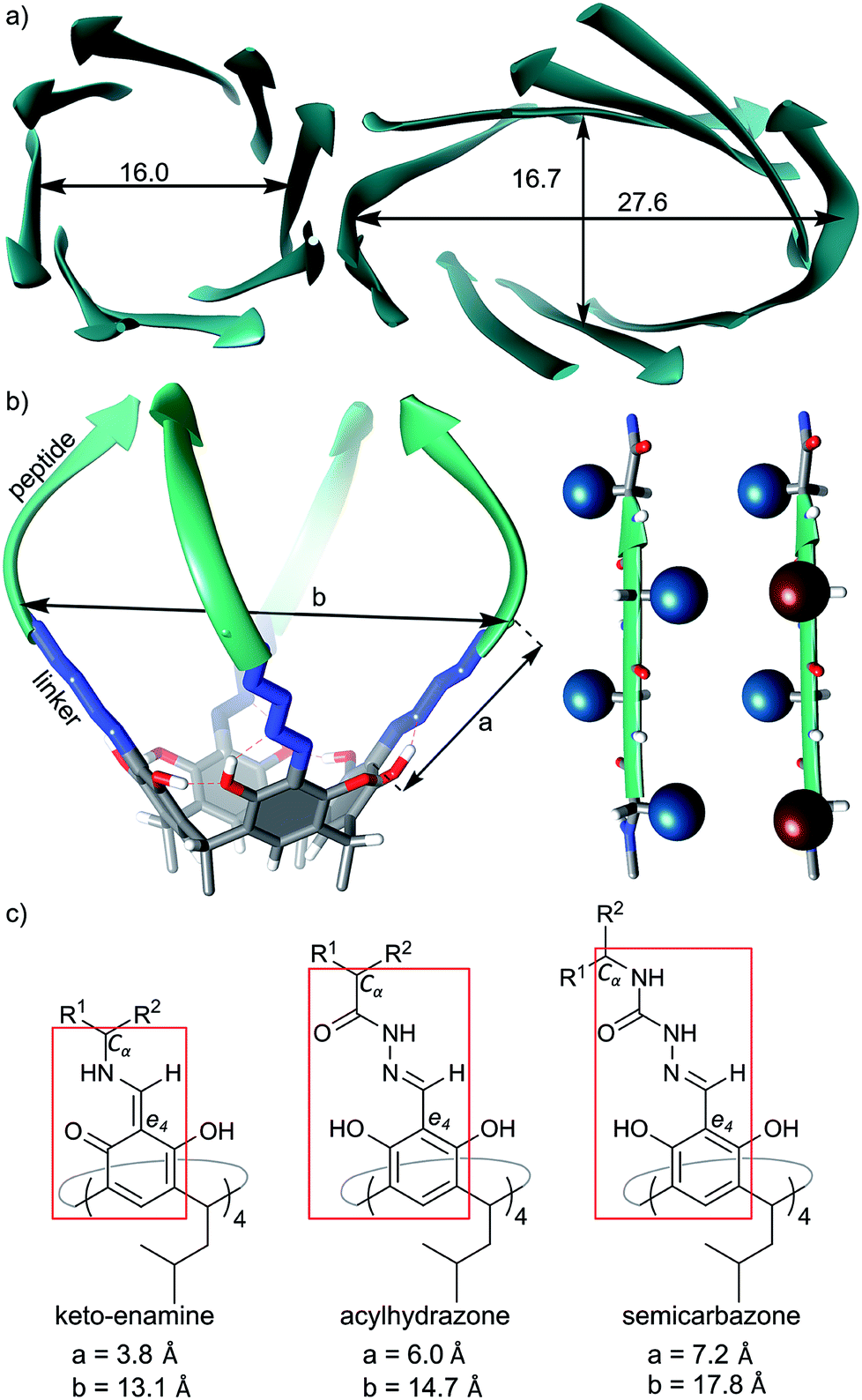 | ||
| Fig. 1 Design principles: (a) structural features of natural eight β-barrels (PDB codes 5BVL and 1GGL), diameters are between opposite Cα carbons given in Å; (b) model of a cavitand (left) and spatial arrangement of side chains in homo- or heterochiral peptides (right); (c) chemical structures of linkers in different cavitands and comparison of their dimensions (a – distance between the aromatic e4 carbon and Cα carbon in the first amino acid; b – distance between two opposite Cα carbons). | ||
It should be noticed that numerous variables may affect this design and introduce new features. Azapeptides17 are able to form additional hydrogen bonds18 and exist in compact cis conformations.19–21 Therefore, besides the desired intermolecular hydrogen bonding motifs,22,23 they may promote the formation of β-turns24,25 and β-hairpins,26,27 which are undesirable features in the current design. Moreover, unforeseen structural features that originate from the flexibility of peptide chains and from heterochirality may appear during self-assembly (the binding motifs of linear heterochiral peptides remain mostly unknown). In order to survey these troublesome features we employed dynamic covalent chemistry which empowers serendipity and enables amplification of the structures neglected by rational design.
Results and discussion
Synthesis
Azapeptides containing single amino acids (4a and 4b) were synthesized using strategy A, which involves the reaction of Cbz-hydrazine active ester 1 with amino acid amides 2a–b (Fig. 2a). Strategy A proved to be ineffective for longer azapeptides. Therefore for azapeptides 6a–c, 7a–d and a reference amine-based azapeptide 9 we used a carbonyldiimidazole (CDI) based procedure (strategy B, Fig. 2b). N-terminal aza fragment 5 was obtained in the reaction of an amino acid tert-butyl ester with carbonyldiimidazole and Cbz-hydrazine. After deprotection of the acid function further elongation was performed using standard peptide coupling protocols (with EDCI, OXYMA coupling reagents).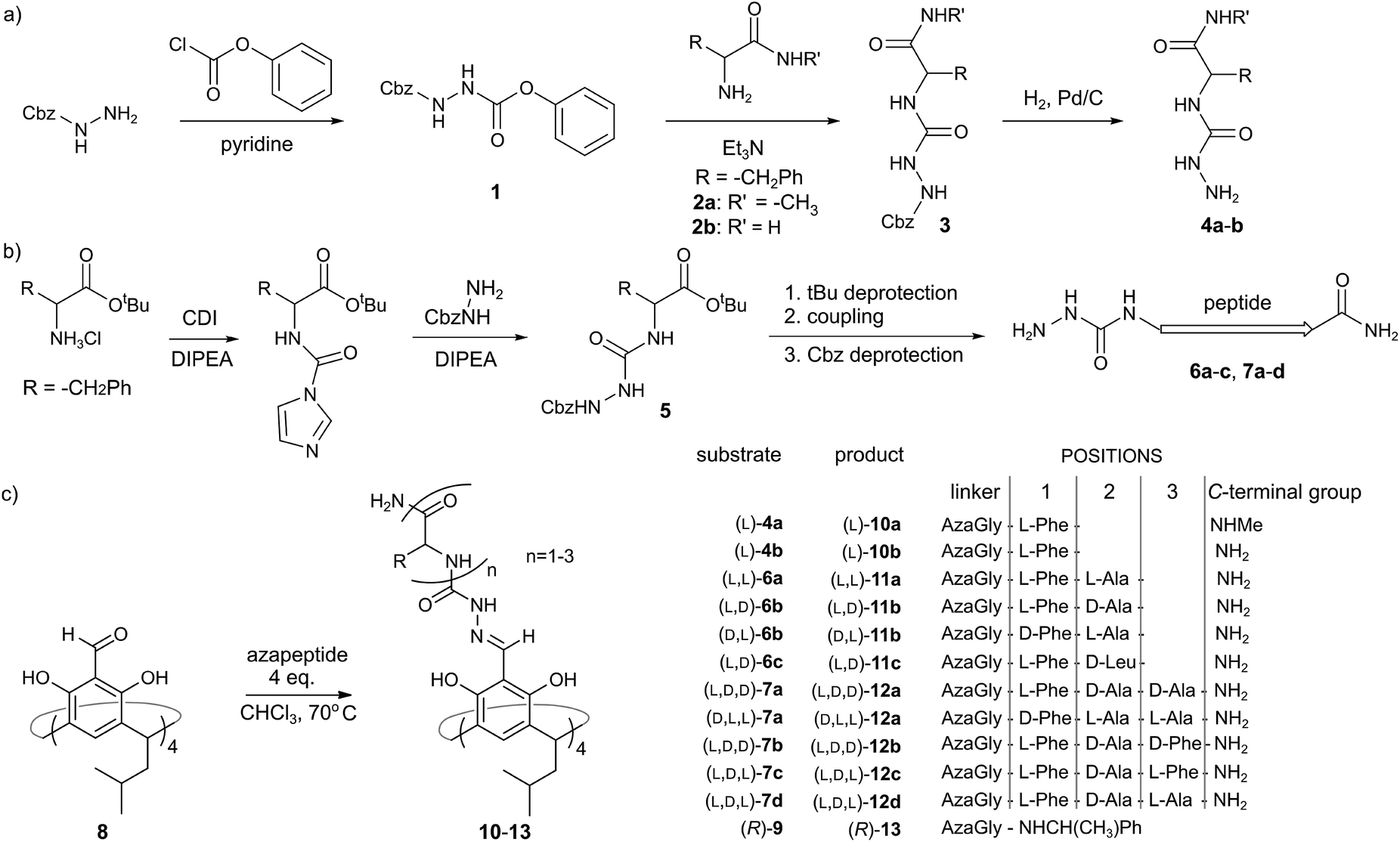 | ||
| Fig. 2 (a and b) Two strategies for the synthesis of azapeptides; (c) cavitand and capsule formation. | ||
First generation – reconnaissance
Reaction of the shortest azapeptide containing L-Phe, 4a, with tetraformylresorcin[4]arene 8 leads to the formation of tetrasubstituted product 10a (Fig. 2c). NMR spectra of 10a in DMSO (Fig. S1–S3†) confirm the C4-symmetric substitution pattern and DOSY indicates that 10a exists as a monomer in DMSO (D = 1.28 × 10−10 m2 s−1, radius 0.86 nm). Crystals suitable for X-ray analysis were obtained by vapour crystallization from a chloroform/methanol mixture. In contrast to the monomeric structure observed in DMSO, the X-ray structure reveals the formation of a dimeric capsule (10a)2 in the solid state (Fig. 3). The capsule core has an approximate C4 symmetry with two hemispheres being symmetrically non-equivalent and adopting substantially different conformations. In one of the hemispheres the semicarbazone linker has a trans conformation of an amide bond (hemisphere trans-A), while in the second hemisphere the semicarbazone linker adopts a cis conformation (hemisphere cis-B). The binding motif involves 11-membered rings formed using peptides' backbones from hemisphere trans-A and the cis-amide group (also part of the backbone) from the hemisphere cis-B. The most pronounced feature of the azapeptide capsule (10a)2, which makes it different from the previously known peptide capsules, is the presence of four phenylalanine side chains inside the cavity. Additionally, there is also enough space in the cavity for encapsulation of two small molecules (here two solvent molecules). The solid state structure of (10a)2 proves that (a) formation of a capsular dimer is possible for azapeptides and (b) widening of the capsules by the application of longer linkers is indeed a promising route towards functionalization of cavities with peptides' side chains. Although this capsular structure is not retained in DMSO and 10a is only sparingly soluble in less polar solvents (e.g. chloroform) which would facilitate self-assembly, the X-ray structure of (10a)2 was taken as a starting point for further optimization. The structure suggests that the terminal –NHMe groups and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups from the neighbouring strands are not involved in the binding motif and therefore the binding is not fully complementary. Therefore, we tested the replacement of the terminal group –NHMe group with the –NH2 group which would potentially allow for its incorporation into the binding motif to improve complementarity.
O groups from the neighbouring strands are not involved in the binding motif and therefore the binding is not fully complementary. Therefore, we tested the replacement of the terminal group –NHMe group with the –NH2 group which would potentially allow for its incorporation into the binding motif to improve complementarity.
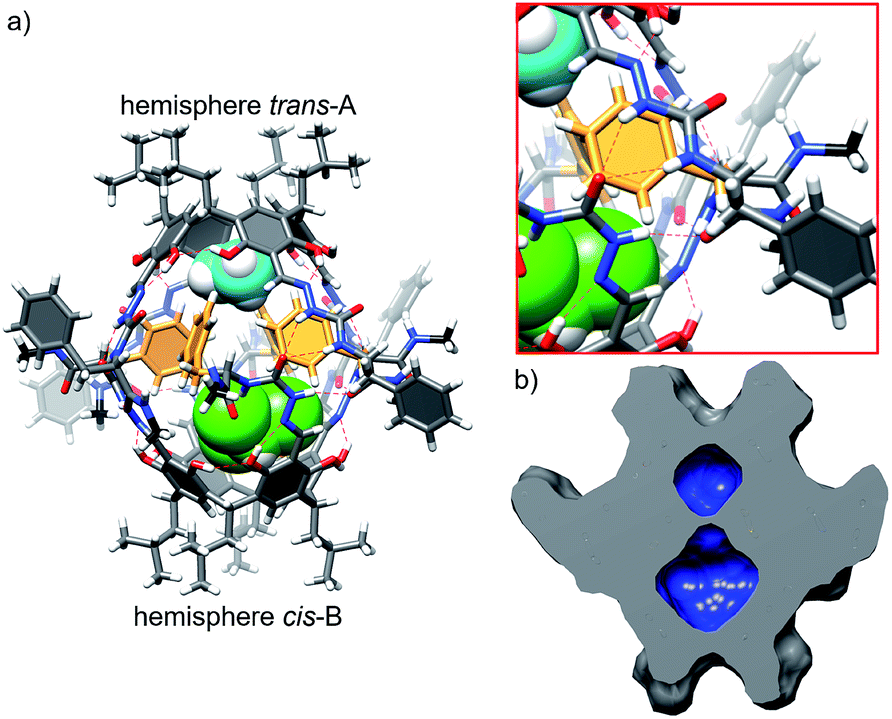 | ||
| Fig. 3 (a) X-ray structure of (10a)2 with an enlarged binding motif (green – encapsulated CHCl3, cyan – encapsulated MeOH, and yellow – encapsulated phenylalanine side chains); (b) molecular surface and volumes (62 Å3 and 172 Å3) of inner cavities in (10a)2 calculated with the Chimera program. | ||
Second generation – improved binding motif
Indeed, the second-generation cavitand 10b (with a terminal –NH2 amide group), despite its apparently higher polarity than 10a, is well soluble in chloroform and forms ordered dimeric capsules in solution. The plausible structure of the capsule (10b)2, which is in agreement with the spectral data, is depicted in Fig. 4d. The diffusion coefficient is 3.35 × 10−10 m2 s−1 (CDCl3, 298 K), which corresponds to a radius of 1.22 nm in agreement with the formation of dimer (10b)2. The formation of the dimer is accompanied by structural ordering which is reflected in the CD spectrum (Fig. 4a). Comparison of the CD spectra of (10b)2 and 13 (a reference cavitand based on a simple chiral amine without dimerization properties) in chloroform shows two orders of magnitude lower intensity for 13 than for (10b)2, while the UV spectra of both compounds remain highly similar. In contrast, in methanol, in which the capsules dissociate, the CD spectra of 10b and reference 13 both show similar and low intensity (Fig. 4b). Theoretical CD spectra (TD DFT) calculated using the plausible model agree well with the experimental spectrum, which validates the model.28 ROESY correlation signals indicate close intramolecular contacts between protons positioned next to the macrocyclic scaffolds (Hf and OH) and protons from the terminal –NH2 group (Hl1 and Hl2) (Fig. 4d and S7†) which suggest an extended shape of the dimeric capsule (10b)2. One of the terminal amide protons –NH2 is downfield shifted by 2.28 ppm with respect to the second one, suggesting its involvement in a hydrogen bonding binding motif. The averaged symmetry of capsule (10b)2 observed in NMR spectra (Fig. 4c and S4, S5†) is D4, thus, both hemispheres and all peptide arms within the hemispheres are symmetrically equivalent and there are no indications of cis/trans azapeptide isomerism. The signs and shapes of CD effects proved to be sensitive only to the inherent chirality of molecules but not to the cis/trans isomerism, and therefore, they do not allow for determination of stereochemistry of the azapeptide bonds in (10b)2. NMR spectra suggest external positioning of phenylalanine side chains (chemical shifts are close to typical values and there are no ROESY correlation signals that can come from internalization). This spectral evidence proves that the binding motif in the second generation capsule (10b)2 is altered compared to the first generation capsule (10a)2. Through-space contacts in (10b)2 are in agreement with an extended conformation of peptide strands as observed in the trans-A hemisphere, and therefore in the plausible model the capsule consists of two trans-A hemispheres with the optimal arrangement of terminal and binding groups, but, as it is expected for this single amino acid sequence, with all side chains positioned externally.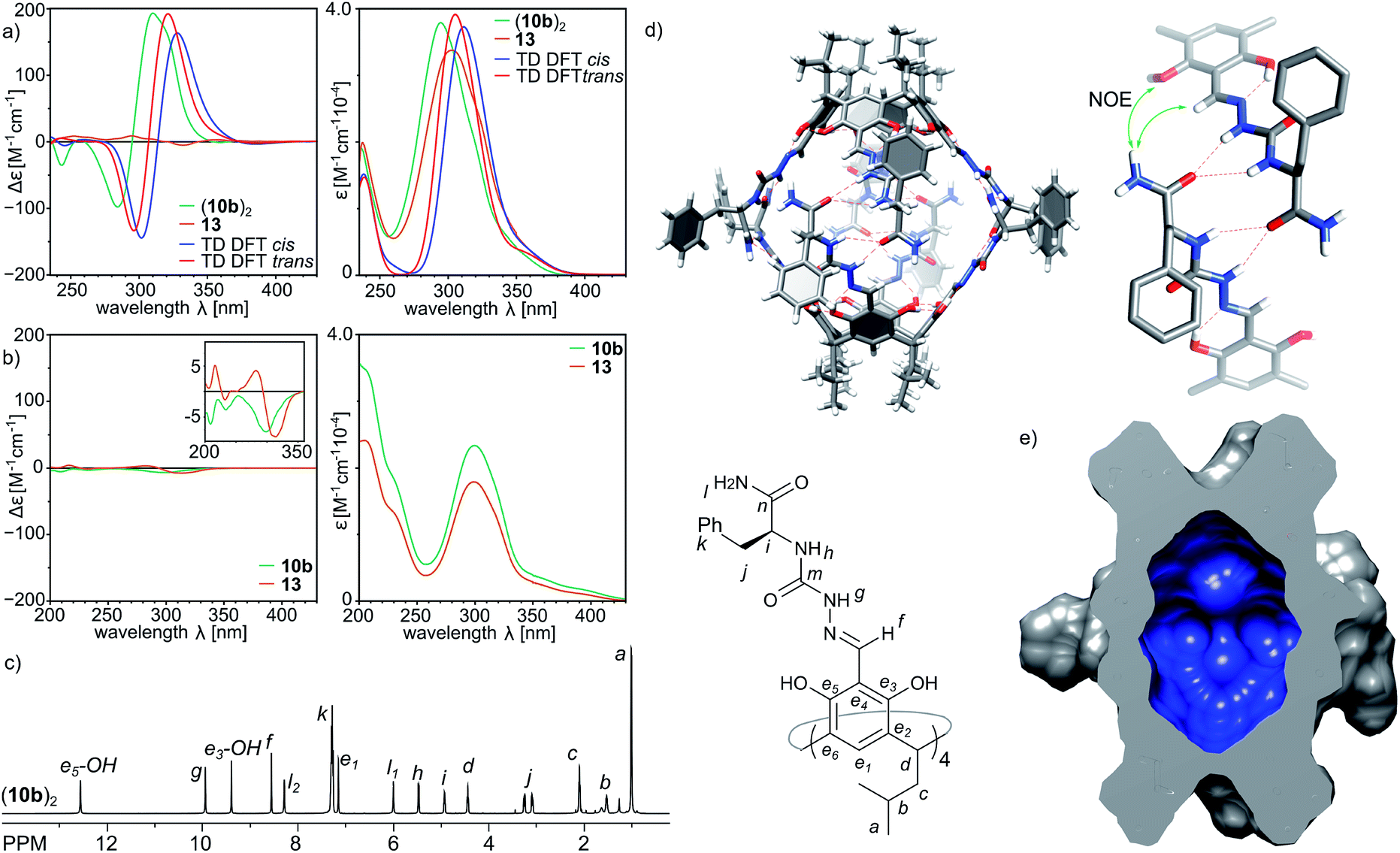 | ||
| Fig. 4 (a and b) Experimental and calculated CD and UV spectra of 10b and 13 (chloroform – top, methanol – bottom, TD DFT B3LYP/6-31g+(d,p), σ = 0.2 eV); (c) 1H NMR spectrum of (10b)2 (CDCl3, 600 MHz, 298 K); (d) modelled structure of the capsule in solution based on 2D NMR spectra (including DOSY and ROESY) composed of two trans-hemispheres of (10b)2; (e) molecular surface and volume (765 Å3) of the inner cavity in (10b)2 calculated with the Chimera program. | ||
Third generation – elongation
The third generation capsules (11a–c) contain the –NH2 terminal amide group and longer peptide sequences consisting of L-Phe at the first position and L-Ala(11a), D-Ala (11b) or D-Leu (11c) at the second position. The sequences were selected in order to check the influence of chirality (LversusD) and size (Ala versus Leu) on the self-assembly and the structure of capsules. Homochiral product 11a exists in DMSO as a monomeric cavitand (D = 1.40 × 10−10 m2 s−1, r = 0.78 nm) and it is poorly soluble in chloroform. Poor solubility in non-polar medium is indirect evidence that 11a does not form a complementary binding motif, and it has numerous solvent-exposed hydrogen bond donors and acceptors. In contrast, heterochiral derivatives 11b and 11c are well-soluble in chloroform and form discrete dimeric species (11b)2 and (11c)2 of D4 symmetry (D = 3.20 × 10−10 m2 s−1, radius 1.27 nm, CDCl3, 298 K). Signals of the terminal amide –NH2 group are differentiated by 2.2 ppm suggesting their involvement in the binding motif, while the presence of ROESY correlation signals between the semicarbazone linker and terminal alanine (side chain (CH3)n and α-CHm) suggests a head-to-head dimer of an extended shape (Fig. 5b and c). For capsules (11b)2 and (11c)2 both side chains are pointing to the same surface of the ribbon in agreement with the structural features of a heterochiral sequence, and therefore, they are expected to be positioned externally. Indeed, the chemical shifts for the signals of terminal side chains, better solubility of (11c)2 compared with (11b)2 and independence of the self-assembly process from the steric effects introduced by the Leu side chain prove that the side chains of the third generation of capsules are positioned outside the cavity.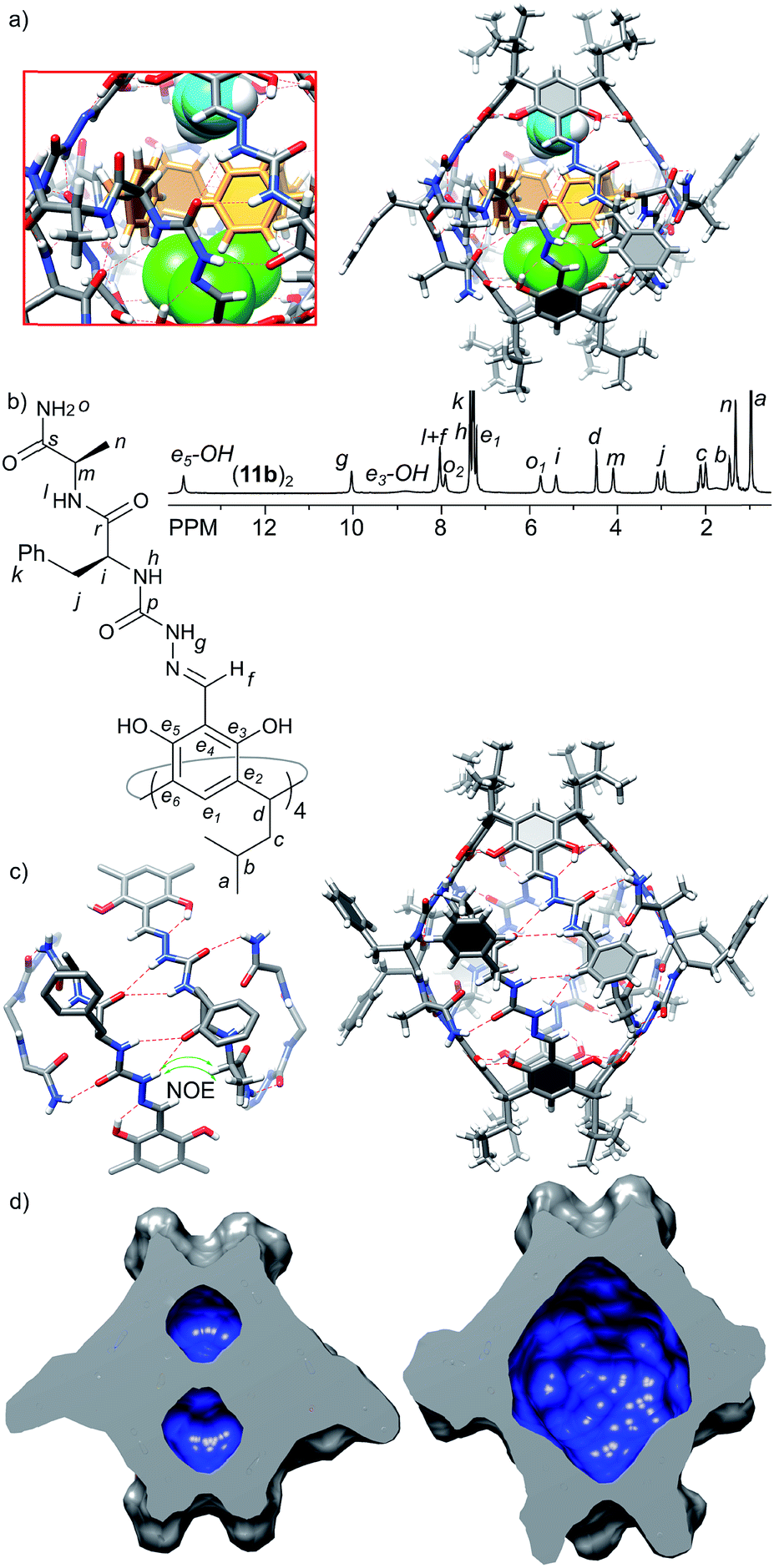 | ||
| Fig. 5 (a) X-ray structure of (11b)2 with an enlarged binding motif (green – encapsulated CHCl3, cyan – encapsulated MeOH, and yellow – encapsulated phenylalanine side chains); (b) 1H NMR spectrum of (11b)2 (CDCl3, 600 MHz, 298 K); (c) modelled structure of the capsule in solution based on 2D NMR spectra (including DOSY and ROESY) composed of two trans-hemispheres of (11b)2; (d) molecular surface and volumes of inner cavities in (11b)2 (left – crystal structure, 82 Å3 and 103 Å3; right – model, 1129 Å3) calculated with the Chimera program. | ||
The X-ray structure of (11b)2 confirms the dimeric nature (Fig. 5a); however, the solid state structure is different than the suggested structure in solution. The approximate symmetry of the capsule core in the solid state is C4 and, as in the case of (10a)2, two different hemispheres trans-A and cis-B are present. In the trans-A hemisphere all peptide strands have extended conformations with side chains positioned externally. The terminal amide –NH2 groups form hydrogen bonds with phenolic OH groups (N⋯O distances are 2.9–3.3 Å) of the second hemisphere (cis-B). The geometry of the trans-A hemisphere and non-covalent contacts are in agreement with the ones postulated in solution. However, the geometry and non-covalent contacts observed for the cis-B hemisphere are substantially different in the solid state than in solution. In the cis-B hemisphere the semicarbazone linker adopts a cis conformation which enforces a folded conformation of the peptide strand. As a consequence the side chain of the first amino acid (Phe) is positioned inside the cavity, and the side chain of the second side amino acid (Ala) is positioned outside the cavity and the terminal amide –NH2 groups are not involved in the intra-capsular binding motif but involved mostly in inter-capsular interactions. Although observed in the solid state, such a conformation is not present in solution (there are no relevant chemical shifts nor ROESY correlation signals). Therefore, we assume that the geometry of the trans-A hemisphere better reflects the approximate geometry in solution and the dimer's structure in solution resembles an extended dimer consisting of two trans-A-like hemispheres (Fig. 5c). The geometry of the cis-B hemisphere could be a result of interactions present only in the solid state: (a) competing inter-capsular interactions that involve the terminal amide groups and (b) preference towards a more compact (and less porous) structure due to spatial confinement. However, this solid state structure reflects the fact that due to the non-perfect complementarity and flexibility the capsule is dynamic.
Fourth generation – internal functionalization
The analysis of the third-generation capsules leads to the conclusion that the homochiral junction between positions 1 and 2 leads to destabilization of the structure. Position 2 is probably still too crowded for placement of a side chain inside the cavity. On the other hand, facile formation of capsules made of heterochiral peptides proves the efficiency of this binding motif. In the next generation capsules we combined these two observations and designed a three amino acid sequence with a heterochiral junction (L-Phe-D-Ala) for effective self-assembly and a homochiral junction (D-Ala-D-Ala) for internal functionalization. Product 12a forms a well-defined dimer (12a)2 of D4 symmetry in chloroform solution. The spatial arrangement in capsule (12a)2 was analysed using ROESY and DOSY and all data are in agreement with the formation of a head-to-head dimer with peptide arms in extended conformations (D = 3.00 × 10−10 m2 s−1, radius 1.36 nm, CDCl3, 298 K). Importantly, 1H NMR shows that the signal of the side chain of the terminal alanine (CH3) is substantially upfield shifted and appears at 0.57 ppm, which indicates that this group is directed towards the interior of the capsule, in close proximity to aromatic walls (Fig. 6). Additionally it shows ROESY correlations with Hg, Hf and Hi. Homologous cavitand 12b, in which the cavity-exposed Ala was replaced by more bulky Phe, also gives capsular species (12b)2, with internally exposed phenylalanine side chains. However, the capsules are much less ordered as indicated by broad signals in the NMR spectrum and less stable, as they disintegrate within days upon standing in solution. Thus, rational structure optimization of azapeptide capsules resulted in inner-cavity functionalization and proved the crucial role of chirality in both efficient self-assembly and functionalization of the inner cavity.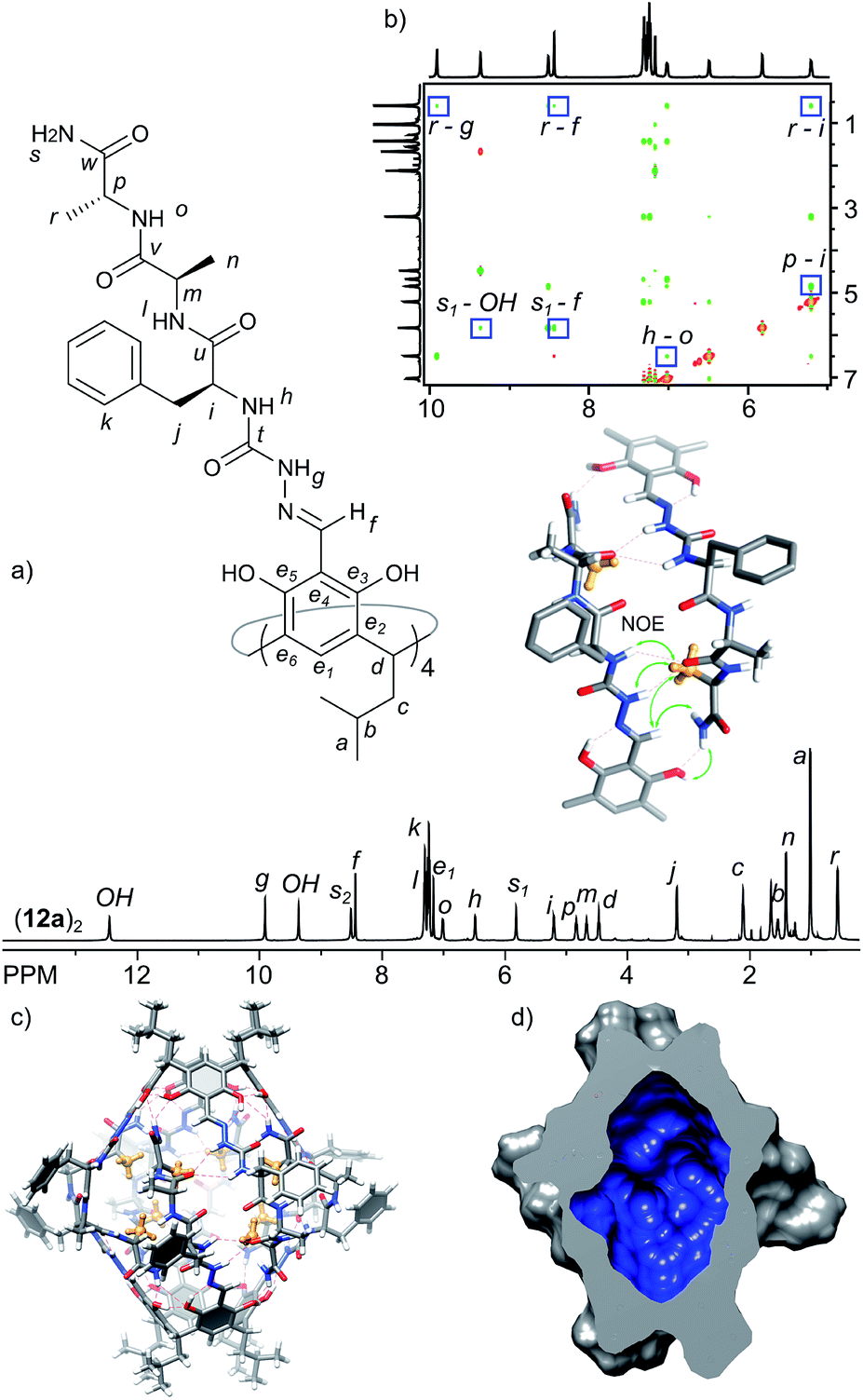 | ||
| Fig. 6 (a) 1H NMR spectrum of (12a)2 (CDCl3, 600 MHz, 298 K); (b) fragment of ROESY spectrum of (12a)2 (most indicative signals are in frames, CDCl3, 600 MHz, 298 K) and the binding motif with NOE correlations; (c) modelled structure of the capsule in solution based on 2D NMR spectra (including DOSY and ROESY) composed of two trans-hemispheres of (12a)2; (d) molecular surface and volume (1098 Å3) of the inner cavity in (12a)2 calculated with the Chimera program. | ||
Control experiments with azapeptides containing three amino acids with two heterochiral junctions (7c and 7d) confirm the crucial role of chirality. Thus the (L, D, L) sequence in 12c and 12d precludes the formation of capsules. The cavitand 12d is insoluble in non-polar solvents. The homologous product 12c, which possesses two hydrophobic phenylalanines in each arm, is well-soluble in chloroform, but it exists as a self-folded C2-symmetric cavitand in this environment (doubled number of NMR signals, D = 3.7 × 10−10 m2 s−1, radius 1.10 nm, CDCl3, 298 K). The shape of the ECD spectrum of 12c and its comparison with the spectra of other cavitands suggest that structural changes lower the symmetry of the resorcinarene core (Fig. 7c and 4a). We assume that the formation of β-turns and a system of intramolecular hydrogen bonds is responsible for this unique conformation of cavitand 12c. The suggested self-folded structure of 12c, which involves β-turns29 and a C2-symmetric boat-like conformation of the macrocycle, is presented in Fig. 7b.
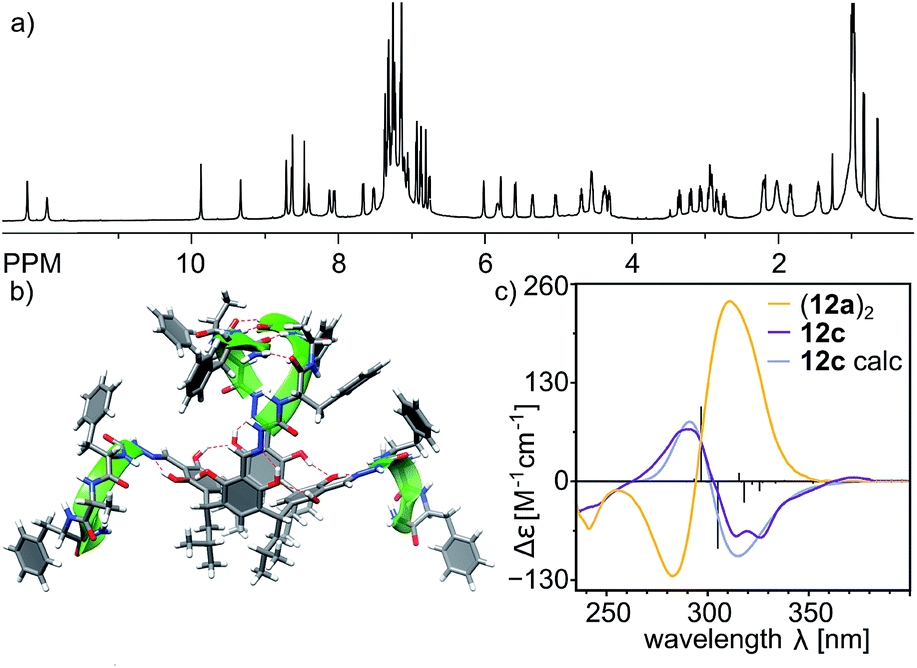 | ||
| Fig. 7 (a) 1H NMR spectrum of 12c (CDCl3, 600 MHz, 298 K); (b) modelled structure of the cavitand in solution; (c) CD spectra of capsule (12a)2 and cavitand 12c in chloroform (calculated spectrum of 12c: TD DFT B3LYP/6-31g+(d,p), σ = 0.2 eV). | ||
Fifth generation – serendipity
The reversible character of reactions between aldehydes and amine-based groups makes them applicable in dynamic covalent chemistry (DCC).30 We have previously exploited the DCC approach for the synthesis of various homo- and heterochiral capsules based on imine or acylhydrazone linkers and proved that they amplify and self-sort out of the mixtures of racemic peptides.13,14 However, the dynamic covalent chemistry of azapeptides is not known. What is more, reversibility of the reaction between semicarbazide groups (being part of the azapeptide functionality) and aldehydes has only been studied for the simplest semicarbazide in a catalytic variant.31With a set of various azapeptides in hand we explored the reversibility and possibility of amplification of self-assembled capsules from mixtures of azapeptides. First, we determined the kinetics of the non-catalytic reactions of azapeptides with 8. The time-dependent experiments indicate that the reactions in chloroform at 70 °C require several to twenty four hours to reach the stationary state (Fig. S9, S20 and S39†). This period of time depends on the peptide length and solubility. Next, we studied the reaction between 8 (C = 30 mM) and two azapeptides of different sequences but the same length (C(aza1) = C(aza2) = 60 mM) in CDCl3 by NMR (Fig. 8a). It should be noted that in most cases such reaction mixtures cannot be analysed by HPLC or MS due to the instability of self-assembled capsules on a chromatographic support or in the gas phase or due to identical mass (for substrates that are stereoisomers). Therefore, due to the complexity and resolution limits of NMR in each reaction we used simultaneously only two azapeptides. The reaction of 8 with rac-7a reaches equilibrium within 2 days (Fig. S40†) and the spectrum of the racemic mixture is virtually identical to that of the mixture with the enantiomerically pure substrate 7a (Fig. 8b) indicating very efficient chiral self-sorting. In the case of rac-6b the capsular product (11b)2 visibly predominates, but self-sorting isn't quantitative. This result was further confirmed by a self-sorting experiment with a pseudoracemic mixture of (L, D)-6b and (D, L)-6c which could be analysed by MS (Fig. S27†). Peaks from enantiomerically pure C4-symmetrical cavitands are the highest, while peaks from mixed products are also present which indicates non-quantitative chiral self-sorting. These results prove that: (1) azapeptides react with aldehydes in a reversible way and therefore they can be used as substrates in dynamic covalent chemistry and (2) self-assembly is a powerful factor that stabilises capsular structures and therefore leads to their amplification.
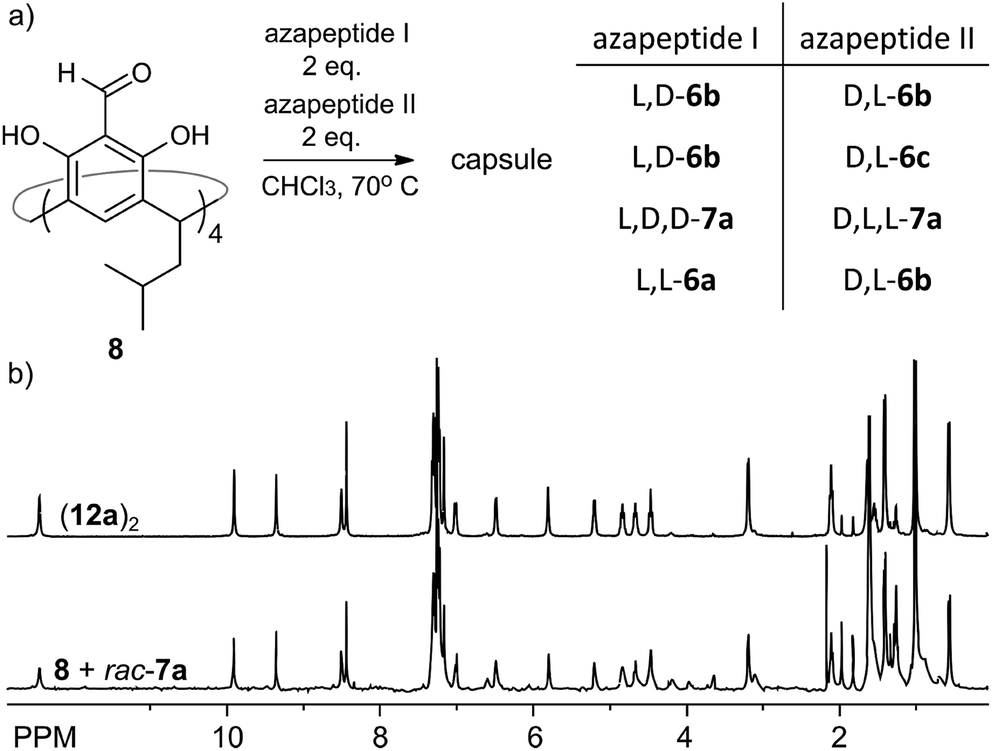 | ||
| Fig. 8 (a) Self-sorting reaction; (b) 1H NMR spectra of (12a)2 and self-sorting reaction of 8 with rac-7a (CDCl3, 600 MHz, 298 K). | ||
With positive results for the dynamic character of azapeptide–aldehyde reactions and encouraging amplification of self-assembled structures we sought after new, hybrid products that can be obtained only from the multisubstrate mixtures. The reaction of 8 with a mixture of (L, L)-6a and (D, L)-6b leads to complete consumption of the substrates within 2 days and the initial formation of homodimeric capsules (capsule (11b)2 remains in solution, while product (11a)2 is poorly soluble, Fig. 9a). However, with time an additional set of signals start to emerge that was not present for neither of the reactions containing single azapeptides as substrates (Fig. 9a and S33†). After 6 days the initially formed homodimeric products were quantitatively transformed into new species. Analysis of NMR spectra indicates that the final product is a capsular heterodimer (L, L-11a)(D, L-11b) in a head-to-head arrangement (correlation signals between terminal NH2 and Hf protons) and of roughly similar size to the previously obtained capsules (D = 3.25 × 10−10 m2 s−1, radius 1.25 nm). Terminal alanine residues from one hemisphere are located inside the cavity (0.62 ppm for CH3 protons) and have correlation signals in the ROESY spectrum with protons from the semicarbazone linker region (Hg, Hf, Hh and Hi) from the other hemisphere. The CD effects for the lowest energy bands in the CD spectrum of (11a)(11b) have almost diminished, and the spectrum is almost an algebraic sum of the two components which indicates that the capsule consists of two hemispheres that are twisted in the opposite directions (Fig. 9c). The plausible structure of the capsule (11a)(11b) that meets all these experimental constraints is presented in Fig. 9b.
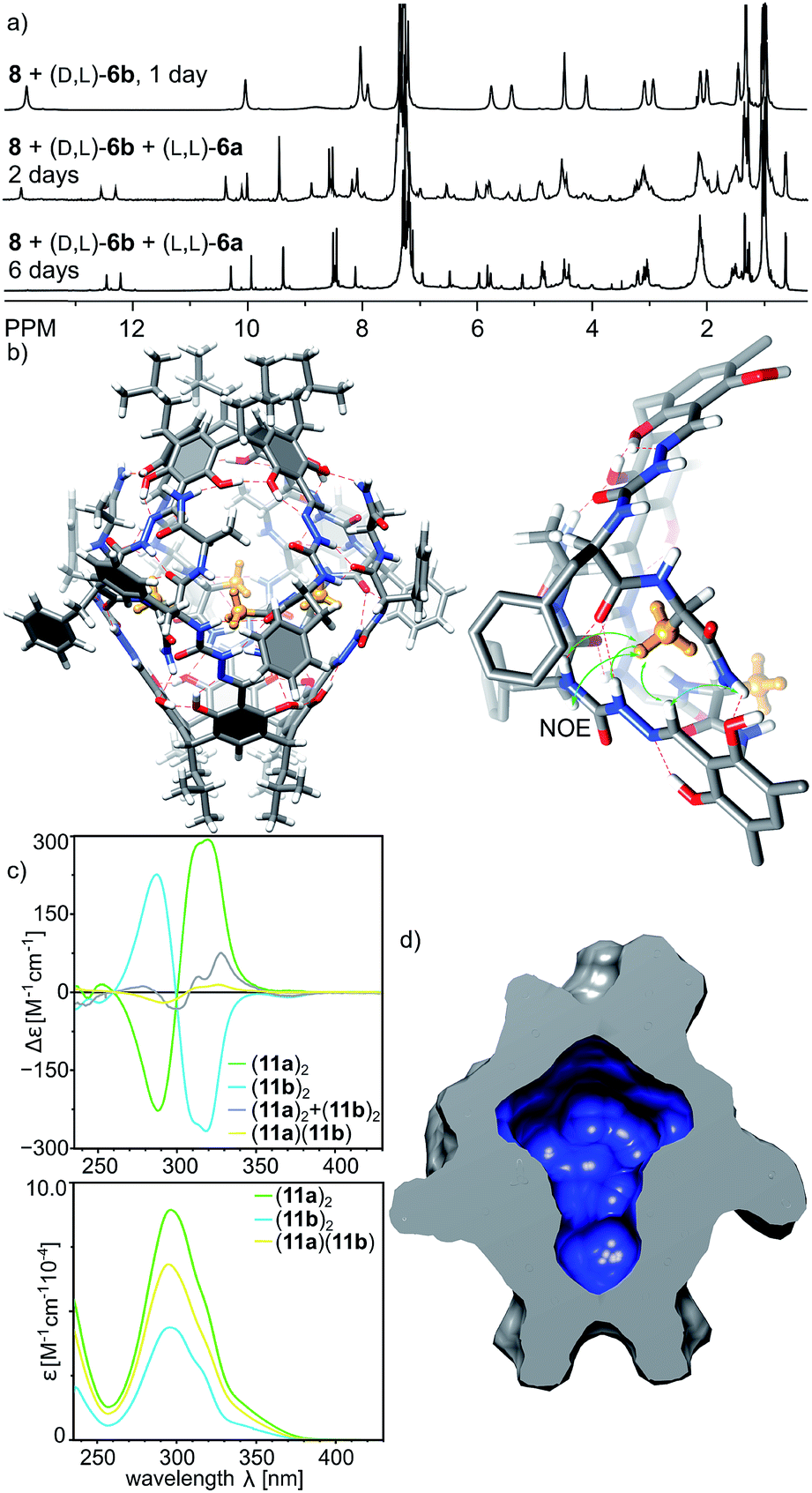 | ||
| Fig. 9 (a) 1H NMR spectra of reactions of 8 with (D, L)-6b and with a mixture of (D, L)-6b and (L, L)-6a (CDCl3, 600 MHz, 298 K); (b) modelled structure of the capsule (11a)(11b) in solution based on 2D NMR spectra (including DOSY and ROESY) composed of two trans-hemispheres; (c) CD and UV spectra of capsules in chloroform ((11a)2 is not observed in the NMR spectra because of poor solubility but it is soluble enough to measure the ECD spectrum); (d) molecular surface and volume (535 Å3) of the inner cavity in (11a)(11b) calculated with the Chimera program. | ||
With a set of peptide capsules of various geometries and chiralities we aimed at the determination of their thermal stability. Temperature-dependent CD measurements for homodimeric capsules (L-10b)2, (L, L-11a)2, and (L, D-11b)2, and heterodimeric capsule (L, L-11a)(D, L-11b) in tetrachloroethane (TCE was used instead of chloroform because it has higher boiling point, and ECD spectra in TCE are analogous to the ones in chloroform, S23†) show that all spectra of homodimeric capsules remain invariable (Fig. 10a, S10, S15, S23 and S42†). Because for non-assembled cavitands the intensities of the ECD bands are two orders of magnitude lower, it can be concluded that homodimeric capsules are thermally stable over the entire temperature range tested (20–100 °C). For the heterodimeric capsule (L, L-11a)(D, L-11b) the intensities of the ECD band are initially two orders of magnitude lower (due to hemispheres of different chiralities) and they decrease with temperature indicating possible instability of the assemblies at higher temperatures (Fig. 10b and S34†).
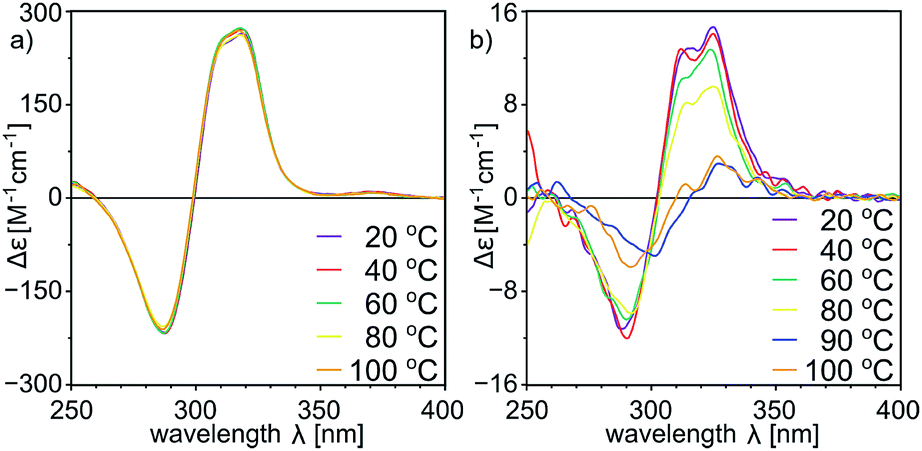 | ||
| Fig. 10 Temperature-dependent ECD spectra of (a) (L, D-11b)2 and (b) (L, L-11a)(D, L-11b) (in tetrachloroethane, normalized using UV spectra, for UV see the ESI†). | ||
Conclusions and outlook
In summary, we have elaborated a strategy for the synthesis of capsules with functionalized internal cavities based on azapeptides. The strategy involves rational design of a macrocyclic compound and a semicarbazone linker, optimization of the binding motif (modification of the C-terminus) and modulation of chirality of peptides towards proper positioning of the side chains (homo- and heterochiral sequences of peptides). As a result we obtained capsules that self-assemble using hydrogen bonding interactions between their backbones and have side chains positioned in their cavities.Additionally, we have proven the reversible character of reactions between azapeptides and aldehydes. This finding opens a way towards the application of azapeptides in dynamic covalent chemistry and towards the synthesis of new capsules in a combinatorial way. Indeed, from a set of combinatorial experiments an unexpected hybrid capsule was obtained, which wasn't designed in a rational way nor it could be obtained from the reaction employing only a single azapeptide as a substrate.
Further self-sorting experiments of azapeptide mixtures indicate that amplification of well-defined products is effective only in cases when capsules are formed. These results prove that self-assembly is an effective stabilization factor that affects thermodynamic equilibria and leads to self-sorting even for highly similar and conformationally labile substrates. This observation is in contrast to the common opinion that rigidity and orthogonality of interactions are indispensable features for selective self-sorting.
The design presented in this paper is closely inspired by natural eight-stranded β-barrels. However, such β-barrels, due to their small internal diameter, have limited abilities to host molecules. This limitation can also be a drawback of the azapeptide capsules. However, the current strategy allows for elongation and alternation of the positions of side chains, and the dynamic nature of the cores enables expansion. The capsules are not fully sealed due to imperfect complementarity of binding motifs, so they can have portals for guest entrance/release. Thus, in contrast to the crowded interiors of natural β-barrels, interiors of these artificial capsules are more accessible. Another limitation of eight-stranded β-barrels is that bulky polar side chains are avoided inside the barrels. In fact, also in these seminal examples only the smallest side chain (methyl group from Ala) was accommodated in the proximity of a hydrophobic pocket. However, the strategy suggested in this paper allows for elongation of the peptides and changes in the position of side chains, and therefore, different locations of internal side chains are also possible. What is more, the formation of hybrid capsules opens up possibilities of diversification of hemispheres and, for example, encapsulation of acid–base pairs at alternating positions mimicking natural catalytic sites (functional dyads, triads, etc.).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Center – Poland (HJ grant no. 2014/15/N/ST5/02019, AS grant no. 2016/20/W/ST5/00478) and Wroclaw Centre for Networking and Supercomputing (grant no. 299). X-ray diffraction data were collected at the EMBL beamline P13 of the Petra III synchrotron in Hamburg (DESY) and beamline 1911, MaxLab in Lund.Notes and references
- E. Berardo, L. Turcani, M. Miklitz and K. E. Jelf, Chem. Sci., 2018, 9, 8513 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Natl. Sci. Rev., 2017, 4, 426 CrossRef CAS.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC , references 62–81.
- I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127 RSC.
- J. Bonnefoy, A. Legrand, E. A. Quadrelli, J. Canivet and D. Farrusseng, J. Am. Chem. Soc., 2015, 137, 9409 CrossRef CAS PubMed.
- C. Martí-Gastaldo, D. Antypov, J. E. Warren, M. E. Briggs, P. A. Chater, P. V. Wiper, G. J. Miller, Y. Z. Khimyak, G. R. Darling, N. G. Berry and M. J. Rosseinsky, Nat. Chem., 2014, 6, 343 CrossRef PubMed.
- A. P. Katsoulidis, K. S. Park, D. Antypov, C. Martí-Gastaldo, G. J. Miller, J. E. Warren, C. M. Robertson, F. Blanc, G. R. Darling, N. G. Berry, J. A. Purton, D. J. Adams and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2014, 53, 193 CrossRef CAS PubMed.
- L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261 RSC.
- P. Ballester and G. Gil-Ramírez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10455 CrossRef CAS PubMed.
- A. Szumna, Chem.–Eur. J., 2009, 15, 12381 CrossRef CAS PubMed.
- H. Jędrzejewska, M. Wierzbicki, P. Cmoch, K. Rissanen and A. Szumna, Angew. Chem., Int. Ed., 2014, 53, 13760 CrossRef PubMed.
- M. Szymański, M. Wierzbicki, M. Gilski, H. Jędrzejewska, M. Sztylko, P. Cmoch, A. Shkurenko, M. Jaskólski and A. Szumna, Chem.–Eur. J., 2016, 22, 3148 CrossRef PubMed.
- A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862 CAS.
- B. Kuberski and A. Szumna, Chem. Commun., 2009, 1959 RSC.
- J. Gante, Angew. Chem., Int. Ed. Engl., 1970, 9, 813 CrossRef CAS PubMed.
- Y. Zhang, R. M. Malamakal and D. M. Chenoweth, J. Am. Chem. Soc., 2015, 137, 12422 CrossRef CAS PubMed.
- H.-J. Lee, H. J. Jung, J. H. Kim, H.-M. Park and K.-B. Lee, Chem. Phys., 2003, 294, 201 CrossRef CAS.
- H.-J. Lee, J.-W. Song, Y.-S. Choi, S. Rob and C.-J. Yoon, Phys. Chem. Chem. Phys., 2001, 3, 1693 RSC.
- H.-J. Lee, J.-W. Song, Y.-S. Choi, H.-M. Park and K.-B. Lee, J. Am. Chem. Soc., 2002, 124, 11881 CrossRef CAS PubMed.
- R.-O. Moussodia, S. Acherar, E. Romero, C. Didierjean and B. Jamart-Grégoire, J. Org. Chem., 2015, 80, 3022 CrossRef CAS PubMed.
- M. I. A. Ibrahim, Z. Zhou, C. Deng, C. Didierjean, R. Vanderesse, J. Bodiguel, M.-C. Averlant-Petit and B. Jamart-Grégoire, Eur. J. Org. Chem., 2017, 4703 CrossRef CAS.
- X.-S. Yan, H. Luo, K.-S. Zou, J.-L. Cao, Z. Li and Y.-B. Jiang, ACS Omega, 2018, 3, 4786 CrossRef CAS.
- R. Chingle, C. Proulx and W. D. Lubell, Acc. Chem. Res., 2017, 50, 1541 CrossRef CAS PubMed.
- J. S. Nowick, K. S. Lam, T. V. Khasanova, W. E. Kemnitzer, S. Maitra, H. T. Mee and R. Liu, J. Am. Chem. Soc., 2002, 124, 4972 CrossRef CAS PubMed.
- R. J. Woods, J. O. Brower, E. Castellanos, M. Hashemzadeh, O. Khakshoor, W. A. Russu and J. S. Nowick, J. Am. Chem. Soc., 2007, 129, 2548 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- V. Brenner, F. Piuzzi, I. Dimicoli, B. Tardivel and M. Mons, Angew. Chem., Int. Ed., 2007, 46, 2463 CrossRef CAS PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003 RSC.
- J. M. Sayer, M. Peskin and W. P. Jencks, J. Am. Chem. Soc., 1973, 95, 4277 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, full NMR spectra, crystallographic data and details of DFT calculations. CCDC 1883372 and 1883373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05455j |
| This journal is © The Royal Society of Chemistry 2019 |