
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNH2-MIL-125(Ti)-derived porous cages of titanium oxides to support Pt–Co alloys for chemoselective hydrogenation reactions†
Zhizhi
Gu
,
Liyong
Chen
 *,
Xuezhao
Li
,
Lin
Chen
,
Yingyue
Zhang
and
Chunying
Duan
*
*,
Xuezhao
Li
,
Lin
Chen
,
Yingyue
Zhang
and
Chunying
Duan
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China. E-mail: lychen@dlut.edu.cn; cyduan@dlut.edu.cn
First published on 12th December 2018
Abstract
The change of atom density induced structural collapse in the transformation process from metal–organic frameworks (MOFs) to their inorganic counterparts is a major challenge to the achievement of porous hollow structures. Herein, we develop an amino acid-mediated strategy for transformation of NH2-MIL-125(Ti) to successfully synthesize well-defined porous cages of titanium oxides (PCT) due to sheets serving as structural scaffolds. On this basis, PCT supported Pt-based nanoparticles are generated via a similar synthetic route, and are utilized to study the selective hydrogenation of carbonyl groups in α,β-unsaturated aldehydes, benefiting from the specific structures of PCT and tunable electronic structures of Pt mainly affected by doping with metal species such as Co. In this case, Pt–Co/PCT composites give 96% selectivity for cinnamyl alcohol at 100% conversion of cinnamaldehyde under 0.2 MPa H2 and 80 °C for 3 h. This research would offer a promising strategy for important organic transformations in academic and industrial research to selectively synthesize high-value-added products.
Introduction
Porous hollow structures, especially assembled with nanosheets, possess large surface area, a distinctive micro-environment between their interiors and exteriors, and specific channels for guest species shuttling.1–3 Thus, porous hollow structures have been widely used in the fields of drug delivery and gas adsorption, as well as catalysis, as supports for highly dispersive catalyst nanoparticles (NPs) that can afford superior catalytic activity for organic transformations.4–8 Template-mediated synthesis has been considered as one of the most intriguing methods for generation of well-defined porous hollow structures.9,10 As of now, metal–organic frameworks (MOFs) with tunable metal ions and organic ligands have become fully fledged self-sacrificial templates for fabrication of metal/carbon-based materials with desirable properties.11 Nonetheless, achievement of porous hollow structures by transformation of MOFs still faces a big challenge in that the ultralow atom density of MOFs easily leads to the collapse of hollow structures in the transformation process.12–14 To overcome this hurdle, it is desirable to produce structural scaffolds to achieve the hollow architectures. Sheet-like NPs possessing large lateral surface area are generally employed as structural scaffolds, and thereby some successful examples have been reported on MOF derived porous hollow structures of inorganic compounds assembled with sheets.15,16 Owing to the more favourable hydrolysis and condensation of titanium(IV) ions compared to other metal ions,17 porous hollow titanium compounds are produced with difficulty by transformation of Ti-MOFs in solution. Actually, calcination is a conventional method to convert Ti-MOFs into solid titanium compounds.18–23 Based on the fact that layered titanates easily form sheet-like structures,24 herein, we developed a novel amino acid-assisted solvothermal strategy to accomplish transformation from polyhedral NH2-MIL-125(Ti) with fcu topology to titanate/TiO2 (defined as titanium oxide) nanosheets; these nanosheets were assembled into porous cages (denoted as PCT) based on polyhedral templates.25 Furthermore, by introducing metal sources, PCT supported metal NPs could be prepared (Fig. 1).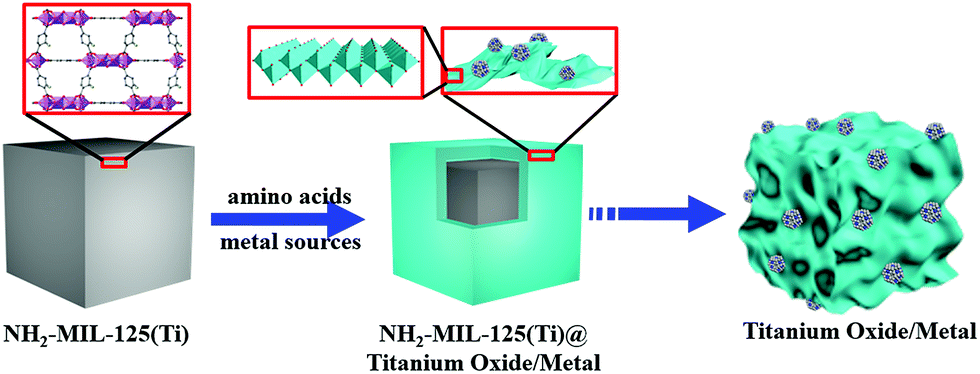 | ||
| Fig. 1 Schematic of porous cages of titanium oxide anchored metal NPs by an amino acid-assisted transformation of NH2-MIL-125(Ti). | ||
Many efforts have focused on engineering metal-based catalysts to address the issue of intramolecular hydrogenation competition between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds.26–28 Pt NPs as a universal catalyst have been applied for hydrogenation of a broad range of bonds, although giving rise to little chemoselectivity.29–31 Based on component-regulated electronic structures and defects, the capability of Pt NPs for selective adsorption and activation of different groups can be adjusted by doping with Fe, Sn, Co and so on.32–36 Meanwhile, to improve stability and activity against complex catalytic systems, Pt NPs are generally immobilized on the supports.37 Combination of metal dopants and porous hollow supports for construction of anchored Pt–metal (Pt–M) alloys may be an intriguing strategy to upgrade the selectivity and reactivity of Pt NPs towards hydrogenation. Some pioneer studies have been devoted to utilization of titanium compounds as supports to improve the catalytic hydrogenation performance of metal catalysts.38–44 Following these concepts, we engineer Pt–M alloy NPs on PCT for highly chemoselective hydrogenation of α,β-unsaturated aldehydes to unsaturated alcohols that are high-value-added fine chemicals.45 Finally, Pt–Co NPs anchored by PCT (Pt–Co/PCT) exhibit exceptionally high reactivity (100%) and selectivity (∼96%) for hydrogenation of carbonyl groups in cinnamaldehyde under mild conditions even without obvious variation after six successive cycles. The Pt–Co/PCT also gives high selectivity for carbonyl groups of other α,β-unsaturated aldehydes at high conversion levels. The enhanced selective hydrogenation performance of the Pt–Co/PCT mainly benefits from dopant mediated microstructures of Pt and porous hollow support regulated mass transport.
C and CO bonds.26–28 Pt NPs as a universal catalyst have been applied for hydrogenation of a broad range of bonds, although giving rise to little chemoselectivity.29–31 Based on component-regulated electronic structures and defects, the capability of Pt NPs for selective adsorption and activation of different groups can be adjusted by doping with Fe, Sn, Co and so on.32–36 Meanwhile, to improve stability and activity against complex catalytic systems, Pt NPs are generally immobilized on the supports.37 Combination of metal dopants and porous hollow supports for construction of anchored Pt–metal (Pt–M) alloys may be an intriguing strategy to upgrade the selectivity and reactivity of Pt NPs towards hydrogenation. Some pioneer studies have been devoted to utilization of titanium compounds as supports to improve the catalytic hydrogenation performance of metal catalysts.38–44 Following these concepts, we engineer Pt–M alloy NPs on PCT for highly chemoselective hydrogenation of α,β-unsaturated aldehydes to unsaturated alcohols that are high-value-added fine chemicals.45 Finally, Pt–Co NPs anchored by PCT (Pt–Co/PCT) exhibit exceptionally high reactivity (100%) and selectivity (∼96%) for hydrogenation of carbonyl groups in cinnamaldehyde under mild conditions even without obvious variation after six successive cycles. The Pt–Co/PCT also gives high selectivity for carbonyl groups of other α,β-unsaturated aldehydes at high conversion levels. The enhanced selective hydrogenation performance of the Pt–Co/PCT mainly benefits from dopant mediated microstructures of Pt and porous hollow support regulated mass transport.
Results and discussion
Preparation and characterization of porous cages of titanium oxides (PCT) and PCT supported metal nanoparticles (NPs)
Nanosized polyhedral NH2-MIL-125(Ti) ∼500 nm in size was prepared via a modified synthetic route,19 confirmed by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images (Fig. S1(a) and S1(b)†) and an X-ray diffraction (XRD) pattern (Fig. S1(c)†). NH2-MIL-125(Ti) as the precursor template was converted into porous hollow structures via an alcoholic thermal process at 176 °C under auxiliary of amino acid molecules, including L-alanine, glycine, L-lysine and DL-serine. As shown in Fig. 2, the morphologies of all samples were the sheet-assembled porous cage-like structures with a diameter of 500 to 800 nm. To understand the components and fine structures, the porous cages prepared by the L-alanine assisted route as a representative sample were further characterized. Layered titanate H2Ti8O17 (JCPDS no. 36-0656) was confirmed by the XRD pattern (Fig. S2†), wherein the diffraction peaks of anatase TiO2 (JCPDS no. 21-1272) also appeared, revealing the dual-component porous cages. The thickness of the sheets ranged from 10 to 30 nm, characterized by a TEM image of the curled edge of a sheet (Fig. S3(a)†) and an AFM image of separated sheets (Fig. S3(b)†). The transformation of Ti(IV) ions was very susceptible to temperature and water. Only anatase TiO2 diffraction peaks in the XRD patterns (Fig. S4(c)†) appeared at 200 °C or upon introducing a small amount of water (50 μL) into the reaction system (5 mL). Porous cage-like structures were not found but a large amount of discrete TiO2 NPs (Fig. S4(a) and (b)†). Thus, the layered titanate sheets play the crucial role of structural scaffolds to prevent damage to the porous cages.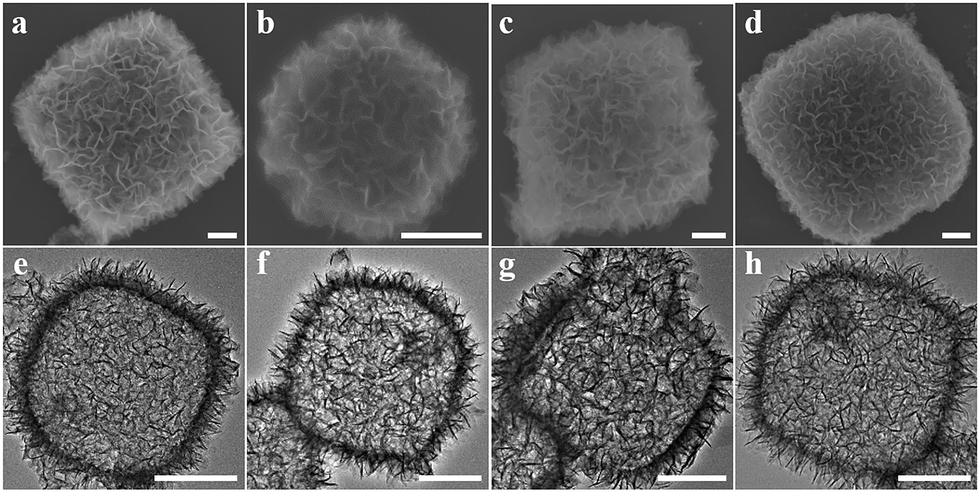 | ||
| Fig. 2 SEM (top) and TEM (bottom) images of samples prepared by transformation of NH2-MIL-125(Ti) with the help of amino acid molecules. (a and e) L-alanine, (b and f) glycine, (c and g) L-lysine and (d and h) DL-serine. All scale bars are 200 nm. | ||
To further understand the components and structures of PCT, Raman spectrum, XPS spectrum and nitrogen adsorption isotherm measurements were carried out. As shown in Fig. S5† (black curve), the PCT gave rise to a Raman active peak at around 149.9 cm−1, assigned to the Eg active mode of anatase TiO2, under irradiation with a 633 nm laser.46 Other Raman active phonons were not clearly detected within 100 to 800 cm−1. The high-resolution Ti 2p XPS spectrum in Fig. S6(a)† showed two predominant peaks at approximately 458.60 and 464.30 eV. Both can be identified as the binding energy of Ti(IV) 2p3/2 and 2p1/2 from TiO2 or layered titanate.46 Nitrogen adsorption measurement at 77 K showed the presence of a significant hysteresis loop at P/P0 larger than 0.4 between the adsorption and desorption curves (Fig. S7(a)†). The type IV isotherm with H2-type hysteresis reveals the mesoporosity of the cages, together with pore size distribution ranging from 3 to 7 nm (Fig. S7(c)†). The high Brunauer–Emmett–Teller (BET) specific surface area estimated to be ca. 375 cm2 g−1 is attributed to titanium oxide sheet assembled hollow structures.
The transformation from polyhedral NH2-MIL-125(Ti) to porous cages of titanium oxides possibly involves the nanoscale Kirkendall effect associated with Ti(IV) ion-related dissolution and recrystallization (Fig. S8†).10 Ti(IV) ions were firstly dissolved from NH2-MIL-125(Ti) nanocrystals by coordination of Ti(IV) with amino acids (L-alanine).47 The solvothermal alcoholysis of the dissolved Ti(IV) took place, leading to formation of sheet-like titanium oxide NPs on the NH2-MIL-125(Ti) nanocrystals.48 With the continuous transformation, successive shells of titanium oxides were generated around NH2-MIL-125(Ti) nanocrystals. Owing to the porous structures of shells, reaction species can go into the interiors to continue dissolving Ti(IV) ions from NH2-MIL-125(Ti) cores. Finally, the cores were completely consumed, and transferred into the completed porous cages. In the transformation process, amino acid molecules in ethanol can effectively control the release of Ti(IV) ions and subsequent crystallization, resulting in the formation of layered titanates. In contrast, without introducing L-alanine, large nanosheets were produced by alcoholysis of Ti(IV) ions, and NH2-MIL-125(Ti) nanocrystals were engulfed by these nanosheets, further suppressing the dissolution of NH2-MIL-125(Ti) nanocrystals (Fig. S9†). Moreover, when a small amount of water was added into the synthetic system, hydrolysis of Ti(IV) possibly was a prominent process, resulting in the formation of anatase TiO2 NPs (Fig. S4†). Thereby, the presence of sheet-like layered titanates acting as morphological scaffolds is a decisive factor to keep the polyhedral cages unchanged. Moreover, the type of Ti-MOF is another key factor in the formation of completed cages. For example, if MIL-125(Ti) was used as the precursor, completed porous cages of titanium oxides were generated with difficulty (Fig. S10†).
Amino acid molecules generally as a class of mild reducing agents are used to prepare metal NPs.49,50 Herein, we chose the synthetic system of L-alanine to prepare various hybrids of Pt and Pt–M NP loaded PCT while introducing H2PtCl6 and other metal salts. Pt–Co/PCT composites were successfully prepared in an ethanolic solution (5 mL) including NH2-MIL-125(Ti) (10 mg), Co(NO3)2 (10 μmol), H2PtCl6 (3.5 μmol), and L-alanine (0.533 mmol) at 176 °C for 24 h. The morphology of the as-made porous cages loaded by metal NPs did not exhibit any obvious difference compared to that of PCT, as shown in Fig. 3(a) and (c). The diffraction peaks corresponding to layered titanate and anatase TiO2 were found in the XRD pattern (Fig. S11†). The Raman spectrum (Fig. S5,† red curve) and the XPS spectrum of Ti 2p (Fig. S6(b)†) further revealed that the porous cages were composed of titanium oxides. The slight downshift of the Ti 2p3/2 from 458.60 to 458.50 eV reveals the interaction between Pt–Co NPs and PCT.51 In comparison to the {111} facets of the face-centered cubic (fcc) structured Pt3Co (JCPDS no. 29-0499) located at 40.53°, the broad diffraction peak in the XRD pattern was slightly shifted to 40.70°, suggesting an atomic ratio of Pt/Co of less than 3 in the as-formed Pt–Co alloy. Monodisperse Pt–Co NPs less than 10 nm in size and their aggregates were observed in a magnified TEM image (Fig. S12†). The high-resolution TEM (HRTEM) image (Fig. 3(b)) of a Pt–Co NP shows two sets of lattice fringes corresponding to {111} and {200} facets.52,53 Defects of grain boundaries appeared in the HRTEM image of Pt–Co NPs (Fig. S13†). The high-angle annular dark-field scanning TEM (HAADF-STEM) image showed that high-contrast Pt–Co NPs were dispersed in the porous shells (Fig. 3(d)). The distribution of Pt, Co, Ti and O elements characterized by energy-dispersive X-ray spectroscopy (EDXS) elemental mapping of a porous cage (Fig. 3, bottom) further confirms the formation of Pt–Co/PCT. The content of Pt was about 3.16 wt% estimated by inductively coupled plasma atomic emission spectrometry (ICP-AES); the Pt/Co atomic ratio was around 2.72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, consistent with the XRD result. The well-defined mesoporous structures of the Pt–Co/PCT were estimated using the N2 adsorption isotherm that exhibited a type IV isotherm with H3-type hysteresis at P/P0 > 0.4 (Fig. S7(b)†), and a broad pore size distribution between 3.5 and 11 nm was observed (Fig. S7(d)†). As expected, the BET specific surface area of the sample decreased from 375 to 309 cm2 g−1 after loading of Pt–Co NPs.
:1, consistent with the XRD result. The well-defined mesoporous structures of the Pt–Co/PCT were estimated using the N2 adsorption isotherm that exhibited a type IV isotherm with H3-type hysteresis at P/P0 > 0.4 (Fig. S7(b)†), and a broad pore size distribution between 3.5 and 11 nm was observed (Fig. S7(d)†). As expected, the BET specific surface area of the sample decreased from 375 to 309 cm2 g−1 after loading of Pt–Co NPs.
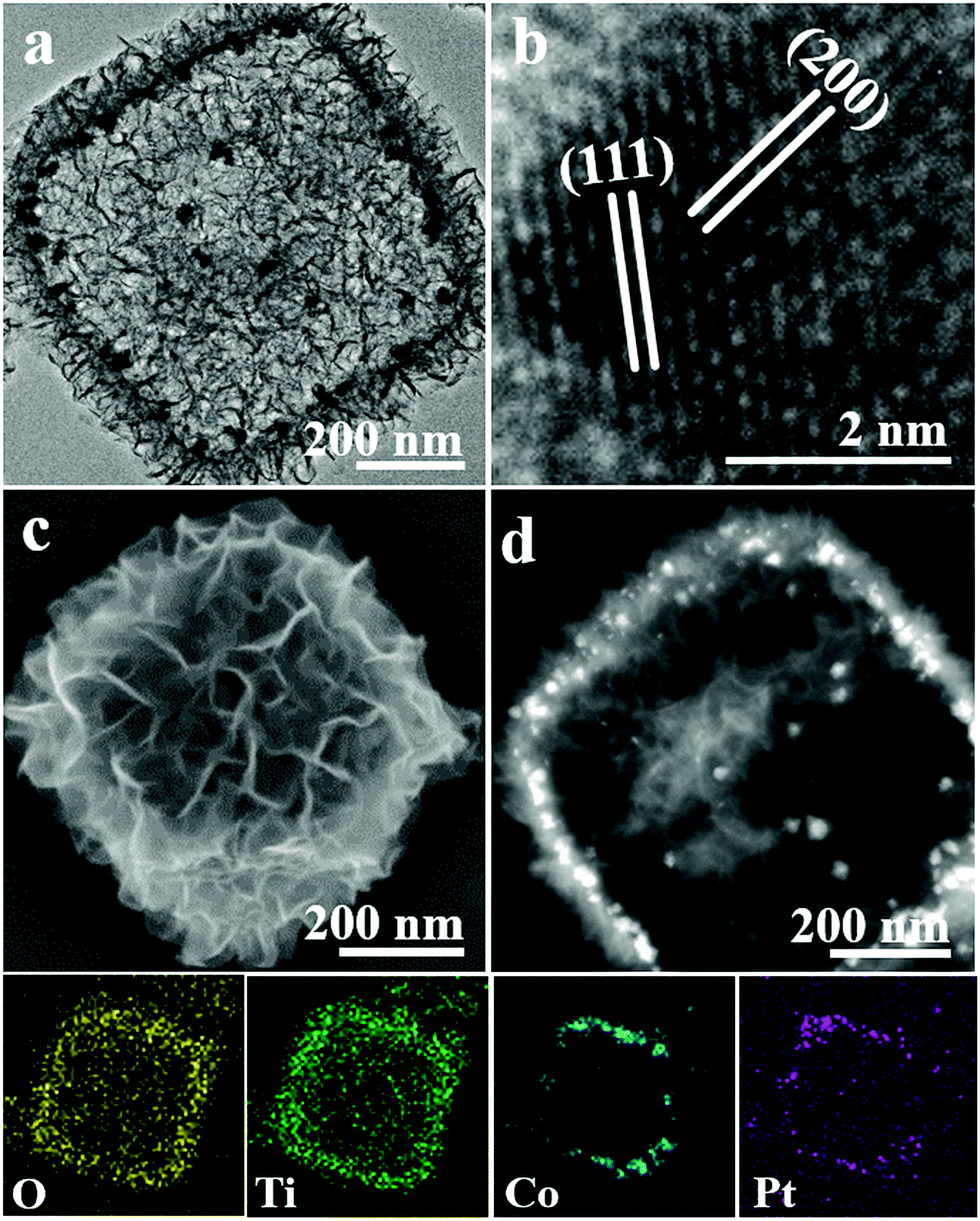 | ||
| Fig. 3 (a) TEM, (c) SEM and (d) HAADF-STEM images of Pt–Co/PCT; (b) HRTEM image of a Pt–Co NP; O, Ti, Co, and Pt EDXS elemental mapping of Pt–Co/PCT (bottom). | ||
By adjusting the amount of H2PtCl6 and Co(NO3)2, the as-synthesized composites with different components and structures were found (Fig. S14†). From these experimental observations, NH2-MIL-125(Ti) is unfavourably transferred to titanium oxides with a decrease of the molar ratio of H2PtCl6 to Co(NO3)2. The atomic ratio of Pt to Co in their alloys may be controlled by adjusting the amount of metal sources. For example, Pt3Co NP loaded PCT (Pt3Co/PCT; Pt: 3.62 wt%) was successfully synthesized by introducing Co(NO3)2 (3.5 μmol) and H2PtCl6 (3.5 μmol) to the synthetic system. In addition, other alloys, such as PtFe NP loaded porous cages of titanium oxides (PtFe/PCT; Pt: 3.43 wt%) were also synthesized by using Fe(NO3)3 to replace Co(NO3)2via the same solvothermal treatment process (Fig. S15†).
As shown in Fig. S16,† without introducing other metal precursors of Fe and Co to the synthetic system, large Pt NPs (∼15 nm) on porous cages of titanium oxides (Pt/PCT_1; Pt: 3.31 wt%) were produced at 176 °C for 12 h, verified by the relatively strong diffraction peaks of fcc structured Pt (JCPDS no. 04-0802). Thus, doping with Co or Fe facilitates generation of the reduced size Pt NPs. The reason could be attributed to formation of stable nuclei of the Pt alloy with a smaller diameter than those formed from pure Pt, and strain energy caused by the difference in size between metal atoms of Pt and Co (or Fe).54 The porous cages of titanium oxides assembled by sheets were not obviously changed after loading Pt NPs. The HRTEM image (Fig. S16(c)†) showed {111} facets of cubic Pt. The inset is a fast Fourier transform (FFT) pattern of two sets of diffraction spots corresponding to {111} and {200} facets of Pt with an interfacial angle of ∼54°.
In contrast, small Pt NP (<10 nm) loaded porous cages of titanium oxides (Pt/PCT_2, Pt: 4.01 wt%) could be prepared at lower reaction temperature, such as 165 °C (Fig. S17†). However, at the low temperature of 165 °C, the NH2-MIL-125(Ti) nanocrystals were transferred to titanium oxides with difficulty even after prolonging the reaction time to 24 h in the synthetic system including NH2-MIL-125(Ti), Co(NO3)2, H2PtCl6, and L-alanine (Fig. S18†). Although introducing metal species into the synthetic system affected the transformation of NH2-MIL-125(Ti), the formation of porous cages of titanium oxide anchored Pt, Pt–Co, Pt3Co and PtFe NPs was mainly related to L-alanine-assisted dissolution and recrystallization of Ti(IV) ions and reduction of metal ions (Fig. S19†).
Chemoselective hydrogenation of α,β-unsaturated aldehydes
The hydrogenation transformations over metal NP loaded PCT heterostructures were carried out under 0.2 MPa H2 at 80 °C (Fig. 4) with an equivalent amount of Pt (0.16 mg). The Pt–Co/PCT catalyst towards hydrogenation of cinnamaldehyde (CAL) displayed a considerably high selectivity of around 96.0% for cinnamyl alcohol (COL) at 100% conversion efficiency after 3 h (Fig. 4(a)). A small amount of saturated alcohol hydrocinnamyl alcohol (HCOL) was detected, and saturated aldehyde hydrocinnamaldehyde (HCAL) was not found (Fig. S25(a)†). CAL hydrogenation did not occur when PCT itself was used as the catalyst. Hence, the supported Pt–Co NPs possess excellent hydrogenation activity for CAL with high selectivity for the carbonyl bond compared with the olefinic bond. To fully understand the catalytic performance of Pt–Co/PCT, time-dependent catalytic experiments, which could reflect the variation trend of the selectivity of Pt–Co/PCT in the catalytic process, were performed (Fig. 4(b) and S26†). Upon shortening the reaction time to 0.5 h, 74.8% conversion of CAL with as high as 97.2% selectivity for COL was observed. With increasing reaction time, conversion of CAL was gradually increased, and selectivity for COL was slightly decreased. Upon prolonging the reaction time to 24 h, the selectivity for COL is partly decreased to 89.5%. The dramatic variation of selectivity for COL did not occur with the change of reaction time.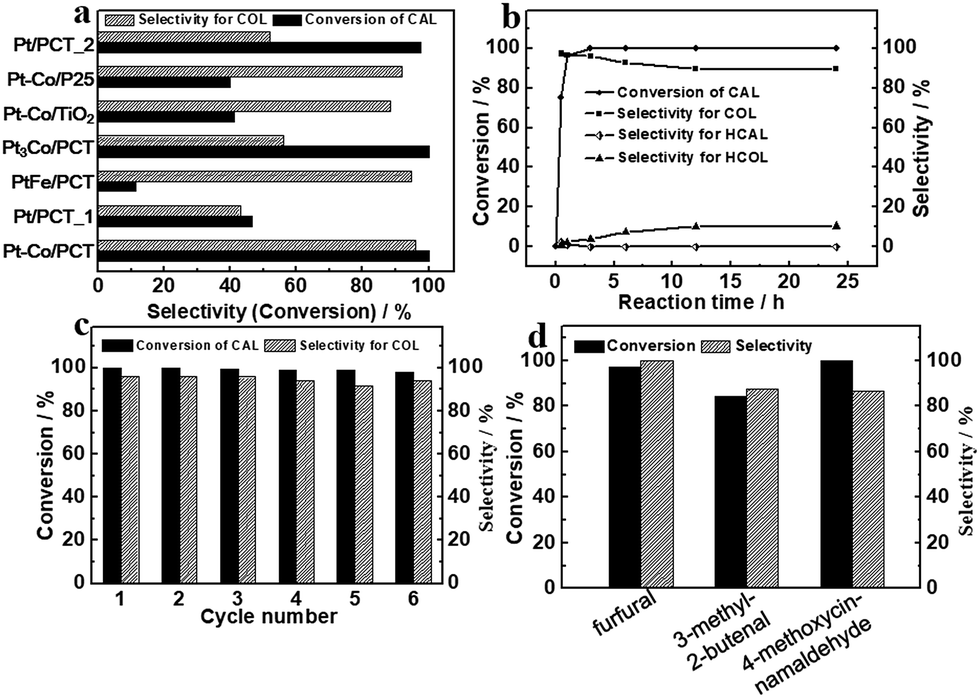 | ||
| Fig. 4 (a) Selective hydrogenation of cinnamaldehyde with different catalysts; (b) curves of conversion of CAL and selectivity for specific hydrogenation products using Pt–Co/PCT as catalyst; (c) recyclability tests of Pt–Co/PCT for catalytic hydrogenation of CAL; (d) selective hydrogenation of different α,β-unsaturated aldehydes by Pt–Co/PCT. | ||
Apart from reactivity and selectivity, recyclability is a crucial feature for heterogeneous catalysts. In the hydrogenation of the CAL process, Pt–Co/PCT shows high stability. No significant loss of the conversion efficiency of CAL or the selectivity for COL can be found after six successive catalytic cycles (Fig. 4(c) and S27†). Structural characterization of Pt–Co/PCT after the sixth cycling run reveals porous cages of titanium oxides without obvious damage and Pt–Co NPs without aggregation (Fig. S20†). Moreover, the Pt content (∼3.04 wt%) and the Pt/Co atomic ratio (∼2.78:1) did not conspicuously change after cyclic catalytic hydrogenation. The stable Pt–Co/PCT heterostructures can afford long-term high-efficiency catalytic performance of CAL hydrogenation.
In comparison with the supported Pt–Co NPs that showed high activity and selectivity for CAL hydrogenation, the Pt/PCT_1 catalysts exhibited low activity with ∼46.5% conversion of CAL for 3 h under the same catalytic condition of 0.2 MPa H2 and 80 °C (Fig. 4(a) and S25(b)†). The product selectivity for COL, HCAL and HCOL was about 42.9, 40.3 and 13.8%, respectively. The experimental observation is different from previous reports on high catalytic activity of Pt NPs, including hydrogenation of olefinic bonds based on an energetically favorable process.55 We infer that the low catalytic activity of Pt/PCT is mainly due to Pt NPs with large size.56 The Pt/PCT_2 catalyst was used for hydrogenation of CAL with 51.9% selectivity for COL at 97.8% conversion, further suggesting the size effect of Pt NPs in the catalytic hydrogenation process. Based on these experimental results, we propose that Co doping is supposed to greatly improve the selective hydrogenation performance of Pt NPs towards carbonyl groups.
To further understand the effect of dopants on catalytic performance of Pt NPs, PtFe/PCT and Pt3Co/PCT were used to study CAL hydrogenation. In contrast to the Pt–Co/PCT catalyst, PtFe/PCT heterostructures exhibited ultra-low conversion of CAL but comparable selectivity for COL under the same conditions (Fig. 4(a) and S25(c)†); Pt3Co-PCT hybrids gave similar catalytic activity (98.9%) towards CAL hydrogenation but a low selectivity of 56.3% for COL (Fig. 4(a) and S25(d)†). Thus, the amount and type of metal dopant played a significant role in determining the hydrogenation reactivity of olefinic and carbonyl bonds over Pt–metal alloys.
The fundamental purpose of engineering porous hollow supports is to solve the issue of mass transport for the enhancement of catalytic activity. As shown in Fig. S21,† commercial P25 and TiO2 were used as supports to prepare heterostructures of P25 or TiO2 anchored Pt–Co NPs (defined as Pt–Co/P25 and Pt–Co/TiO2, respectively). Note that TiO2 originated from NH2-MIL-125(Ti) by calcination in an air atmosphere. The content of Pt in the two heterostructures, slightly higher than that in Pt–Co/PCT, was around 3.92 and 4.27 wt%, respectively. The heterostructures gave low catalytic activity of CAL and acceptable selectivity for COL (Fig. 4(a), and S25(e) and S25(f)†). Therefore, by comparison with hydrogenation performance of Pt–Co alloys loaded on PCT, P25 and TiO2 supports, PCT shows an obvious advantage in the improvement of catalytic activity of Pt–Co alloys. The enhanced hydrogenation activity of Pt–Co/PCT is attributed to the support with porous hollow structures and favourable mass transport of substrates and products. In addition, we expected that the use of other non-Ti-based materials, such as porous SiO2, SBA-15, and cobalt layered double hydroxides, as supports would further increase the effect of supports, but Pt–Co alloys cannot be achieved via the amino acid-assisted route (Fig. S22†). From this perspective, introducing Ti-based supports is possibly beneficial for the production of Pt–Co alloys in L-alanine-related synthetic media.
Therefore, apart from improvement of mass transport by porous hollow structures, the excellent catalytic hydrogenation performance of Pt–Co/PCT hybrids was mainly attributed to the change of electronic structures of Pt, as well as introduction of defects to Pt. The defects were characterized using HRTEM images (Fig. S13†). To reveal the electronic structures of Pt, high-resolution XPS measurements were performed. As shown in Fig. 5(a) and (b), the Pt 4f region of core level binding energies was deconvoluted into two sets of spin–orbit doublet peaks in both Pt/PCT and Pt–Co/PCT_1. They can be assigned to multiple oxidation states of Pt0 and Pt2+. Pt0 4f7/2 and 4f5/2 peaks at 70.25 and 73.45 eV, and Pt2+ 4f7/2 and 4f5/2 peaks at 72.70 and 75.90 eV were observed for Pt/PCT_1. In contrast, Pt0 4f7/2 and 4f5/2 were positively shifted to 70.60 and 73.80 eV, revealing the reduced electron density of Pt in the Pt–Co/PCT hybrids; Pt2+ 4f7/2 and 4f5/2 were similar to those in Pt/PCT_1. The difference of Pt0 4f binding energy between Pt/PCT_1 and Pt–Co/PCT mainly originates from Co doping.52,57 Nonetheless, the binding energy of Pt 4f in Pt–Co/TiO2 and Pt–Co/P25 is still slightly different from that in Pt–Co/PCT (Fig. 5(c) and (d)). Thus, the electronic structures of Pt are affected by both dopants and supports.
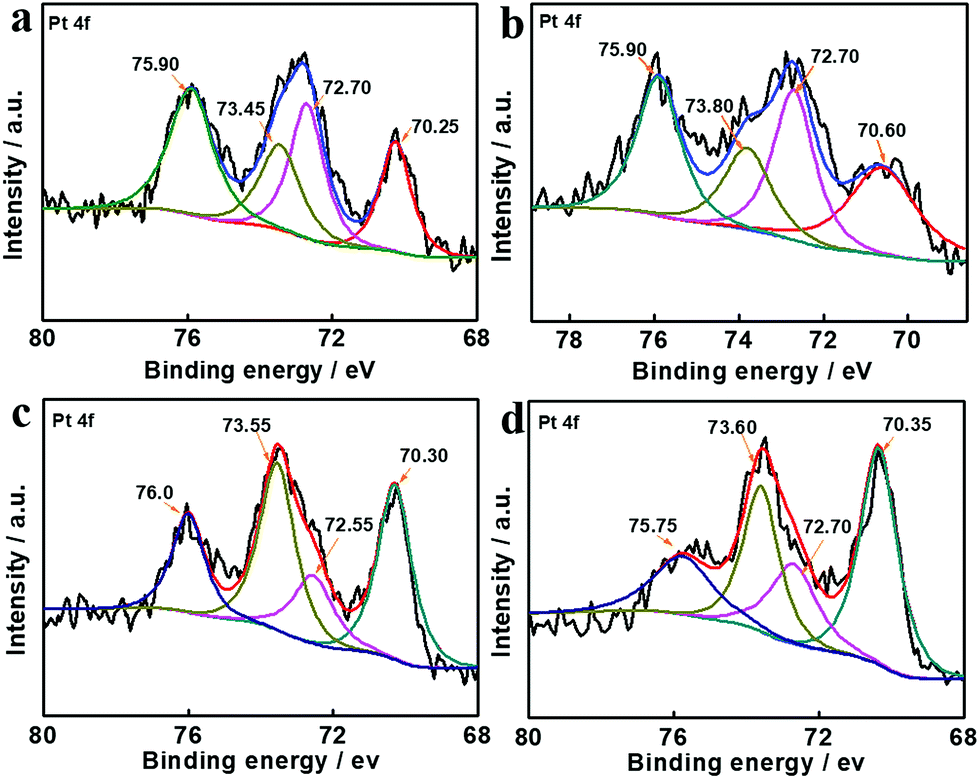 | ||
| Fig. 5 Core level Pt 4f XPS spectra of (a) Pt/PCT_1, (b) Pt–Co/PCT, (c) Pt–Co/TiO2, and (d) Pt–Co/P25. | ||
The electron density of Pt can affect their hydrogenation activity and regulate selectivity for carbonyl groups as well.26,57 The low electron density induced by the Co dopant and the titanium oxide support leads to a smaller contribution of back-donation of Pt 5d electrons to adsorbed molecules, and thus electron-rich O atoms of carbonyl groups are favourably adsorbed on the surface of Pt. Meanwhile, electron donation from carbonyl groups to Pt is increased with the increased d-vacancy in Pt–Co alloys, resulting in activation of carbonyl groups. FTIR spectra (Fig. S23†) showed that the stretching vibration of the carbonyl group in CAL at 1676 cm−1 was shifted to 1709 cm−1 after mixing with the Pt–Co/PCT, but no obvious change of the olefinic bond stretching vibration at 1630 cm−1 occurred. The result reveals that the carbonyl group possibly prefers to adsorb on Pt–Co NPs. Thereby, the significant enhancement of selectivity for COL is mainly due to the low electronic density of Pt0 in Pt–Co/PCT, accounting for its preferential interaction with carbonyl bonds and subsequent hydrogenation (Fig. S24†).
To further demonstrate the universal selective hydrogenation capabilities of Pt–Co/PCT, we expanded the substrate scope of α,β-unsaturated aldehydes to furfural, 3-methyl-2-butenal, and 4-methoxycinnamaldehyde under 0.2 MPa H2 and at 80 °C for 3 h (Fig. 4(d) and S28†). As expected, preferential hydrogenation of carbonyl bonds over olefinic bonds in all catalytic reactions was found when Pt–Co/PCT was used as the catalyst. Furfural alcohol was the sole product at 97.2% conversion of furfural, signifying the ultra-high chemoselectivity of Pt–Co/PCT for the carbonyl bond of furfural. In contrast to CAL, the yield of selective hydrogenation of carbonyl groups was decreased to 86.6% when 4-methoxycinnamaldehyde was completely converted. As for the α,β-unsaturated aliphatic aldehyde 3-methyl-2-butenal, Pt–Co/PCT gave a relatively low catalytic activity (conversion: 84.4%) with 87.7% selectivity for hydrogenation of carbonyl groups. Their selectivity for the products of unsaturated alcohols in the hydrogenation process of 3-methyl-2-butenal and 4-methoxycinnamaldehyde was slightly decreased with longer reaction time. These results reveal that the Pt–Co/PCT is one of the high-efficiency catalysts for chemoselective hydrogenation of α,β-unsaturated aldehydes based on the synergistic effect of the Co dopant and the porous cages of the titanium oxide support.
Conclusions
In summary, we developed an amino acid-mediated one-pot strategy to successfully synthesize the supported Pt or Pt–M alloy NPs on porous cages of titanium oxide while using NH2-MIL-125(Ti) as a self-sacrificial template. The as-formed nanosheets of layered titanates are the key factor for the formation of cage-like structures. The Pt–Co/PCT composite exhibits superior reactivity for catalytic hydrogenation of α,β-unsaturated aldehydes with high selectivity for unsaturated alcohols under mild conditions. The high-efficiency catalytic performance of the supported Pt–Co NPs was mainly attributed to two factors: (1) the Co dopant/titanium oxide support induced the formation of low electron density Pt accounting for its preferential interaction with carbonyl bonds, and (2) porous hollow supports facilitated the mass transport of substrates and products across the shells assembled by sheets. Our findings would help guide endeavours to engineer other novel composite catalysts by transformation of MOFs for achievement of many challenging organic transformations with high performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (Grant No. 21671030), the Key Program of the National Natural Science Foundation of China (Grant No. 21531001), and the 111 Project (Grant No. B16008).References
- Y. Chen and J. Shi, Adv. Mater., 2016, 28, 3235 CrossRef CAS PubMed
.
- R. E. Morris and J. Cejka, Nat. Chem., 2015, 7, 381 CrossRef CAS PubMed
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734 CrossRef CAS PubMed
- P. Zhang, L. Wang, S. Yang, J. A. Schott, X. Liu, S. M. Mahurin, C. Huang, Y. Zhang, P. F. Fulvio, M. F. Chisholm and S. Dai, Nat. Commun., 2017, 8, 15020 CrossRef PubMed
- N. Zhang and Y.-J. Xu, Chem. Mater., 2013, 25, 1979 CrossRef CAS
- Z. Zhang, Y. Chen, X. Xu, J. Zhang, G. Xiang, W. He and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 429 CrossRef CAS PubMed
- H.-W. Liang, X. Zhuang, S. Bruller, X. Feng and K. Mullen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed
- P.-C. Chen, X. Liu, J. L. Hedrick, Z. Xie, S. Wang, Q.-Y. Lin, M. C. Hersam, V. P. Dravid and C. A. Mirkin, Science, 2016, 352, 1565 CrossRef CAS PubMed
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983 CrossRef CAS PubMed
- L. Yu, H. Hu, H. B. Wu and X. W. Lou, Adv. Mater., 2017, 29, 1604563 CrossRef
- H. Zhang, T. Wang, J. Wang, H. Liu, D. Thang Duy, M. Li, G. Liu, X. Meng, K. Chang, L. Shi, T. Nagao and J. Ye, Adv. Mater., 2016, 28, 3703 CrossRef CAS
- W.-T. Koo, S.-J. Choi, S.-J. Kim, J.-S. Jang, H. L. Tuller and I.-D. Kim, J. Am. Chem. Soc., 2016, 138, 13431 CrossRef CAS PubMed
- H.-x. Zhong, J. Wang, Y.-w. Zhang, W.-l. Xu, W. Xing, D. Xu, Y.-f. Zhang and X.-b. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235 CrossRef CAS PubMed
- L. Chen, Y. Guo, H. Wang, Z. Gu, Y. Zhang, X. Li, H. Wang and C. Duan, J. Mater. Chem. A, 2018, 6, 4636 RSC
- Z. Jiang, Z. Li, Z. Qin, H. Sun, X. Jiao and D. Chen, Nanoscale, 2013, 5, 11770 RSC
- D. Fattakhova-Rohlfing, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487 CrossRef CAS PubMed
- Z. Wang, X. Li, H. Xu, Y. Yang, Y. Cui, H. Pan, Z. Wang, B. Chen and G. Qian, J. Mater. Chem. A, 2014, 2, 12571 RSC
- K. Khaletskaya, A. Pougin, R. Medishetty, C. Rosler, C. Wiktor, J. Strunk and R. A. Fischer, Chem. Mater., 2015, 27, 7248 CrossRef CAS
- W. S. Chi, D. K. Roh, C. S. Lee and J. H. Kim, J. Mater. Chem. A, 2015, 3, 21599 RSC
- X. Shi, Z. Zhang, K. Du, Y. Lai, J. Fang and J. Li, J. Power Sources, 2016, 330, 1 CrossRef CAS
- A. V. Vinogradov, H. Zaake-Hertling, E. Hey-Hawkins, A. V. Agafonov, G. A. Seisenbaeva, V. G. Kessler and V. V. Vinogradov, Chem. Commun., 2014, 50, 10210 RSC
- Z. Hong, M. Kang, X. Chen, K. Zhou, Z. Huang and M. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 32071 CrossRef CAS PubMed
- L. Wang and T. Sasaki, Chem. Rev., 2014, 114, 9455 CrossRef CAS
- C. Zlotea, D. Phanon, M. Mazaj, D. Heurtaux, V. Guillerm, C. Serre, P. Horcajada, T. Devic, E. Magnier, F. Cuevas, G. Ferey, P. L. Llewellyn and M. Latroche, Dalton Trans., 2011, 40, 4879 RSC
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76 CrossRef CAS
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295 CrossRef CAS
- B. Wu, H. Huang, J. Yang, N. Zheng and G. Fu, Angew. Chem., Int. Ed., 2012, 51, 3440 CrossRef CAS
- F. Meemken and A. Baiker, Chem. Rev., 2017, 117, 11522 CrossRef CAS PubMed
- C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816 CrossRef CAS PubMed
- C. V. Deraedt, R. Ye, W. Ralston, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2017, 139, 18084 CrossRef CAS PubMed
- Z. Niu, N. Becknell, Y. Yu, D. Kim, C. Chen, N. Kornienko, G. A. Somorjai and P. Yang, Nat. Mater., 2016, 15, 1188 CrossRef CAS
- W. Sang, T. Zheng, Y. Wang, X. Li, X. Zhao, J. Zeng and J. G. Hou, Nano Lett., 2014, 14, 6666 CrossRef CAS
- S. C. Tsang, N. Cailuo, W. Oduro, A. T. S. Kong, L. Clifton, K. M. K. Yu, B. Thiebaut, J. Cookson and P. Bishop, ACS Nano, 2008, 2, 2547 CrossRef CAS PubMed
- A. B. Merlo, B. F. Machado, V. Vetere, J. L. Faria and M. L. Casella, Appl. Catal., A, 2010, 383, 43 CrossRef CAS
- X. X. Han, R. X. Zhou, B. H. Yue and X. M. Zheng, Catal. Lett., 2006, 109, 157 CrossRef CAS
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780 CrossRef CAS
- A. Corma, P. Serna, P. Concepcion and J. Juan Calvino, J. Am. Chem. Soc., 2008, 130, 8748 CrossRef CAS PubMed
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, ACS Catal., 2012, 2, 671 CrossRef CAS
- S. Kenjo and H. Yoshitake, Microporous Mesoporous Mater., 2017, 237, 12 CrossRef CAS
- E. Bailon-Garcia, F. Carrasco-Marin, A. F. Perez-Cadenas and F. J. Maldonado-Hodar, J. Catal., 2016, 344, 701 CrossRef CAS
- T. Ekou, L. Ekou, A. Vicente, G. Lafaye, S. Pronier, C. Especel and P. Marecot, J. Mol. Catal. A: Chem., 2011, 337, 82 CrossRef CAS
- G. Kennedy, L. R. Baker and G. A. Somorjai, Angew. Chem., Int. Ed., 2014, 53, 3405 CrossRef CAS PubMed
- H. Liang, B. Zhang, H. Ge, X. Gu, S. Zhang and Y. Qin, ACS Catal., 2017, 7, 6567 CrossRef CAS
- P. Claus, Top. Catal., 1998, 5, 51 CrossRef CAS
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321 CrossRef
- T. Marino, M. Toscano, N. Russo and A. Grand, J. Phys. Chem. B, 2006, 110, 24666 CrossRef CAS
- J. Zhu, S. Wang, Z. Bian, S. Xie, C. Cai, J. Wang, H. Yang and H. Li, CrystEngComm, 2010, 12, 2219 RSC
- Z. Gu, L. Chen, H. Wang, Y. Guo, M. Xu, Y. Zhang and C. Duan, Int. J. Hydrogen Energy, 2017, 42, 26713 CrossRef CAS
- H. Pang, F. Gao and Q. Lu, CrystEngComm, 2010, 12, 406 RSC
- A. Lewera, L. Timperman, A. Roguska and N. Alonso-Vante, J. Phys. Chem. C, 2011, 115, 20153 CrossRef CAS
- B. Y. Xia, H. B. Wu, N. Li, Y. Yan, X. W. Lou and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 3797 CrossRef CAS
- M. U. Khan, L. Wang, Z. Liu, Z. Gao, S. Wang, H. Li, W. Zhang, M. Wang, Z. Wang, C. Ma and J. Zeng, Angew. Chem., Int. Ed., 2016, 55, 9548 CrossRef
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143 CrossRef CAS
- S. Song, X. Liu, J. Li, J. Pan, F. Wang, Y. Xing, X. Wang, X. Liu and H. Zhang, Adv. Mater., 2017, 29, 1700495 CrossRef
- E. Bailon-Garcia, F. Carrasco-Marin, A. F. Perez-Cadenas and F. J. Maldonado-Hodar, Catal. Commun., 2016, 82, 36 CrossRef CAS
- H. Igarashi, T. Fujino, Y. M. Zhu, H. Uchida and M. Watanabe, Phys. Chem. Chem. Phys., 2001, 3, 306 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, XRD, TEM, SEM, AFM, XPS, Raman spectra, FT-IR spectra, GC-MS analysis and the nitrogen adsorption isothermal curve. See DOI: 10.1039/c8sc05450a |
| This journal is © The Royal Society of Chemistry 2019 |