
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure revision of cryptosporioptides and determination of the genetic basis for dimeric xanthone biosynthesis in fungi†
Claudio
Greco
 a,
Kate
de Mattos-Shipley
a,
Andrew M.
Bailey
b,
Nicholas P.
Mulholland
c,
Jason L.
Vincent
c,
Christine L.
Willis
a,
Russell J.
Cox
*ade and
Thomas J.
Simpson
*a
a,
Kate
de Mattos-Shipley
a,
Andrew M.
Bailey
b,
Nicholas P.
Mulholland
c,
Jason L.
Vincent
c,
Christine L.
Willis
a,
Russell J.
Cox
*ade and
Thomas J.
Simpson
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, UK BS8 1TS. E-mail: russell.cox@oci.uni-hannover.de; tom.simpson@bristol.ac.uk
bSchool of Biological Sciences, 24 Tyndall Avenue, Bristol, BS8 1TQ, UK
cSyngenta, Jealott's Hill International Research Centre, Bracknell, RG42 6EY, UK
dInstitute for Organic Chemistry, Leibniz University of Hannover, Schneiderberg 1B, 30167, Hannover, Germany
eBMWZ, Leibniz University of Hannover, Schneiderberg 38, 30167, Hannover, Germany
First published on 21st January 2019
Abstract
Three novel dimeric xanthones, cryptosporioptides A–C were isolated from Cryptosporiopsis sp. 8999 and their structures elucidated. Methylation of cryptosporioptide A gave a methyl ester with identical NMR data to cryptosporioptide, a compound previously reported to have been isolated from the same fungus. However, HRMS analysis revealed that cryptosporioptide is a symmetrical dimer, not a monomer as previously proposed, and the revised structure was elucidated by extensive NMR analysis. The genome of Cryptosporiopsis sp. 8999 was sequenced and the dimeric xanthone (dmx) biosynthetic gene cluster responsible for the production of the cryptosporioptides was identified. Gene disruption experiments identified a gene (dmxR5) encoding a cytochrome P450 oxygenase as being responsible for the dimerisation step late in the biosynthetic pathway. Disruption of dmxR5 led to the isolation of novel monomeric xanthones. Cryptosporioptide B and C feature an unusual ethylmalonate subunit: a hrPKS and acyl CoA carboxylase are responsible for its formation. Bioinformatic analysis of the genomes of several fungi producing related xanthones, e.g. the widely occurring ergochromes, and related metabolites allows detailed annotation of the biosynthetic genes, and a rational overall biosynthetic scheme for the production of fungal dimeric xanthones to be proposed.
Introduction
Anthraquinones and xanthones are among the most common and earliest discovered fungal secondary metabolites,1 and they often have interesting and useful bioactivities. These compounds are polyketides, the skeletons of which are produced from acetyl and malonyl CoA by non-reducing polyketide synthases (nr-PKS).2 Examples include: the anthraquinones emodin 1 and its reduced congener chrysophanol 2 which is a precursor of shamixanthone 3,3 and monodictyxanthone 44 from Aspergillus nidulans.5,6 Emodin 1 is a precursor of the antifungal agent geodin 5,7 which while not strictly a xanthone, belongs to the same biosynthetic family. We have also recently isolated other xanthones such as agnestin A 6 from Paecilomyces variotii and linked it to its biosynthetic genes.8 Cladofulvin 79 from the tomato pathogen Cladosporium fulvum is a dimeric anthraquinone also derived from 2. Dimeric xanthones are also known, and they often have varied biological activities.10 They include the antimalarial ascherxanthone A 8;11 the antimicrobial and anticancer dicerandrol C 9;12 and the mycotoxins secalonic acids B 10 and D 11 which are also known as ergochromes (Fig. 1).13 Although many of the dimeric xanthones, particularly the widespread secalonic acid family, have been studied for many years,14 key aspects of their biosynthesis such as the mechanisms and timing of the dimerisation process, and isolation and analysis of their biosynthetic gene clusters remain largely unknown.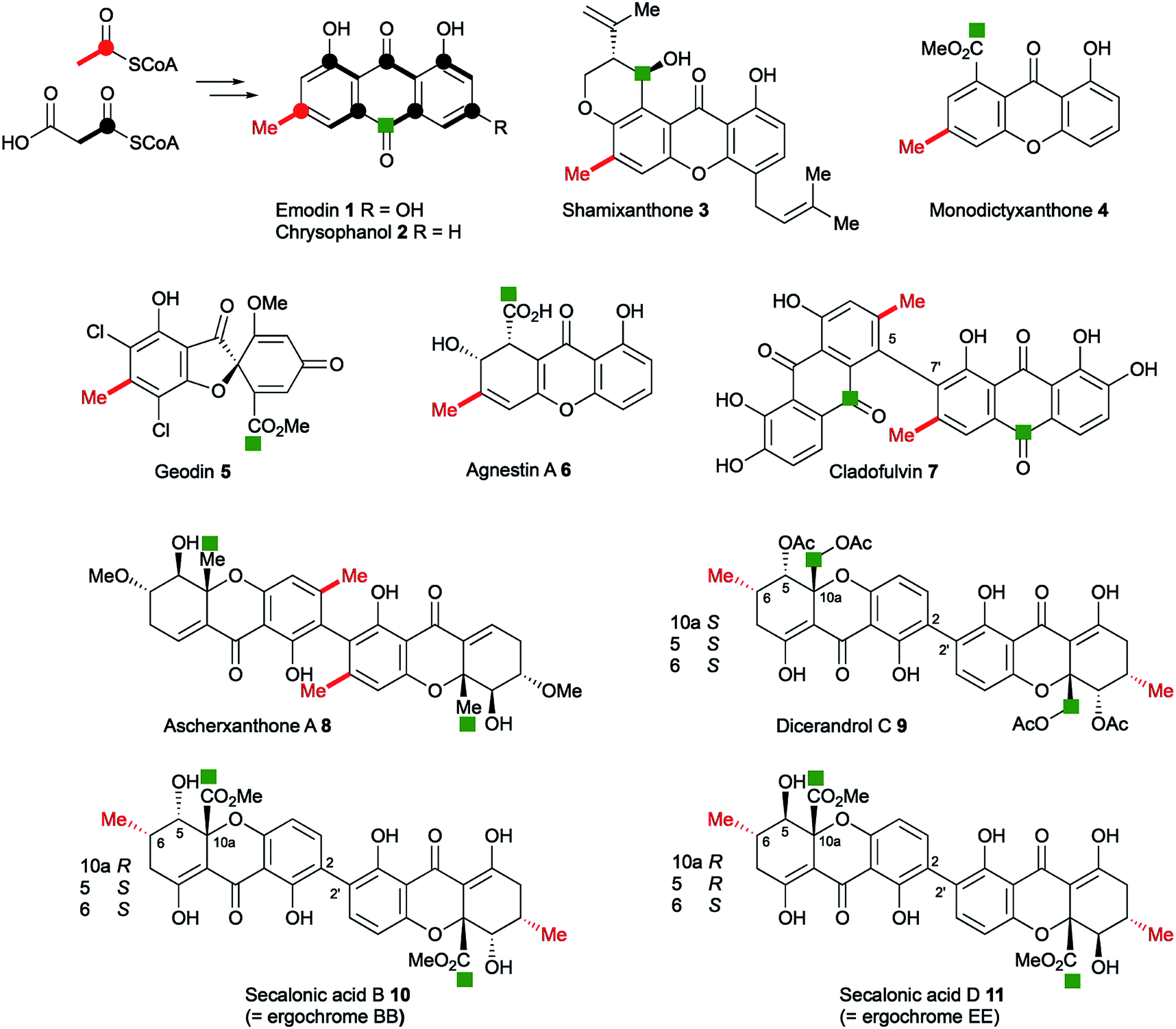 | ||
| Fig. 1 Typical fungal anthraquinone and xanthone metabolites. Isotope labelling pattern is shown for emodin/chrysophanol with the polyketide starter unit indicated in red. The green square labels (e.g. C-12 of the secalonic acids) indicate equivalent carbons derived from the C-10 carbonyl of emodin/chrysophanol. | ||
In the course of our ongoing studies on fungal maleidride biosynthesis15 we analysed fermentations of the endophytic fungus Cryptosporiopsis sp. 8999, which produces the unusual octadride, viburspiran.16 This strain was also reported17 to produce a metabolite named cryptosporioptide, which was assigned the monomeric xanthone methyl ester structure 12 (Fig. 2). This structure contains an unusual ring-contracted xanthone which is difficult to rationalise biosynthetically, and an unprecedented N-malonyl aminal bridge. Further studies18 of this strain also led to the isolation of the corresponding free acid, named cryptosporioptide A 13, and cryptosporioptide B 14, which lacks the malonic acid ester amide bridge.
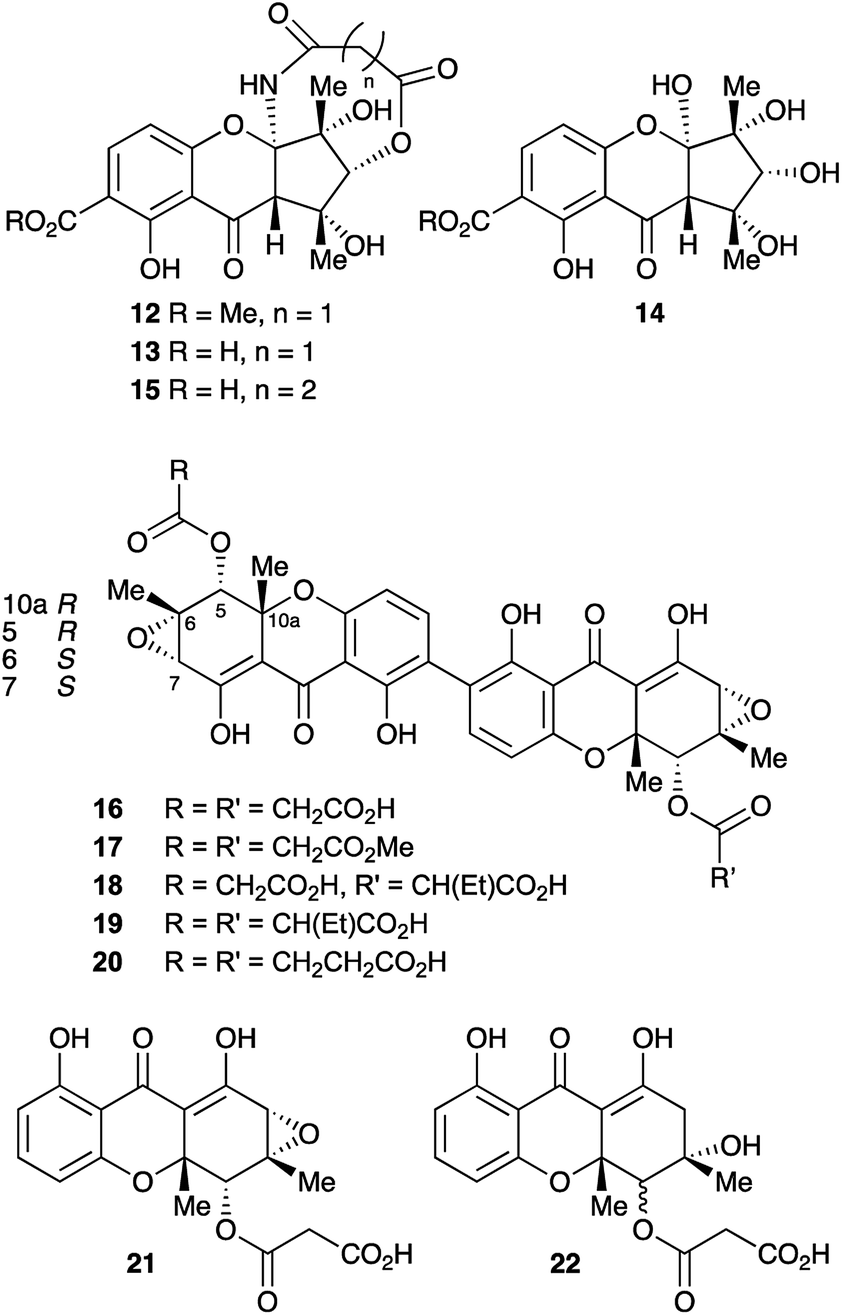 | ||
| Fig. 2 Previous (12–15) and reassigned (16–20) structures of the cryptosporioptides. | ||
Subsequently, isolation of an analogue 15, in which the malonate bridge has been replaced by a succinate, was reported from the insect parasite Cordyceps gracilloides.19 Confusingly, this was also named cryptosporioptide A.
The structure of the parent cryptosporioptide 12 was assigned on the basis of FAB-HRMS which indicated a molecular formula C19H19NO10. Detailed analysis of 1D and 2D 1H and 13C NMR spectra gave the connectivities and relative stereochemistry on which the structures were assigned. The absolute configuration was assigned by circular dichroism allied to time-dependent density functional theory (TDDFT) computational procedures.11 The related structures 13 and 15 were assigned on the basis of the similarity of their NMR spectra and optical rotations to those originally reported for cryptosporioptide.
We now report the isolation and structure elucidation of a novel dimeric tetrahydroxanthone metabolite 16, methylation of which gives a compound with identical NMR properties to those reported17 for cryptosporioptide, along with related dimeric structures, 17–19 (Fig. 2). Following sequencing of the biosynthetic gene cluster and targeted gene knock-outs, the monomeric structures, 21 and 22 were also isolated. Comparison with genome sequences of fungi known to produce xanthones and related structures, allows identification and annotation of the biosynthetic gene clusters (BCGs) for several monomeric and dimeric xanthones, inter alia the secalonic acids.
Results
Cryptosporiopsis sp. 8999 was grown on a range of different media. Three related metabolites were produced in good yields when the fungus was grown on brown rice (e.g.Fig. 4B). The major metabolite (900 mg kg−1) had almost identical IR, UV and 1H and 13C NMR data apart from the lack of a methoxy group when compared to that reported for cryptosporioptide A 13.17 When esterified using TMS-diazomethane, the resulting methyl ester had identical NMR data to those reported for cryptosporioptide 12 by Saleem et al.,17 for which the reported FAB-HRMS data was 444.0781 [M + Na]+ (C19H19NO10Na).17 However, when the molecular weight for the derivatised compound was redetermined using FAB-HRMS and ESI-HRMS, we obtained HRMS of 801.1506 [M + Na]+, and 778.1828 [M]+ respectively, both consistent with a molecular formula C38H34O18, and notably lacking nitrogen. The 1H NMR spectrum showed 9 hydrogen environments, but 19 different carbon environments were observed in the 13C NMR spectrum consistent with a symmetrical dimer.The presence of a tetrasubstituted aromatic ring was indicated by ortho-coupled hydrogens (δH 7.70 and 6.43 ppm, J = 8.5 Hz) in the 1H NMR spectrum, which also showed two methyl singlets (δH 1.65 and 1.59 ppm), two oxygen bearing methine singlets (δH 3.42 and 5.64 ppm), two low field exchangeable hydrogens (δH 11.69 and 14.02 ppm) and geminally coupled methylenes (δH 3.39 and 3.44 ppm, J = 15.8 Hz). The 13C NMR spectrum also showed other signals attributable to a phenolic ring (δC 159.2, 157.2, 140.6 and 106.1 ppm), four ester/enolic carbons (δC 165.5, 166.7, 170.0 and 104.4 ppm) and a benzophenone carbonyl (δC 187.9 ppm). Signals at δC 56.0, 58.8, 73.9 and 78.9 ppm are consistent with the presence of an epoxide, secondary alcohol and ether. The connectivities were determined from extensive HMBC correlations which confirmed the presence of the aromatic ring, and a highly substituted cyclohexene (Fig. 3), the key signal being H-5 (5.64 ppm) which correlates with a total of eight carbons (6, 7, 8a, 9, 10a, 11, 12 and 13 at δC 58.8, 56.0, 170.0, 187.9, 78.9, 28.6, 18.2 and 165.5 ppm respectively). The H-5/C-13 correlation establishes the position of attachment of the malonate moiety, the other malonate carbonyl (C-15) correlating with the methoxyl (δH 3.65, δC 52.7 ppm). The connectivities between the two carbocyclic rings were confirmed by observation of HMBC correlations (see ESI Fig. S15†) from both H-5 (5.64 ppm) and H-4 (6.43 ppm) to the C-9 ketone (δC 187.9 ppm), and nOes between the 12-methyl and H-4 in the aromatic ring. The relative stereochemistry C-5 to C-7 and C-11 was established by nOes between H-5 and H-7 and the 11- and 12-methyls.
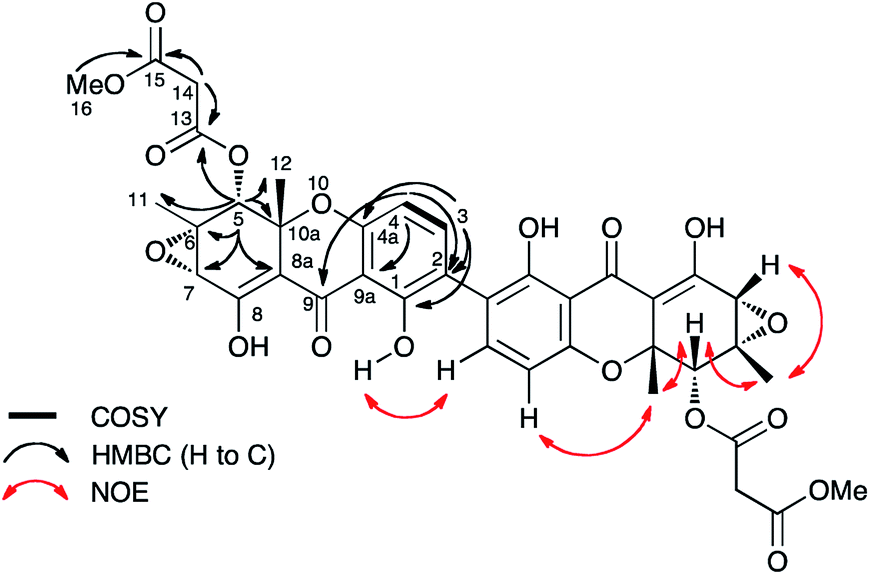 | ||
| Fig. 3 Selected correlations observed in 2D NMR spectra of cryptosporioptide A dimethyl ester 17. Secalonic acid numbering used. See Table S7 (ESI†) for full details. | ||
While the absolute configuration of the original cryptosporioptide structure was established by comparison of the measured circular dichroism (CD) with that predicted by computational methods, the complete revision of the structure renders this assignment invalid, and indeed suggests caution should be exercised when applying these methods which are being increasingly used for configurational assignment. The absolute configuration at the C-10a stereogenic centres of secalonic acid B 10 and related structures, e.g. dicerandrol C 9 have been assigned on the basis of a positive n–π* CD band at 330–340 nm as 10aR and 10aS respectively (note change in CIP designation due to priority inversion for CO2Me and CH2OAc).20–22 The absolute configuration of dicerandrol C 9 has also been confirmed by total synthesis.23 Cryptosporioptide A dimethyl ester 17 has a reported CD (341 nm, Δε = + 5.0)17 which is consistent with the 10aR configuration. Thus cryptosprioptide can be designated as 5R,6S,7S,10aR.
Finally, the connectivity between the individual xanthone monomers was established by an intra-dimer nOe between 1-OH and H-3. The point of dimerisation was confirmed by isolation of monomers 21 and 22 following KO of DmxR5, the cytochrome P450 responsible for oxidative coupling (see below). Thus the previously reported structures 12, and 13 have been revised to 17 and 16 respectively, and we propose renaming them cryptosporioptide A dimethyl ester, and cryptosporioptide A respectively. We have not observed any trace of the methyl ester 17 in any of our extracts, and the reported isolation of 17 as a natural product is possibly an artefact of the purification involved (Sephadex LH20 eluted with methanol).
The remaining two dimeric metabolites, cryptosporioptides B 18 and C 19, differ from cryptosporioptide A 16 in the malonyl subunit, their UV and NMR spectra being otherwise identical (see ESI Tables S1 and S2†). HRMS showed their molecular formulae to be C38H34O18 and C40H38O18 respectively. The NMR spectra showed the latter 19 to be also essentially symmetrical with signals attributable to the ethylmalonate moieties (δH 3.29, 2H, m; 1.81, 4H, m; and 0.85, 6H, m). Doubling of some signals was observed, probably due to facile epimerisation of the ethylmalonyl substituent, giving diastereomers at C-13. The spectra for 18 are more complex with many signals doubled due to the presence of only one ethylmalonate moiety removing the symmetry. Again on the basis of the close similarities of the NMR spectra, the C. gracilloides metabolite1915 has been renamed as cryptosporioptide D 20.
While the biosynthetic pathways towards monomeric xanthones and related compounds such as geodin 5 have been investigated previously, there is much less information on the dimeric systems despite these having been the subject of intensive study over many years.10 The main question is the timing of ring cleavage to give the xanthone moiety relative to dimerisation. The recently reported20 co-occurrence of the monomeric blennolides (e.g. ergochrome B) with their symmetrical dimer, secalonic acid B 10 supports, but does not unequivocally prove that during secalonic acid biosynthesis, xanthone formation precedes dimerisation. In those systems where the anthraquinonoid carbonyl (i.e. C-10) is retained, it is often found as a methyl ester as in the secalonic acids (note C-12 in SA numbering), or partially reduced to a hydroxymethyl equivalent, e.g. in dicerandrol C 9, or fully reduced to a methyl group as in the ascherxanthones, e.g.8 and cryptosporioptides 16–20. Other questions include: which of the original anthraquinone rings retains the cleaved carbonyl C-10 (C-12 in SA nomenclature) – that containing the polyketide starter unit as in secalonic acids, e.g.10 and cryptosporioptides or the non-starter ring as in, e.g. ascherxanthones 8; and the location of the site of dimerisation (Fig. 1).
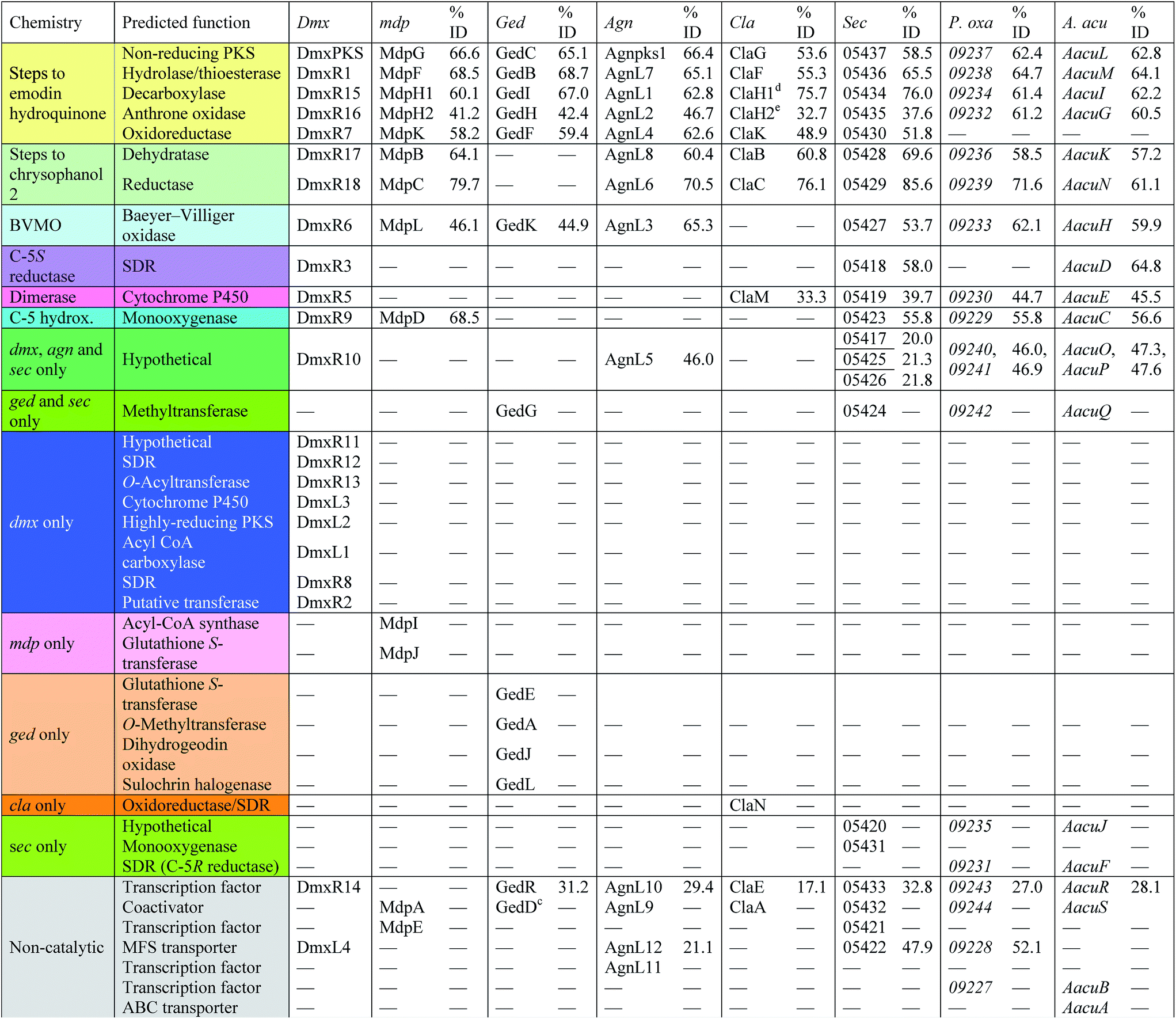
With these questions in mind, the 54 Mbp draft genome of Cryptosporiopsis sp. 8999 was obtained using Illumina MiSeq and assembled and annotated using Newbler v29. The total number of contigs was 2166 with an N50 of 204 Kb. Bioinformatic studies using antiSMASH24 identified ten BGCs containing non-reducing polyketide synthase (nr-PKS) genes. A putative gene cluster for xanthone biosynthesis25 was readily identified by BLAST analysis using protein sequences from: the shamixanthone 3/monodictyphenone 4 BGC from Aspergillus nidulans;5,6,26 the agnestin 6 BGC from Paecilomyces variotii;8 the recently (but partially, vide infra) described secalonic acid 10 BGC from Claviceps purpurea;27 the geodin 5 BGC from Aspergillus terreus;7 and the cladofulvin 7 BGC from Cladosporium fulvum.9 Development of a transformation system, targetted knockout of the putative cryptosporioptide (dmx) PKS using the bipartite method of Neilsen and coworkers,28 and subsequent observation of abolition of all cryptosporioptide production (Fig. 4) proved this assignment to be correct. We then used the cryptosporioptide BGC to screen the genomes of other fungi for similar clusters. Penicillium oxalicum29 and Aspergillus aculeatus30 are both known to produce secalonic acid D 11,31 and their genomes have been sequenced.32 In both cases we found a BGC (P. oxa, A. acuTable 1) featuring homologs of many of the genes in the dmx BGC.
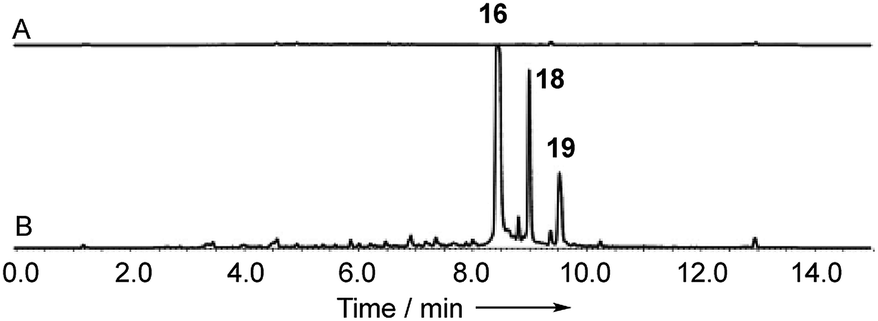 | ||
| Fig. 4 Knockout of dmxpks1. (A) LCMS analysis (DAD) of organic extract of Cryptosporiopsis sp. 8999 Δdmxpks1 strain fermented on brown rice; (B) LCMS analysis (DAD) of organic extract of wild type Cryptosporiopsis sp. 8999 fermented on brown rice. LCMS method 50–90% CH3CN:H2O gradient, 15 min. | ||
|
The dmx BGC spans approximately 84 Kb surrounding dmxPKS which encodes a fungal nr-PKS homologous to the monodictyphenone PKS (MdpG, 67% identity) from A. nidulans6 and the analogous polyketide synthases encoded by the geodin 5 (ged), agnestin 6 (agn), cladofulvin 7 (cla) and secalonic acid 10, 11 (sec) clusters (Table 1, Fig. 5). Homologies to all genes which encode proteins required for the synthesis of emodin 1 (dmxpks, dmxR1, dmxR15 and dmxR16) are present, as are the genes required for the synthesis of chrysophanol 2 (dmxR7, dmxR17 and dmxR18). The geodin pathway, which does not proceed via chrysophanol 2, lacks homologs of dmxR17 and dmxR18.
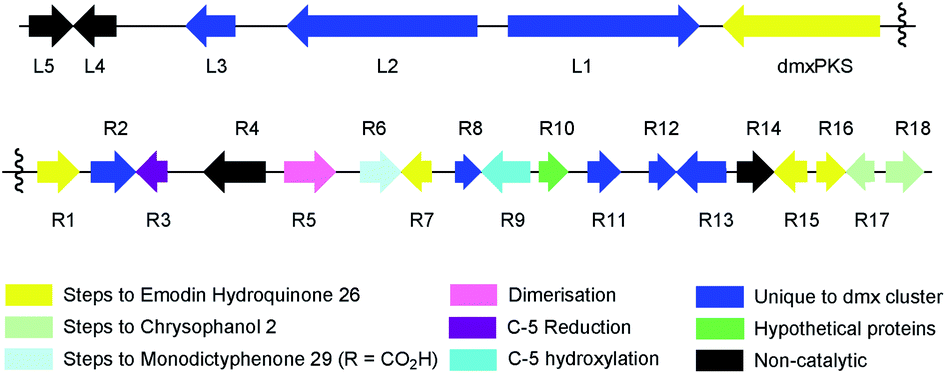 | ||
| Fig. 5 Map of the cryptosporioptide dimeric xanthone (dmx) BGC coloured by proposed function (see Table 1). Not to scale. | ||
Oxidative ring-opening of chrysophanol 2 is specified by dmxR6 which encodes a Baeyer–Villiger monooxygenase (BVMO) homologous to those encoded by the mdp, ged, agn and sec clusters where this transformation is required. Notably cladofulvin, which is a dimeric anthraquinone, does not require this chemistry and the cla cluster lacks a dmxR6 homolog. Beyond this point the content of the clusters varies significantly.
The dmx BGC has a series of unique genes not present in the other clusters such as dmxR2, dmxR8, dmxR11-13 and dmxL1-3. In particular dmxL2 encodes a highly reducing PKS (hr-PKS) homologous to the lovastatin diketide synthase33 and squalestatin tetraketide synthase.34 The gene dmxL1 encodes an acyl-CoA carboxylase. The gene dmxR5 encodes a cytochrome P450 enzyme homologous to ClaM which is known to dimerise nataloe-emodin to form cladofulvin 7.35,36 This gene is missing from the clusters of the monomeric compounds, but is present in the sec cluster (CPUR_05419). Finally, dmxR13 encodes an O-acyl transferase, and again this is absent from the other clusters.
In order to gain evidence for the function of these genes we devised knockout experiments. Disruption of dmxR6 (BVMO) gave high titres of chrysophanol 2 as expected, showing that dimerisation occurs after anthraquinone ring cleavage and xanthone formation (Fig. 6). Deletion of dmxR5 (putative dimerase) abolished dimer production and two novel metabolites with similar UV spectra to the cryptosporioptides but with molecular weights consistent with monomers were isolated (Fig. 7). Full NMR analysis (see ESI†) confirmed that these were hemi-cryptosporioptide 21 (C19H16O9), and an analogue 22 (C19H18O9) containing a tertiary alcohol at C-6 which was isolated as a mixture of epimers at C-5.
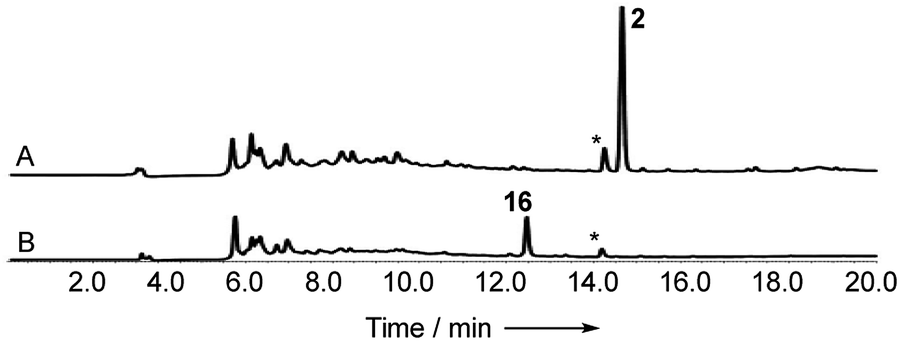 | ||
| Fig. 6 Knockout of dmxR6 (BVMO). (A) LCMS analysis (DAD) of organic extract of Cryptosporiopsis sp. 8999 ΔdmxR6 fermented on brown rice; (B) LCMS analysis (DAD) of organic extract of wild type Cryptosporiopsis sp. 8999 fermented on brown rice. LCMS method 50–90% CH3CN:H2O gradient, 20 min. * = unrelated compound. | ||
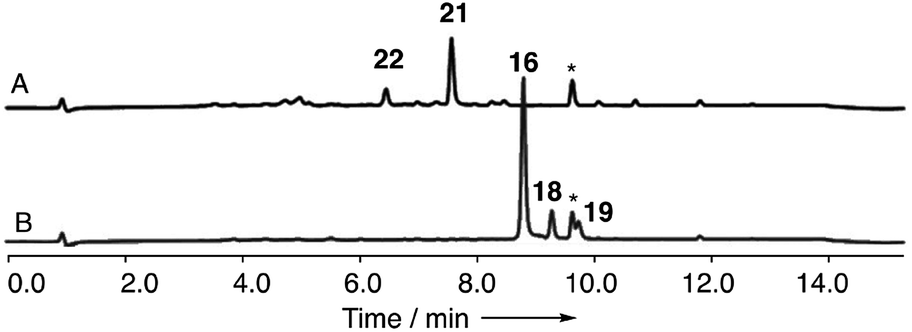 | ||
| Fig. 7 Knockout of dmxR5 (P450). (A) LCMS analysis (DAD) of organic extract of Cryptosporiopsis sp. 8999 ΔdmxR5 fermented on brown rice; (B) LCMS analysis (DAD) of organic extract of wild type Cryptosporiopsis sp. 8999 fermented on brown rice. LCMS method 50–90% CH3CN:H2O gradient, 15 min. * = unrelated compound. | ||
When monomer 21, but not 22, was re-fed to the ΔdmxPKS mutant, cryptosporioptide A 16 production was restored (Fig. 8), confirming this as a pathway intermediate.
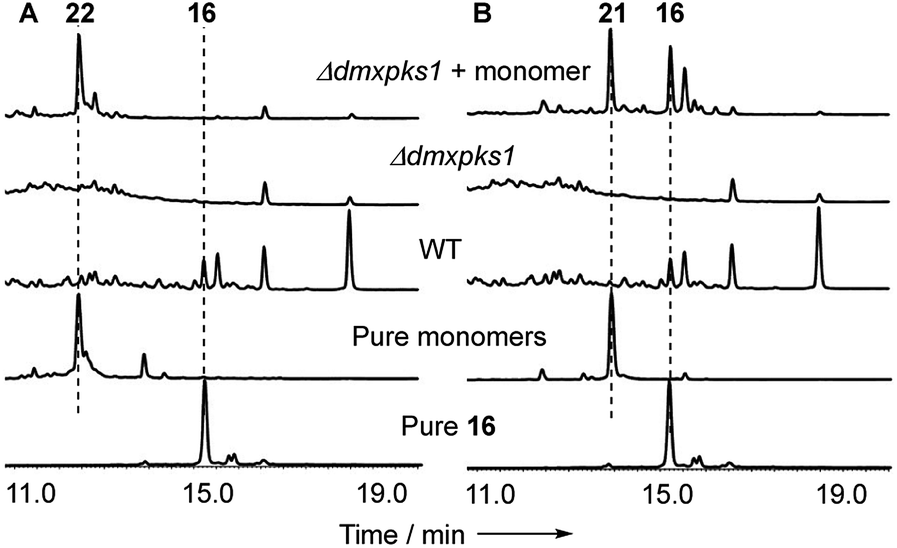 | ||
| Fig. 8 Experiments showing that 21 is a precursor of 16: (A) feeding experiments using purified monomer 22; (B) feeding experiments using purified monomer 21. Fermentations were performed in liquid PDB media. LCMS method 10–90% CH3CN:H2O gradient, 20 min. DAD scans shown. | ||
Disruptions of dmxL2 and dmxL1 both abolished production of cryptosprioptide B 18 and C 19, to give exclusively cryptosporioptide A 16 (Fig. 9B and C). Knockout of dmxR13 gave a similar result, abolishing cryptosporioptide C 18 and D 19 production but retaining cryptosporioptide A 16 in smaller quantities than previously (Fig. 9D). A number of new metabolites were produced in small quantities that precluded structure elucidation although mass spectrometry indicated molecular weights consistent with monomeric xanthones, in particular non-malonylated monomeric xanthone 35 (Scheme 1).
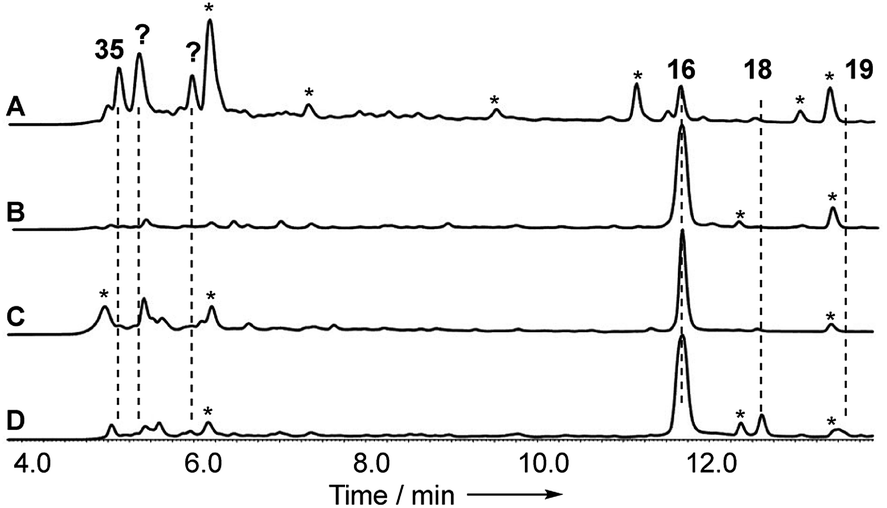 | ||
| Fig. 9 Analysis of knockout experiments: (A) LCMS analysis (DAD) of organic extracts from ΔdmxR13 (O-acetyl transferase); (B) LCMS analysis (DAD) of organic extracts from ΔdmxL1 (acyl CoA carboxylase); (C) LCMS analysis (DAD) of organic extracts from ΔdmxL2 (hrPKS); (D) LCMS analysis (DAD) of organic extracts from WT (Cryptosporiopsis sp. 8999). All strains were grown on brown rice. LCMS method 10–90% CH3CN:H2O gradient, 15 min. * = unrelated metabolite. | ||
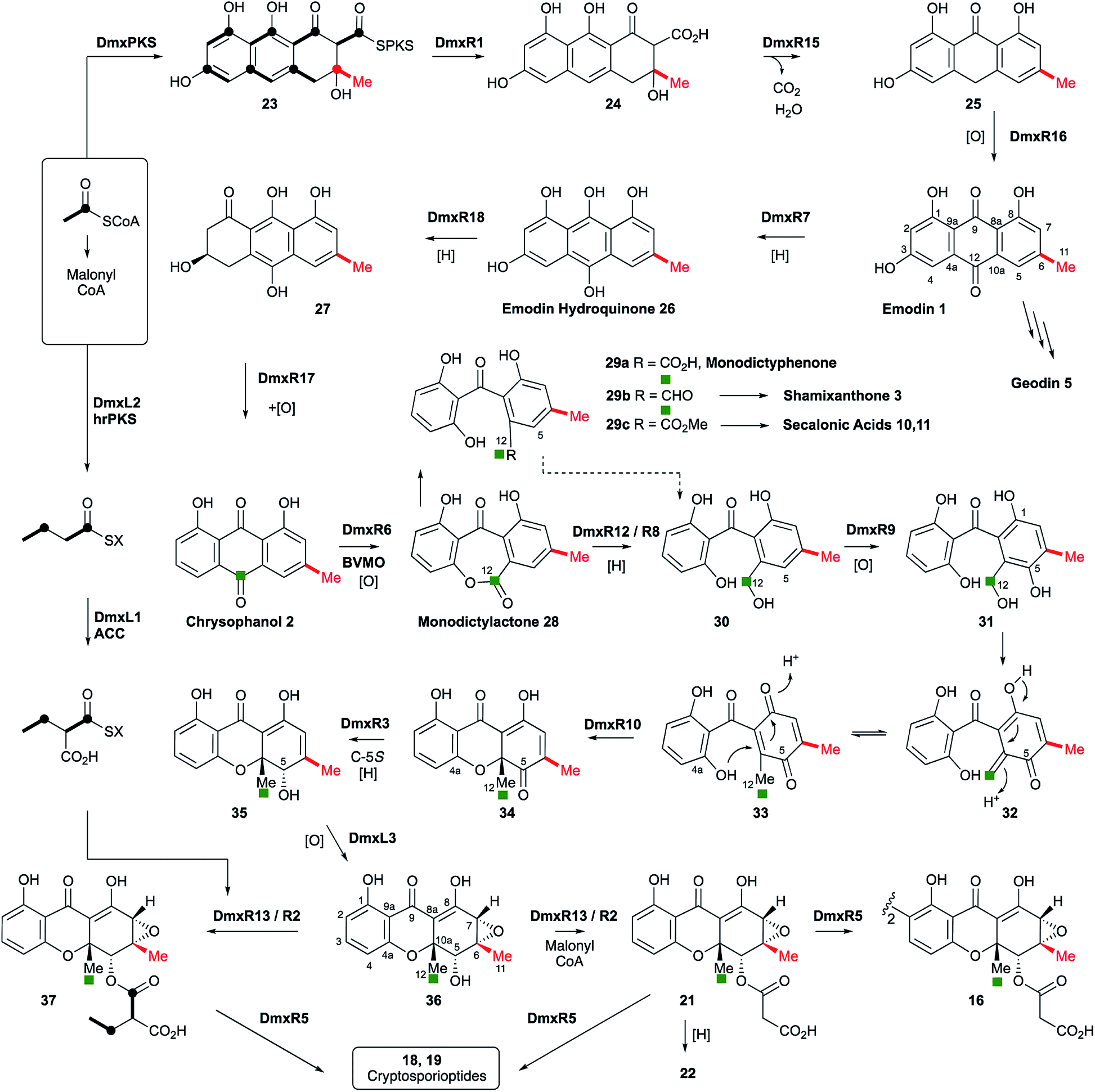 | ||
| Scheme 1 Proposed cryptosporioptide biosynthesis and relationships among monomeric and dimeric xanthone metabolites. Secalonic acid numbering used throughout.20 Red bond indicates polyketide starter unit. Green atom derived from C-10 of chrysophanol (=C-12 secalonic acid numbering). | ||
Discussion
From these results, a biosynthetic sequence for cryptosporioptides can be proposed (Scheme 1) and more general conclusions for the formation of dimeric xanthones made. The BGCs encoding the biosynthesis of shamixanthone 3 (mdp),5,6 geodin 5 (ged),7 agnestin 6 (agn)8 and cladofulvin 79 (cla) are all known. Tudzynski and coworkers recently revealed a putative secalonic acid (sec) BGC in Claviceps purpurea.27 In their analysis fifteen genes were described, but here, by comparison to the other known BGCs we extend this analysis to include six more genes in the sec cluster and two additional gene clusters from Penicillium oxalicum and Aspergillus aculeatus (Table 1 and see ESI†).All analysed clusters encode proteins with high homologies (41–76% orf for orf) to proteins known to be involved in the biosynthesis of emodin hydroquinone 26, and all BGCs except that for geodin contain genes which advance the pathway to chrysophanol 2 (Table 1). Thus, the nrPKS (DmxPKS) produces an enzyme-bound octaketide 23 which is released (DmxR1, giving 24), decarboxylated (DmxR15, giving 25) and oxidised (DmxR16) to give emodin 1, followed by reduction (DmxR7) to emodin hydroquinone 26. These steps are fully consistent with previous experimental results.8 A-ring reduction (DmxR18 giving 27), dehydration (DmxR17) and probable spontaneous re-oxidation, results in overall deoxygenation to chrysophanol 2, again consistent with previous results.8 Baeyer–Villiger oxidation (DmxR6) would then be expected to give monodictylactone 28 in equilibrium with monodictyphenone 29a (R = CO2H) as we recently observed in the agnestin pathway.8 All pathways encode this BVMO except the cla cluster which does not form xanthones. We have previously8 demonstrated the existence of BVMO enzymes which have complementary regioselectivity, and such a BVMO is likely to be operating in the ascherxanthone 8 pathway in which C-12 (C-10 AQ numbering) becomes attached to the non-starter unit ring (Fig. 1).
At this stage we propose a branch-point in the pathway. Conversion of monodictyphenone 29a (R = CO2H) to a methyl ester 29c would direct the intermediates towards the secalonic acids which feature a distinctive C-12 methyl ester. Recent results from Matsuda and coworkers support this conclusion by showing that a specific methyltransferase NsrG directs monodictyphenone 29a towards the biosynthesis of the heterodimeric xanthone neosartorin.37 This is consistent with the presence of methyltransferase-encoding genes (CPUR_05424, P_oxa-09242 and aacuQ) in the secalonic acid BGCs but not the others (Table 1). Alternatively, reduction at C-12 to form an aldehyde 29b would direct the pathway towards the shamixanthone group. In the case of the cryptosporioptides, however, reduction of C-12 to an alcohol 30 and hydroxylation at C-5 (likely DmxR9, see below) could give the electron-rich aromatic 31 which could eliminate H2O to form the ortho-quinonemethide 32, followed by tautomerisation to para-quinone 33 and complete the formal reduction to produce the 10-methyl group.
C-5 hydroxylation is required in the cryptosporioptide 16, shamixanthone 3 and secalonic acid 10, 11 pathways. In shamixanthone 3, this is proposed to be carried out by the monooxygenase MdpD, which shows 68% homology to DmxR9, while CPUR_05423 encodes a homolog in the sec cluster. Homologs of DmxR9 are also encoded in the P. oxalicum and A. aculeatus clusters. Notably homologs of dmxR9 are missing from the ged, agn and cla clusters where this chemistry is not required.
In an early application of doubly 13C-labelled acetate feeding experiments, Vining and coworkers showed38 that during secalonic acid biosynthesis in Pyrenochaeta terrestris, the initial product of anthraquinone ring cleavage must have a symmetrical 1,3-dihydroxyphenyl ring as in 29–33. Our suggested pathway is in agreement with this observation.
We propose that conjugate addition of C-4a-OH to the resulting para-quinone 33 then gives cyclohexadienone 34, which is then reduced at C-5 to give the dihydroxanthone 35. The ring closing reaction may be performed by DmxR10. This protein has no close BLAST hits with proteins of known function, but structural analysis using Phyre-2 (ref. 39) (see ESI†) shows that it contains a SnoAL domain40 and is related to proteins such as PhzB41 and Trt14 (ref. 42) known to be involved in secondary metabolite ring-forming reactions.
In this step the stereochemistry at C-10a is set, and it is known that both epimers at this position can be formed in the cases of the secalonic acids (e.g. see Fig. 10). It is interesting to note that the three secalonic acid clusters examined here (Table 1) appear to encode multiple copies of this protein, conceivably explaining the presence of both 10a-epimers in these systems. However further experimental work will be required to confirm this hypothesis.
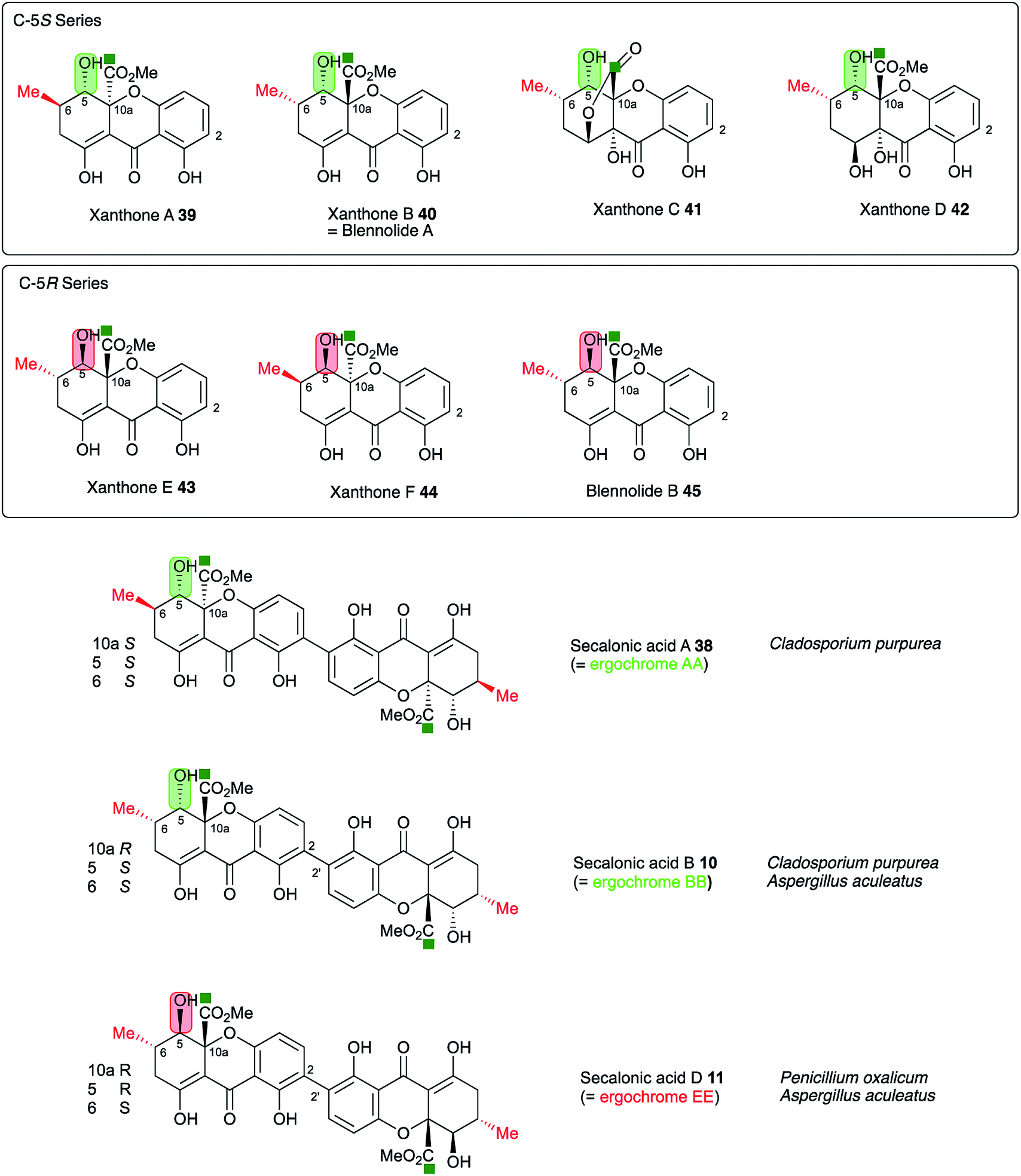 | ||
| Fig. 10 Stereochemical analysis of the known secalonic acids and correlation with producing organisms Penicillium oxalicum, Aspergillus aculeatus and Claviceps purpurea. Atom labelling according to Fig. 1. | ||
The 6,7-epoxide in the cryptosporioptides could be introduced by the cytochrome P450 monooxygenase encoded by dmxL3, which is unique to the dmx cluster, to give 36. Our results suggest that the hrPKS (DmxL2) manufactures butyrate. This is consistent with domain analysis of DmxL2 (NCBI CDD)43 which shows it to consist of N-terminal KS and AT domains, followed by a DH domain. Highly-reducing PKS often feature a C-methyltransferase domain but this appears to be absent or inactive in DmxL2, consistent with the lack of methylation observed. Canonical C-terminal ER, KR and ACP domains make up the rest of the hrPKS. Butyrate is then carboxylated (DmxL1) to form ethylmalonate. It is not yet clear whether the carboxylation occurs while the butyrate is attached to the ACP of DmxL2, but this unusual fungal metabolite could then be esterified to O-5 by DmxR13.
In the absence of ethylmalonate or the acyl transferase DmxR13, malonyl CoA can be used, possibly transferred by DmxR2. Finally, dimerisation (DmxR5) gives the observed dimers 16, 18 and 19 as the final products of the pathway. A homolog of dmxR5 is found in the C. purpurea sec cluster (CPUR_05419, again previously unrecognised) and P. oxalicum and A. aculeatus clusters, and the cladofulvin cluster where claM has been already shown to direct oxidative dimerisation.9 However, dmxR5 is missing from the agn, mdp and ged clusters as expected. The lower relative homology of the cladofulvin dimerase may reflect the fact that cladofulvin is a dimeric anthraquinone rather than a dimeric xanthone, and that cladofulvin is an asymmetric dimer in contrast to the symmetric cryptosporioptide and secalonic acid dimers.
A key step in the pathways to the dimeric xanthone metabolites is reduction at C-5. This could result in either 5S or 5R products depending on the selectivity of the reductase. Both types of xanthones are known44 (Fig. 10) and are, as shown above, likely to be direct precursors of the secalonic acids. Indeed, the ergochrome nomenclature explicitly recognises this, such that secalonic acid A 38 is also known as ergochrome AA showing that it is a dimer of two xanthone A 39 units. Xanthones A 39, B 40, C 41 and D 42 have C-5S stereochemistry, while xanthones E 43 and F 44 and blennolide B 45 have 5R stereochemistry. C. purpurea produces secalonic acids which are derived from xanthones A–D and it thus must reduce to give exclusively 5S stereochemistry. In contrast P. oxalicum produces secalonic acid D 11 which derives from xanthone E 43 which has 5R stereochemistry. Aspergillus aculeatus, however, produces both secalonic acids B 10and D 11 which are made from xanthones B 40and E 43 and it must therefore be able to reduce at C-5 to give both possible stereoisomers. In accord with this observation the sec cluster encodes a single SDR, which is homologous to DmxR3 (58%). The cryptosporioptides 16, 18 and 19 and secalonic acids A 38 and B 10 possess 5S stereochemistry, so we propose that DmxR3 is a 5S reductase. The P. oxalicum cluster possesses a different SDR encoded by P_oxa-09231 and we propose that this is a 5R selective reductase. In agreement with these ideas the A. aculeatus cluster encodes two SDRs, one of which (AacuD) is homologous to the 5S reductase DmxR3, while the other (AacuF) is homologous to the 5R selective P_oxc-09231. Homologous SDRs are not encoded by the agn, geo or cla clusters where the pathways do not require C-5 reduction, again consistent with this hypothesis.
Conclusions
Here we have shown that the originally reported monomeric structures of the cryptosporioptides must be reassigned to a series of dimeric xanthones featuring unusual malonate substituents and full reduction at C-10 (=C-12 in SA nomenclature). Isolation of the cryptosporioptide BGC and its verification by directed knockout experiments allowed the unusual biosynthesis and attachment of the ethylmalonyl substituents to be determined. Furthermore, comparison with known, but partially characterised fungal gene clusters, has allowed a fuller hypothesis regarding the biosynthesis of the dimeric xanthones in general, and the secalonic acids in particular, to be developed. Questions remain surrounding the proposed reduction of monodictylactone 28 in the cryptosporioptide and shamixanthone pathways where evidence for enzyme candidates is lacking. Similarly the catalyst for xanthone formation in the shamixanthone pathway is unknown, although the cryptosporioptide, agnestin and secalonic acid pathways appear to use homologs of DmxR10 for this step. Missing catalysts in the shamixanthone pathway may be related to the fact that the shamixanthone BGC is split, with outlying prenyltransferase genes and at least one redox-encoding gene located in a BGC distant from the core PKS-encoding BGC so the ‘missing’ genes may be elsewhere on the A. nidulans genome. However, the remaining very close genetic, and therefore chemical, homologies of the pathways revealed here should allow further targeted engineering in known dimeric xanthone clusters to be designed and performed, and it should also allow clusters encoding new dimeric xanthones to be more rapidly recognised and discovered.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank BBSRC (BB/J006289/1, BB/L01386X/1) and Syngenta for funding. LCMS instruments were provided by EPSRC (EP/F066104/1) and DFG (INST 187/621). 500 MHz NMR (EP/L011999/1) was provided by EPSRC. Cryptosporiopsis sp. 8999 was a kind gift from Dr Barbara Schulz and its genome was sequenced at the University of Cambridge Sequencing Centre. Markiyan Samborskyy is thanked for assistance with bioinformatics. Katherine Williams and Zhongshu Song are thanked for technical assistance.Notes and references
- R. Schor and R. J. Cox, Nat. Prod. Rep., 2018, 35, 230–256 RSC.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC.
- K. K. Chexal, C. Fouweather, J. S. E. Holker, T. J. Simpson and K. Young, J. Chem. Soc., Perkin Trans. 1, 1974, 1584–1593 RSC.
- A. Krick, S. Kehraus, C. Gerhäuser, K. Klimo, M. Nieger, A. Maier, H.-H. Fiebig, I. Atodiresei, G. Raabe, J. Fleischhauer and G. König, J. Nat. Prod., 2007, 70, 353–360 CrossRef CAS PubMed.
- Y.-M. Chiang, E. Szewczyk, A. Davidson, R. Entwistle, N. Keller, C. Wang and B. Oakley, Appl. Environ. Microbiol., 2010, 76, 2067–2074 CrossRef CAS PubMed.
- J. Sanchez, R. Entwistle, J.-H. Hung, J. Yaegashi, S. Jain, Y.-M. Chiang, C. Wang and B. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS PubMed.
- M. T. Nielsen, J. B. Nielsen, D. C. Anyaogu, D. K. Holm, K. F. Nielsen, T. O. Larsen and U. H. Mortensen, PLoS One, 2013, 8, e72871 CrossRef CAS PubMed.
- A. J. Szwalbe, K. Williams, Z. Song, K. de Mattos-Shipley, J. L. Vincent, A. M. Bailey, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 1227–1231 RSC.
- S. Griffiths, C. H. Mesarich, B. Saccomanno, A. Vaisberg, P. De Wit, R. J. Cox and J. Collemare, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6851–6856 CrossRef CAS PubMed.
- (a) T. Wezeman, S. Bräse and K.-S. Masters, Nat. Prod. Rep., 2015, 32, 6–28 RSC; (b) K.-S. Masters and S. Brӓse, Chem. Rev., 2012, 112, 3717–3776 CrossRef CAS PubMed.
- (a) M. Isaka, S. Palasarn, K. Kocharin and J. Saenboonrueng, J. Nat. Prod., 2005, 68, 945–946 CrossRef CAS PubMed; (b) Z. Xiao, Y. Li and S. Gao, Org. Lett., 2017, 19, 1834–1837 CrossRef CAS PubMed.
- M. M. Wagenaar and J. Clardy, J. Nat. Prod., 2001, 64, 1006–1009 CrossRef CAS.
- A. Ciegler, A. W. Hayes and R. F. Vesonder, Appl. Environ. Microbiol., 1980, 39, 285–287 CAS.
- B. Franck, G. Bringmann and G. Flohr, Angew. Chem., 1980, 92, 483–484 CrossRef CAS.
- K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2016, 55, 6784–6788 CrossRef CAS PubMed.
- M. Saleem, H. Hussain, I. Ahmed, S. Draeger, B. Schulz, K. Meier, M. Steinert, G. Pescatelli, T. Kurtán, U. Flörke and K. Krohn, Eur. J. Org. Chem., 2011, 808–812 CrossRef CAS.
- M. Saleem, M. I. Tousif, N. Riaz, I. Ahmed, B. Schulz, M. Ashraf, R. Nasar, G. Pescitelli, H. Hussain, A. Jabbar, N. Shafiq and K. Krohn, Phytochemistry, 2013, 93, 199–202 CrossRef CAS PubMed.
- M. I. Tousif, N. Shazmeen, N. Riaz, N. Shafiq, T. Khatoon, B. Schulz, M. Ashraf, A. Shaukat, H. Hussain, A. Jabbar and M. Saleem, J. Asian Nat. Prod. Res., 2014, 16, 1068–1073 CrossRef CAS PubMed.
- P.-Y. Wei, L.-X. Liu, T. Liu, C. Chen, D.-Q. Luo and B.-Z. Shi, Molecules, 2015, 20, 5825–5834 CrossRef CAS PubMed.
- W. Zhang, K. Krohn, Z. Ullah, U. Flörke, G. Pescitelli, L. Di Bari, S. Antos, T. Kurtán, J. Rheinheimmer, S. Draeger and B. Schulz, Chem.–Eur. J., 2008, 14, 4913–4923 CrossRef CAS PubMed.
- R. Anderson, G. Buchi, B. Kobbe and A. L. Demain, J. Org. Chem., 1977, 42, 352 CrossRef.
- D. Rönsberg, A. Debbab, A. Mándi, V. Vesylyeva, P. Böhler, B. Stork, L. Engelke, A. Hamacher, R. Swadaogo, M. Diederich, V. Wray, W. Lin, M. U. Kassack, C. Janiak, S. Scheu, S. Wesselborg, T. Kurtán, A. H. Aly and P. Proksch, J. Org. Chem., 2013, 78, 12409–12425 CrossRef PubMed.
- D. Ganapathy, J. Reiner, G. Valdomir, S. Senthilkumar and L. Tietze, Chem.–Eur. J., 2017, 23, 2299–2302 CrossRef CAS PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef CAS PubMed.
- The annotated dmx BGC has been uploaded to GenBank with accession number MK182094.
- T. J. Simpson, ChemBioChem, 2012, 13, 1680–1688 CrossRef CAS PubMed.
- L. Neubauer, J. Dopstadt, H.-U. Humpf and P. Tudzynski, Fungal Biol. Biotechnol., 2016, 3, 1–14 CrossRef PubMed.
- M. Nielsen, L. Albertsen, G. Lettier, J. Nielsen and U. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed.
- R. de Vries, R. Riley, A. Wiebenga, G. Aguilar-Osorio, S. Amillis, C. Uchima, G. Anderluh, M. Asadollahi, M. Askin, K. Barry, E. Battaglia, Ö. Bayram, T. Benocci, S. Braus-Stromeyer, C. Caldana, D. Cánovas, G. Cerqueira, F. Chen, W. Chen, C. Choi, A. Clum, R. dos Santos, A. de Damásio, G. Diallinas, T. Emri, E. Fekete, M. Flipphi, S. Freyberg, A. Gallo, C. Gournas, R. Habgood, M. Hainaut, M. Harispe, B. Henrissat, K. Hildén, R. Hope, A. Hossain, E. Karabika, L. Karaffa, Z. Karányi, N. Kraševec, A. Kuo, H. Kusch, K. LaButti, E. Lagendijk, A. Lapidus, A. Levasseur, E. Lindquist, A. Lipzen, A. Logrieco, A. MacCabe, M. Mäkelä, I. Malavazi, P. Melin, V. Meyer, N. Mielnichuk, M. Miskei, Á. Molnár, G. Mulé, C. Ngan, M. Orejas, E. Orosz, J. Ouedraogo, K. Overkamp, H.-S. Park, G. Perrone, F. Piumi, P. Punt, A. Ram, A. Ramón, S. Rauscher, E. Record, D. Riaño-Pachón, V. Robert, J. Röhrig, R. Ruller, A. Salamov, N. Salih, R. Samson, E. Sándor, M. Sanguinetti, T. Schütze, K. Sepčić, E. Shelest, G. Sherlock, V. Sophianopoulou, F. Squina, H. Sun, A. Susca, R. Todd, A. Tsang, S. Unkles, N. van de Wiele, D. van Rossen-Uffink, J. de Oliveira, T. Vesth, J. Visser, J.-H. Yu, M. Zhou, M. Andersen, D. Archer, S. Baker, I. Benoit, A. Brakhage, G. Braus, R. Fischer, J. Frisvad, G. Goldman, J. Houbraken, B. Oakley, I. Pócsi, C. Scazzocchio, B. Seiboth, P. vanKuyk, J. Wortman, P. Dyer and I. Grigoriev, Genome Biol., 2017, 18, 28 CrossRef PubMed.
- N. Yodsing, R. Lekphrom, W. Sangsopha, T. Aimi and S. Boonlue, Curr. Microbiol., 2018, 75, 513–518 CrossRef CAS PubMed.
- P. S. Steyn, Tetrahedron, 1970, 26, 51–57 CrossRef CAS PubMed.
- G. Liu, L. Zhang, X. Wei, G. Zou, Y. Qin, L. Ma, J. Li, H. Zheng, S. Wang, C. Wang, L. Xun, G.-P. Zhao, Z. Zhou and Y. Qu, PLoS One, 2013, 8, e55185 CrossRef CAS PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- C. G. Girol, K. M. Fisch, T. Heinekamp, S. Gunther, W. Huttel, J. Piel, A. A. Brakhage and M. Müller, Angew. Chem., Int. Ed., 2012, 51, 9788–9791 CrossRef PubMed.
- S. E. Bode, D. Drochner and M. Müller, Angew. Chem., Int. Ed., 2007, 46, 5916–5920 CrossRef CAS PubMed.
- Y. Matsuda, C. Gotfredsen and T. Larsen, Org. Lett., 2018, 20, 7197–7200 CrossRef CAS PubMed.
- I. Kurobane, L. C. Vining, A. G. McInnes, J. A. Walter and J. L. C. Wright, Tetrahedron Lett., 1978, 19, 1379–1382 CrossRef.
- L. Kelley, S. Mezulis, C. Yates, M. Wass and M. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- A. Sultana, P. Kallio, A. Jansson, J. Wang, J. Niemi, P. Mäntsälä and G. Schneider, EMBO J., 2004, 23, 1911–1921 CrossRef CAS PubMed.
- W. Blankenfeldt and J. F. Parsons, Curr. Opin. Struct. Biol., 2014, 29, 26–33 CrossRef CAS PubMed.
- T. Mori, T. Iwabuchi, S. Hoshino, H. Wang, Y. Matsuda and I. Abe, Nat. Chem. Biol., 2017, 13, 2443 CrossRef PubMed.
- A. Marchler-Bauer, Y. Bo, L. Han, J. He, C. Lanczycki, S. Lu, F. Chitsaz, M. Derbyshire, R. Geer, N. Gonzales, M. Gwadz, D. Hurwitz, F. Lu, G. Marchler, J. Song, N. Thanki, Z. Wang, R. Yamashita, D. Zhang, C. Zheng, L. Geer and S. Bryant, Nucleic Acids Res., 2017, 45, D200–D203 CrossRef CAS PubMed.
- I. Kurobane, L. C. Vining and A. G. McInnes, J. Antibiot., 1979, 32, 1256–1266 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05126g |
| This journal is © The Royal Society of Chemistry 2019 |