
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWaterproof architectures through subcomponent self-assembly†
Edmundo G.
Percástegui
 ,
Jesús
Mosquera
,
Tanya K.
Ronson
,
Alex J.
Plajer
,
Marion
Kieffer
and
Jonathan R.
Nitschke
*
,
Jesús
Mosquera
,
Tanya K.
Ronson
,
Alex J.
Plajer
,
Marion
Kieffer
and
Jonathan R.
Nitschke
*
Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, UK. E-mail: jrn34@cam.ac.uk
First published on 12th December 2018
Abstract
Metal–organic containers are readily prepared through self-assembly, but achieving solubility and stability in water remains challenging due to ligand insolubility and the reversible nature of the self-assembly process. Here we have developed conditions for preparing a broad range of architectures that are both soluble and kinetically stable in water through metal(II)-templated (MII = CoII, NiII, ZnII, CdII) subcomponent self-assembly. Although these structures are composed of hydrophobic and poorly-soluble subcomponents, sulfate counterions render them water-soluble, and they remain intact indefinitely in aqueous solution. Two strategies are presented. Firstly, stability increased with metal–ligand bond strength, maximising when NiII was used as a template. Architectures that disassembled when CoII, ZnII and CdII templates were employed could be directly prepared from NiSO4 in water. Secondly, a higher density of connections between metals and ligands within a structure, considering both ligand topicity and degree of metal chelation, led to increased stability. When tritopic amines were used to build highly chelating ligands around ZnII and CdII templates, cryptate-like water-soluble structures were formed using these labile ions. Our synthetic platform provides a unified understanding of the elements of aqueous stability, allowing predictions of the stability of metal–organic cages that have not yet been prepared.
Introduction
In analogy to vesicles in natural systems, the well-defined hydrophobic inner pockets of water-soluble metal–organic containers allow guests to be selectively incorporated and released with control over their chemical reactivity.1 For instance, high-energy reactive species may be stabilised,2 reaction rates can be enhanced3 or otherwise inaccessible products may be generated.4 In water, the hydrophobic effect can help to drive encapsulation,5 enabling applications based upon guest binding.6However, the construction of molecular containers that are both soluble and stable in water remains challenging.7,8 Many such structures incorporate organic building blocks containing solubilising functional groups that are polar2c,5a,9 or charged10 to ensure aqueous solubility, often requiring laborious synthetic efforts.
Even if a metallosupramolecular structure is water-soluble, it may not be stable in aqueous solution. Formation equilibria can be driven backwards if organic building blocks are poorly soluble, due to the reversible nature of the metal–ligand bonds that hold these structures together. The assembly of architectures that hold together robustly in water has thus required the use of more costly second-4a,8,11 and third-row metals,12 which are kinetically more inert. This approach may lead to formation of kinetically trapped intermediates,12a,13 which has in turn led to the development of metal-exchange or redox modifications on preformed assemblies in order to kinetically “lock” a structure and prevent its disassembly.14
The subcomponent self-assembly method has been widely used by others15 and us16 to construct metal–organic architectures through the synergistic formation of dynamic covalent (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) imine4b,17 and coordinative N → metal ion linkages. The formation of cages that are soluble and stable in water via subcomponent self-assembly has been possible only when sufficiently water-soluble subcomponents are used.2a,9a,10a Although the exchange of trifluoromethanesulfonate (OTf−) for sulfate anions (SO42−) on preformed FeII cages constructed from water-insoluble subcomponents provided a route to water solubility in certain cases,18 many cages thus prepared disassembled at room temperature. When subcomponents lack sufficient water solubility, the trace amounts of disassembled precursors present at equilibrium can reach saturation and precipitate, thus resulting in the observed disassembly. As a result, the low water solubility of the more accessible hydrophobic ligands appears at first glance to preclude their integration into water-soluble cages.
N) imine4b,17 and coordinative N → metal ion linkages. The formation of cages that are soluble and stable in water via subcomponent self-assembly has been possible only when sufficiently water-soluble subcomponents are used.2a,9a,10a Although the exchange of trifluoromethanesulfonate (OTf−) for sulfate anions (SO42−) on preformed FeII cages constructed from water-insoluble subcomponents provided a route to water solubility in certain cases,18 many cages thus prepared disassembled at room temperature. When subcomponents lack sufficient water solubility, the trace amounts of disassembled precursors present at equilibrium can reach saturation and precipitate, thus resulting in the observed disassembly. As a result, the low water solubility of the more accessible hydrophobic ligands appears at first glance to preclude their integration into water-soluble cages.
The strategies developed herein complement past methods, allowing for many of the limitations discussed above to be overcome. Specific combinations of water-insoluble subcomponents with salts of nickel(II), cobalt(II), zinc(II), and cadmium(II) resulted in the facile assembly of kinetically robust water-soluble structures ranging from face-capped M8L6 cubes and M4L4 tetrahedra, to edge-linked M4L6 tetrahedra, and M2L3 triple helicates.
The use of sulfate as the counterion was essential to bring about water solubility, and it was introduced either by direct assembly from MIISO4 or via anion-exchange from acetonitrile-soluble structures. Stability in aqueous media was achieved using two distinct strategies. Firstly, we took advantage of the diverse degree of kinetic inertness of metal ions with stability order of NiII > FeII > CoII > ZnII > CdII.19 Using NiII as a template maximised the strength of metal–ligand bonding, thus stabilising the product complexes to hydrolysis and decomposition. The second strategy enhanced the binding cooperativity20 around metal templates through (a) the use of subcomponents of increasing topicity (ability to bridge between varying numbers of metal ions), and (b) the augmentation of chelate effects. Hence, subcomponents that form tetratopic ligands, able to bridge four metal ions, were observed to form the most stable cages, followed by tri- and ditopic ones. ZnII and CdII ions did not bind strongly enough, even to tetratopic ligands, to prevent aqueous disassembly. Water-stable cages containing these metal ions could be constructed, however, by incorporating tritopic chelating amines to cap the vertices of ZnII- and CdII-based assemblies. The more densely-connected structures thus obtained were stabilised by extensive chelate effects,20 and represent the first water-soluble subcomponent self-assembled structures containing these labile ZnII and CdII cations, complimenting rare examples of aqueous coordination assemblies that incorporate these metals.21
Results and discussion
Self-assembly reactions to form water-soluble polyhedra as sulfate salts were attempted using subcomponents A–E (Scheme 1) with divalent metal ions (MII = CoII, NiII, ZnII, CdII) as detailed below. We chose these subcomponents to gauge the role of different metal ions on cage stability through comparison with their FeII analogues.18 | ||
| Scheme 1 Subcomponent self-assembly of CoII-capsules. Preparation of acetonitrile-soluble Co-1·X to Co-5·X (X = OTf−, NTf2−, or BF4−) from subcomponents A–E and their anion exchange sequences. | ||
Cobalt(II) architectures
The CoII capsules shown in Scheme 1 were prepared from subcomponents A–E in acetonitrile and subjected to anion metathesis. The formation of these acetonitrile-soluble cages was sensitive to the identity of the counterion, and none could be formed as clean products directly from CoSO4 (ESI Section 2†).The reaction between fourfold-symmetric zinc porphyrin A, 2-formylpyridine, and cobalt(II) trifluoromethanesulfonate in DMF produced the previously unknown cube Co-1·OTf, as revealed by electrospray mass spectrometry (ESI-MS). Addition of tetrabutylammonium (TBA) sulfate to an MeCN solution of Co-1·OTf led to precipitation of Co-1·SO4 as a magenta solid that was soluble in water (Fig. S1 and S2†). The wide-sweep 1H NMR spectrum in D2O was consistent with the O-symmetric structure previously observed for the FeII analogue.18,22 Dispersion of eleven proton signals over the range 245.2 to −63.9 ppm confirmed the presence of paramagnetic CoII ions at the cage vertices. No decomposition of cage Co-1·SO4 in D2O (50.0 μM and 500 μM) was detected by NMR spectroscopy after 3 months at 25 °C. In contrast, heating these solutions to 50 °C led to complete cube decomposition within 8 h as evidenced by precipitation of blue porphyrin A and disappearance of the signals in the 1H NMR spectrum.
The water stability of Co-1·SO4 allowed reversibility of the anion exchange process. Addition of LiOTf to a D2O solution of Co-1·SO4 produced a purple precipitate that was soluble in acetonitrile. NMR and ESI-MS analyses matched those of the initial Co-1·OTf product (Fig. S3 and S4†). The complete Co-1·OTf → Co-1·SO4 → Co-1·OTf anion exchange cycle was carried out twice with 90–94% recovery by weight per cycle.
Slow diffusion of benzene into an acetonitrile solution of Co-1·OTf afforded brown crystals of the complex; Co-1 crystallised in the tetragonal space group P4/n (Fig. 1). All of the CoII centres adopt the same Δ or Λ configuration, and both enantiomers are present in the crystal lattice. The internal cavity volume of 1331 Å3 (ESI Section 7†) is similar to the previously reported FeII analogue (1340 Å3).22 In the solid state this cavity is occupied by disordered acetonitrile and benzene solvent molecules; we infer that solvent molecules are also likely to occupy the cavity in solution.
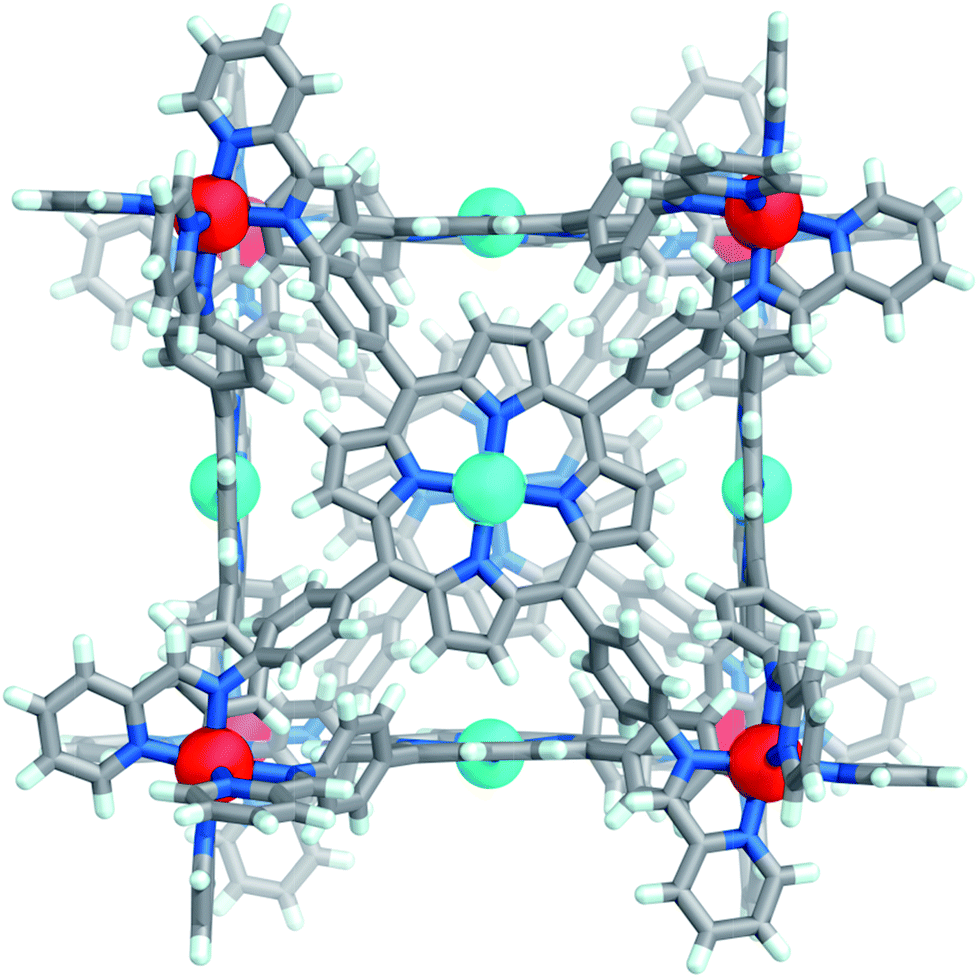 | ||
| Fig. 1 Crystal structure of cube Co-1·OTf. Counterions, solvents and disorder are omitted for clarity. CoII and ZnII centers are coloured dark orange and cyan, respectively. | ||
The reaction of trianiline B with 2-formylpyridine and cobalt(II) sulfate yielded a crude mixture containing the target water-soluble CoII4L4 tetrahedron as the major product and a CoII2L3 helicate (detected by ESI-MS and NMR spectroscopy) analogous to previously-observed FeII congeners;23 these products were not amenable to separation. Conversely, treatment of B with 2-formylpyridine and cobalt(II) bis(trifluoromethanesulfonyl)imide (triflimide, NTf2) in CH3CN produced cleanly the face-capped CoII4L4 tetrahedral capsule Co-2·NTf2. This cage incorporates four CoII centres bridged by four threefold-symmetric ligands (L) resulting from the condensation of aniline B with 3 equivalents of 2-formylpyridine.
Treatment of Co-2·NTf2 with TBA2SO4 in acetonitrile yielded the water-soluble Co-2·SO4 salt as a dark orange precipitate. The wide-sweep 1H NMR spectrum in D2O revealed a highly symmetric product; eight proton signals for the ligand were distributed over the range of 241.2 to −19.6 ppm (Fig. S5†). ESI-MS in aqueous solution further confirmed the CoII4L4 composition of Co-2·SO4 (Fig. S6†). No decomposition was observed for aqueous solutions (250–750 μM) of Co-2·SO4 after two months at 25 °C; on the contrary and regardless of the concentration, complete decomposition within 6 h was observed when these solutions were heated to 50 °C, indicating kinetic but not thermodynamic stability.
The addition of LiNTf2 to an aqueous solution of Co-2·SO4 resulted in precipitation of the Co-2·NTf2 derivative, as confirmed by NMR spectroscopy and ESI-MS (Fig. S7 and S8†). Further addition of TBA2SO4 to this acetonitrile solution of Co-2·NTf2 induced precipitation of Co-2·SO4. The complete Co-2·NTf2 → Co-2·SO4 → Co-2·NTf2 anion exchange cycle was carried out twice, with 90–95% recovery by weight per cycle.
Subcomponent self-assembly of benzidine C with 2-formylpyridine and Co(BF4)2 afforded the previously-unreported tetrahedral cage Co-3·BF4 as the single product (Fig. S9 and S10†); the ESI-MS showed only peaks corresponding to the CoII4L6 complex. The analogous FeII4L6 cage, incorporating aniline C, exists as a system of interconverting diastereomers in solution,24 where BF4− led to the T-symmetric diastereomer as the main species. This anion gave a similar diastereoselectivity in the present case. The wide-sweep 1H NMR spectrum in CD3CN contained clusters of peaks consistent with a mixture of homochiral T (ΔΔΔΔ/ΛΛΛΛ), heterochiral C3 (ΔΔΔΛ/ΛΛΛΔ), and achiral S4 (ΔΔΛΛ) diastereomers as previously observed for the FeII4L6 cage system.24 We could clearly distinguish a set of seven major peaks, consistent with the presence of the corresponding T-symmetric diastereomer as the predominant species.
Anion exchange of OTf− for SO42− induced precipitation of a pale orange solid, which dissolved in D2O. However, disassembly took place within minutes, thus hindering any attempt to regenerate the initial Co-3·BF4 cage. We attribute the marked difference in stability between capsules Co-2·SO4 and Co-3·SO4 to the more robust framework that resulted from self-assembly of the tritopic aniline B.
Next we prepared the new cage Co-4·OTf as well as Co-5·OTf, the framework of which was previously reported incorporating toluidine,25 from tritopic and ditopic aldehydes D and E, respectively, anisidine, and Co(OTf)2. Their identities were confirmed through NMR and ESI-MS analyses. The addition of TBA2SO4 to MeCN solutions of both cages generated orange precipitates that we infer to consist of the corresponding sulfate salts. However, the dissolution of these precipitates in D2O led to decomposition as evidenced by precipitation of subcomponents D and E. NMR analysis of a D2O solution of Co-4·SO4 showed that ca. 80% was hydrolysed after 1 h at room temperature; in contrast, it was not possible to observe any cage peaks when the precipitate of Co-5·SO4 was dissolved in D2O, due to its more rapid disassembly.
Nickel(II) architectures
Although many coordination cages incorporating FeII (ref. 15a–e, 22–24 and 26) and CoII (ref. 3a, 5a, 15f, 25 and 27) have been reported, far fewer have used NiII,28 despite the stronger coordination bonds this metal ion forms with nitrogen ligands.29 Our own enquiries into NiII-based structures have been limited by difficulties in obtaining structural information in solution;28b–c1H NMR signals of octahedral NiII complexes are in many cases unresolvably broad due to unfavourable relaxivity.30The series of new NiII polyhedra (Schemes 2 and 3) reported herein were characterised using ESI-MS and X-ray crystallography in some cases. To supply further NMR structural evidence, 19F NMR spectroscopy was employed to verify the binding of fluorinated guests within the cage cavities (Scheme 2 and ESI Section 3.6†). These guests were observed to bind only in D2O, suggesting that the hydrophobic effect is essential for their encapsulation in water.5 Unlike the iron(II)18 and cobalt(II) systems assembled from subcomponents A–E, it was possible to obtain water-soluble structures from direct assembly with NiSO4 in certain cases. The stronger coordination bonds of the NiII structures led to greater stability in aqueous solution, as detailed below.
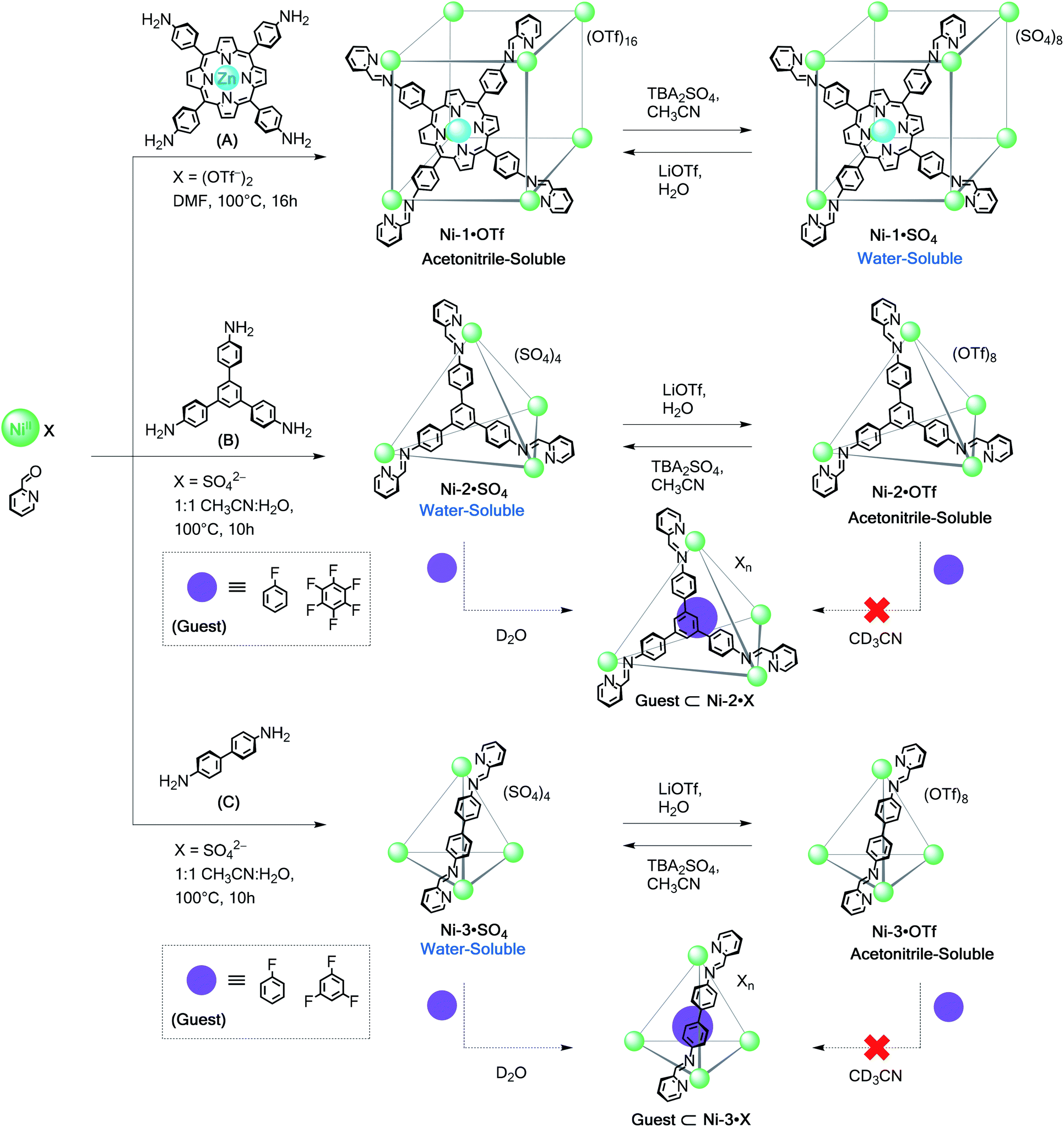 | ||
| Scheme 2 Subcomponent self-assembly of water-soluble Ni-1·SO4 to Ni-3·SO4 cages from anilines A–C, their reversible anion-exchange sequences and encapsulation of selected fluorinated guests. | ||
 | ||
| Scheme 3 Self-assembly reactions of aldehydes D and E with nickel(II) salts. | ||
Reaction of tetratopic aniline A with 2-formylpyridine and Ni(OTf)2 in DMF afforded the cube Ni-1·OTf. Exchange of triflate for sulfate resulted in precipitation of the water-soluble Ni-1·SO4 cage as a red-brown solid; ESI-MS analysis of an aqueous solution of this precipitate corresponded to a NiII8L6 cubic framework (Fig. S14†). Aqueous solutions of Ni-1·SO4 developed no precipitate after 3 months at room temperature; ESI-MS spectra (recorded periodically) of these solutions showed the presence of the cubic cage. Conversely, Ni-1·SO4 decomposed within 16 h at 85 °C, as evidenced by precipitation of the water-insoluble Zn-porphyrin A. Addition of LiOTf to a H2O solution of Ni-1·SO4 resulted in regeneration of Ni-1·OTf, as confirmed by ESI-MS in MeCN. Slow diffusion of benzene into this solution gave dark green-brown crystals of Ni-1·OTf (Fig. 2a), which was isomorphous to cube Co-1·OTf. As in the case of Ni-1·OTf, the internal cavity, which has a volume of 1324 Å3 (ESI Section 7†), is occupied by disordered solvent molecules.
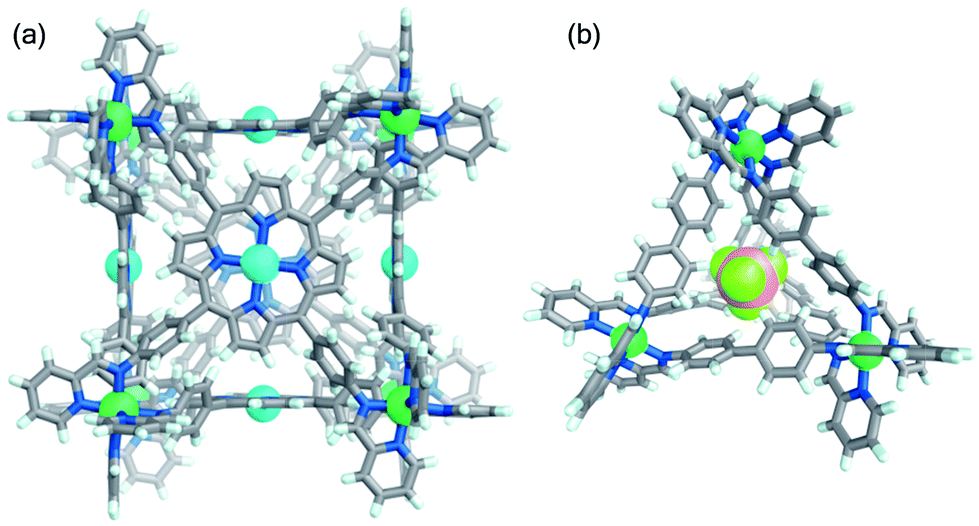 | ||
| Fig. 2 Crystal structures of (a) cube Ni-1 and (b) tetrahedron Ni-3 viewed through an open face and showing the included BF4− anion. Counterions, solvents and disorder are omitted for clarity. Ni, Zn, F, and B atoms are coloured green, cyan, pale green and pink, respectively. | ||
In contrast to our observations for iron(II)18 and cobalt(II), tri- and ditopic anilines B and C reacted readily with NiSO4 to form the Ni-2·SO4 and Ni-3·SO4 tetrahedra in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN:H2O as identified by ESI-MS in H2O (Fig. S16 and S18†). Addition of LiOTf to cages Ni-2·SO4 and Ni-3·SO4 in water resulted in the precipitation of the corresponding Ni-2·OTf and Ni-3·OTf capsules, as verified by ESI-MS analyses in MeCN (Fig. S17 and S19†).
:1 CH3CN:H2O as identified by ESI-MS in H2O (Fig. S16 and S18†). Addition of LiOTf to cages Ni-2·SO4 and Ni-3·SO4 in water resulted in the precipitation of the corresponding Ni-2·OTf and Ni-3·OTf capsules, as verified by ESI-MS analyses in MeCN (Fig. S17 and S19†).
Single-crystal X-ray diffraction on a yellow crystal obtained from slow diffusion of iPr2O into an MeCN solution of Ni-3 in the presence of excess BF4− confirmed its NiII4L6 composition (Fig. 2b). Cage Ni-3·OTf crystallised in the monoclinic space group C2/c. The architecture is tetrahedral with non-crystallographic T point symmetry, with both enantiomers present in the unit cell. A BF4− guest anion was bound in the capsule cavity, benefiting from stabilising CH⋯F non-classical hydrogen-bonding interactions.31 Cages Ni-2·OTf and Ni-3·OTf were converted back into Ni-2·SO4 and Ni-3·SO4 following treatment in MeCN with TBA2SO4. The anion exchange sequence was carried out twice with ±95% cage recovery.
To verify the encapsulation abilities of these complexes, we treated them with fluorobenzene, 1,3,5-trifluorobenzene, and hexafluorobenzene as prospective guests. The binding of each was followed by 19F NMR spectroscopy. No encapsulation was inferred to have taken place with Ni-2·OTf and Ni-3·OTf in CD3CN. Only one 19F NMR signal for the prospective guest was observed in each case at the same chemical shift as in the absence of the host. Conversely, all of these guests were inferred to bind within Ni-2·SO4 and Ni-3·SO4 in D2O because new peaks were observed for the free and encapsulated guests. The peaks for the free fluorinated molecules, which were added in excess, progressively disappeared likely due to separation and evaporation from the aqueous solution given their poor solubility in water. As a result, only the peaks of the host–guest complexes persisted in the 19F NMR spectra over time (Fig. S23–S25†). This difference in binding parallels that observed for related systems using these solvents, and suggests that the hydrophobic effect may be essential for encapsulation in water.5,18
The reaction of tritopic aldehyde D with NiSO4 and anisidine yielded Ni-4·SO4 in aqueous solution (Scheme 3), as revealed by ESI-MS (Fig. S20†). This NiII4L4 framework also remained intact following anion exchanges. The addition of LiOTf produced the water-insoluble congener as characterised by ESI-MS (Fig. S21†). This sequence could be reversed to reform the parent water-soluble sulfate cage upon treatment with TBA2SO4. Similar to capsules Ni-2·SO4 and Ni-3·SO4, tetrahedron Ni-4·SO4 also bound fluorobenzene in D2O, as indicated by the presence of signals corresponding to the free and encapsulated guest in the 19F NMR spectrum (Fig. S24 and S25†).
Conversely, the reaction of ditopic aldehyde E with NiSO4 did not afford the sulfate salt of the cage, instead yielding only intractable products. As previously observed,28cE reacts with Ni(OTf)2 to form a mixture of assemblies. When treated with TBA2SO4 in MeCN, this mixture gave species that were initially soluble in water, but which decomposed by precipitation within minutes.
Self-assembly with labile metal ions: zinc(II) and cadmium(II) architectures
To further extend the scope and value of the subcomponent self-assembly method in water, we have investigated the construction of water-soluble architectures using ZnII and CdII, which are much more labile (KH2O = 107 and 108 s−1, respectively) than their transition metal ion congeners.19 As with FeII, CoII, and NiII, subcomponents A–E formed acetonitrile-soluble capsules with the more labile ions ZnII and CdII from the corresponding triflate salts; Scheme 4 shows these reactions using A and D as examples. Although it was possible to prepare cubes Zn-1·OTf and Cd-1·OTf, as well as tetrahedra Zn-4·OTf and Cd-4·OTf, these capsules exhibited poor stability in aqueous solution (Fig. S26–S32†). Exchange of triflate for sulfate generated precipitates that we inferred to consist of cage sulfate salts. However, dissolution of Zn-1·SO4 and Cd-1·SO4 in D2O resulted in rapid precipitation of subcomponent A. While it was possible to observe low-intensity resonances attributable to cube Zn-1 within the first hour of dissolution, the Cd-1 framework had nearly fully hydrolysed after 10 min. Consequently, it was not possible to recover the triflate cubes through reverse ion exchange. Attempts to form these capsules through direct assembly from ZnSO4 or CdSO4 were unsuccessful.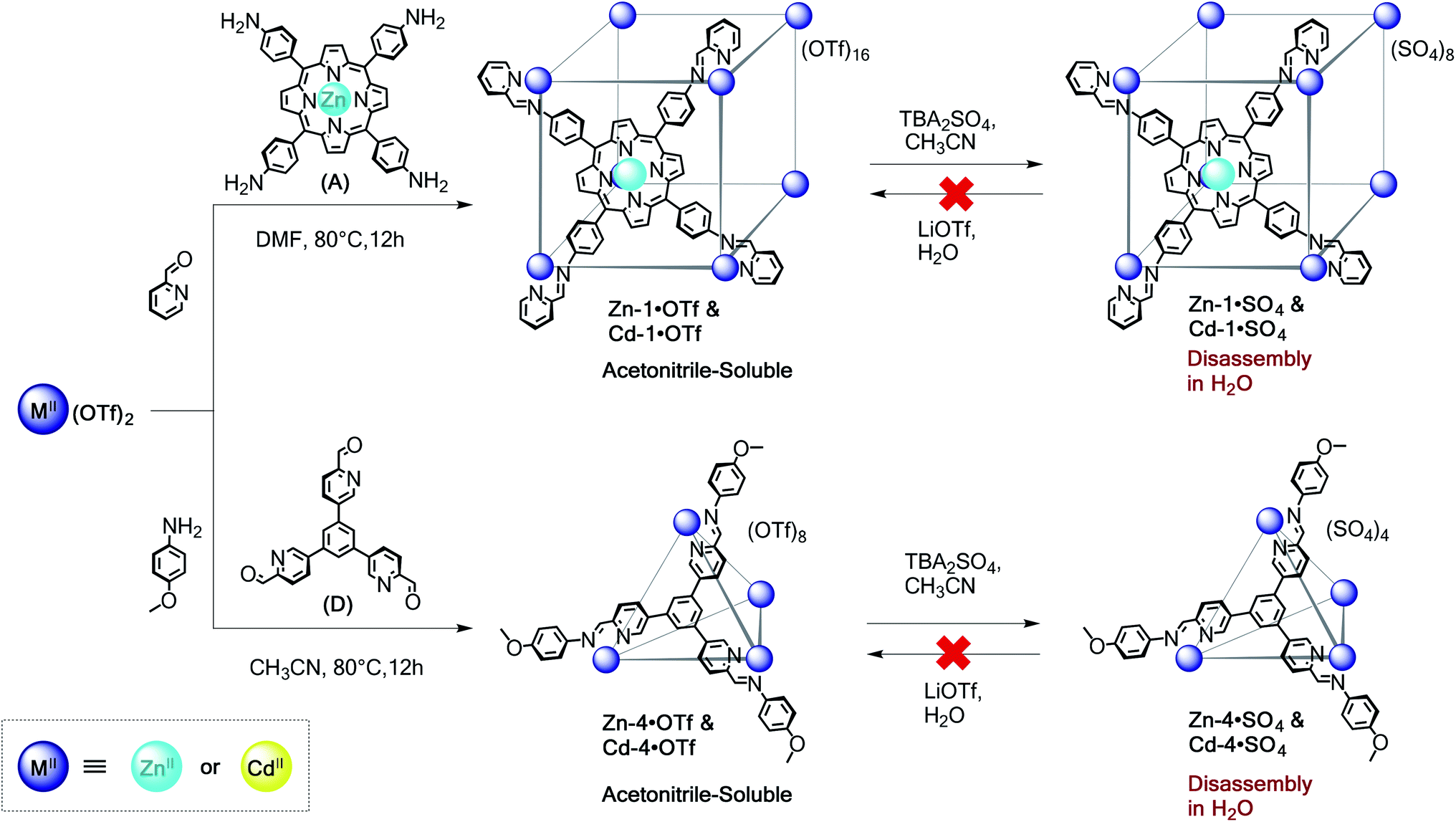 | ||
| Scheme 4 Self-assembly of face-capped cubic 1 and tetrahedral 4 architectures based on zinc(II) and cadmium(II). Sulfate salts of capsules obtained via anion exchange from the triflate congeners were observed to disassemble in water. Direct assembly reactions using ZnSO4 or CdSO4 did not form the target water-soluble products. | ||
Water stability of ZnII and CdII structures through increased chelate effect
In order to prepare water-stable structures containing the more labile ZnII and CdII template ions, we employed the chelate effect.32 Chelating tris(2-aminoethyl)amine (TREN) and tris(3-aminopropyl)amine (TRPN) subcomponents were combined with the ditopic and tritopic subcomponents. This strategy proved fruitful, resulting in the isolation of a collection of water-soluble structures as their sulfate salts; remarkably, they represent the first water-soluble subcomponent self-assembled structures containing labile ZnII and CdII ions. Future work will probe the effects of pH upon the aqueous stability and guest binding properties of these assemblies.As shown in Fig. 3, ditopic aldehydes E–H produced ZnII2L3 triple helicates when treated with TREN and ZnSO4, whereas with TRPN and CdSO4 generated CdII4L6 tetrahedral cages as the sole discrete products. This behaviour parallels what had been observed in the cases of Zn-6·OTf and cage Cd-11·OTf, which were previously obtained only as water-insoluble triflate salts.33
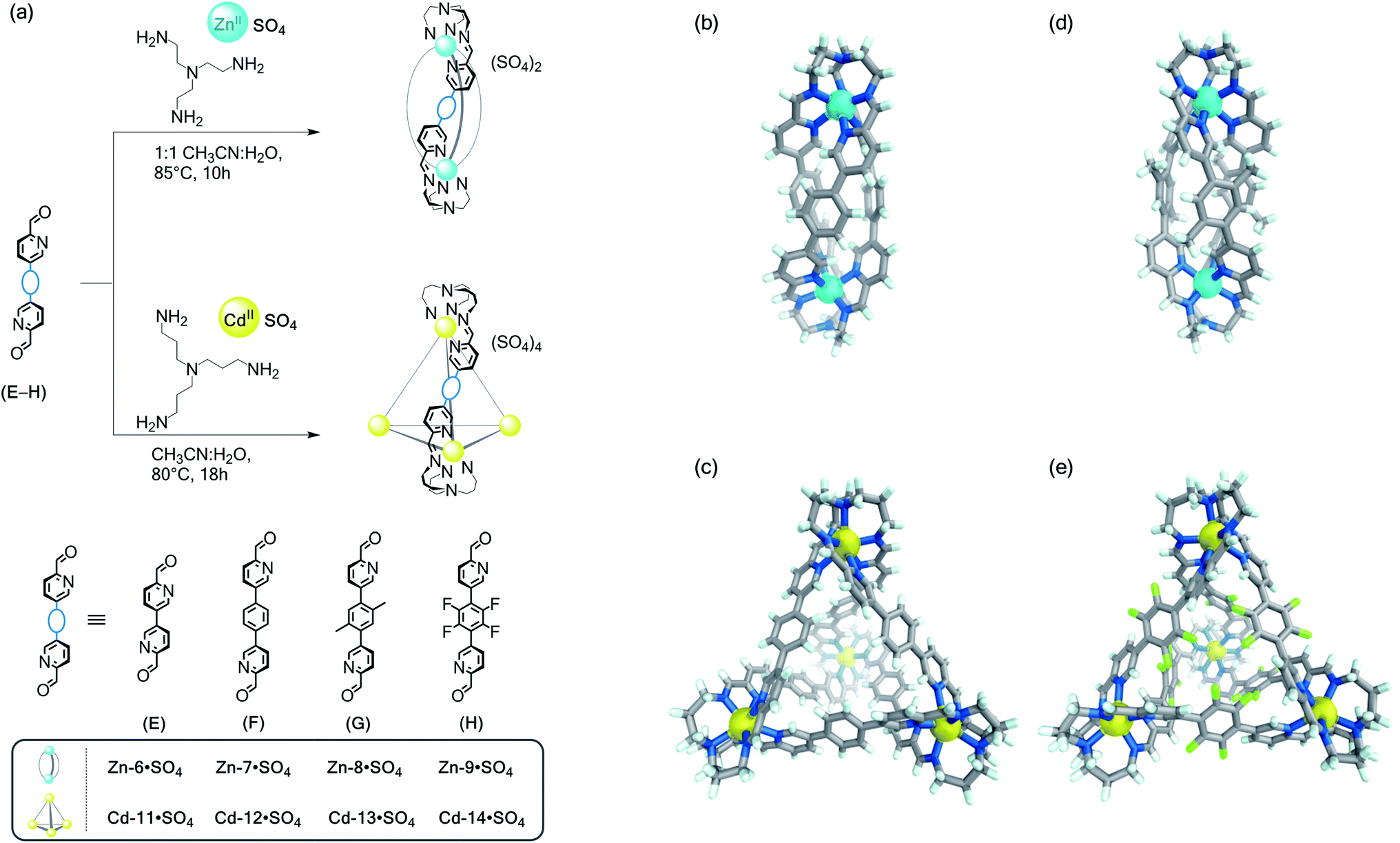 | ||
| Fig. 3 Water-soluble ZnII- and CdII-based architectures. (a) Self-assembly reactions to generate ZnII-helicates and CdII-tetrahedra from C2-symmetric subcomponents E–H and the tritopic amines TREN and TRPN. Structures based upon aldehyde F: cationic portions of the X-ray structures of (b) helicate Zn-7·OTf and (c) tetrahedron Cd-12·OTf. (d) Cationic parts of the X-ray structures of helicate Zn-8·OTf (based upon aldehyde G) and (e) tetrahedron Cd-14·OTf (based upon aldehyde H). Counterions, solvents and disorder are omitted for clarity. ZnII, CdII and fluorine atoms are coloured cyan, yellow, and pale green, respectively. | ||
All structures were characterised by NMR spectroscopy, ESI-MS, and in some cases X-ray crystallography (ESI Section 5†). ESI-MS analyses showed charge states consistent with ZnII2L3 or CdII4L6 formulations in all cases. DOSY 1H NMR spectra were also consistent with the assigned structures. While the measured diffusion coefficients (D) for the ZnII2L3 helicates ranged from 2.98–2.54 × 10−10 m2 s−1, the smaller values for the CdII4L6 tetrahedra (D = 2.13–1.67 × 10−10 m2 s−1) attested to their larger dimensions.
Strikingly, assembly products remained intact in solution at concentrations ranging from 100–2500 μM for more than four months at room temperature, and no decomposition was observed by NMR spectroscopy when heating their D2O solutions to 80 °C. All underwent anion metathesis with LiOTf to produce the corresponding acetonitrile-soluble complexes without decomposition. The chelate effect is inferred to be responsible for the high stability of these cryptate-like structures,34 contrasting with the decomposition observed for ZnII- and CdII-templated structures that do not incorporate chelating amines (Scheme 4).
Crystals of the products of subcomponent self-assembly involving aldehyde F, helical Zn-7 and tetrahedral Cd-12, were grown by diffusion of iPr2O into MeCN solutions of the corresponding triflate derivatives obtained by anion metathesis with LiOTf (Fig. 3b and c). While Zn-7 crystallised in the monoclinic space group C2 and displays a ZnII2L3 structure, CdII4L6 assembly Cd-12 has a tetrahedral framework.
The reaction of F with TRPN and CdSO4 to prepare Cd-12·SO4 displayed concentration dependence (Fig. S45†). When carried out at [F] = 17.6 mM, two products were observed by NMR spectroscopy in a 35:65 ratio, consistent with CdII4L6 cage Cd-12·SO4 and a CdII2L3 helicate. Cage formation was not suppressed by increasing the reactant concentration to [F] = 25.7 mM, and no interconversion between helicate and cage was observed after heating the mixture to 50 °C for seven days. This observation suggested that, once formed, both products were kinetically stable with a high activation barrier preventing interconversion. When the same reaction was carried out at the lower concentration of [F] = 5.7 mM, the helicate was suppressed below the limits of NMR spectroscopy detection and quantitative tetrahedron formation was observed. There is minimal steric clash between the phenylene moieties of F within a CdII2L3 helicate, which we infer to lower the activation enthalpy penalty incurred by steric crowding and structural strain to a point where it matches the activation entropy penalty incurred by incorporating twice as many building blocks, to generate the Cd-12 tetrahedron. This behaviour was only observed for F, with concentration having no perceptible effect on the outcome of self-assembly reactions involving E, G or H, which led to a single helicate or tetrahedron product in each case.
For dialdehydes E–H, however, when CdSO4 was used with TREN, or when ZnSO4 was used with TRPN, mixtures of soluble products with complex NMR and ESI mass spectra were observed, along with insoluble products; signals corresponding to MII2L3 helicates and MII4L6 tetrahedra were not identified. We infer that the incorporation of TRPN into a cadmium vertex leads to a geometry favourable to the formation of a CdII4L6 tetrahedron, as the coordination of the apical nitrogen to CdII (Fig. 3c) acts to cantilever the dialdehyde residues out into a splayed configuration that favours tetrahedron formation over the helicate. Conversely, the tighter wrapping of a TREN residue around the smaller ZnII ion, involving no apical coordination (Fig. 3b), appears to preorganise the system for helicate formation by bringing the dialdehyde residues together.
An X-ray-quality crystal of helicate Zn-8 was grown from vapour diffusion of iPr2O into an MeCN solution of Zn-8·OTf (obtained by anion metathesis with LiOTf) in the presence of excess KSbF6. Diffraction analysis of complex Zn-8 (Fig. 3d) evidenced its helical structure, with the required bending of its G moieties distributed across the three aromatic rings of each residue, as with Zn-7.
We recently reported that in contrast to the nonfluorinated ligand F, the perfluorinated subcomponent H preferentially adopted meridional (mer) over facial (fac) stereochemistry around hexacoordinated FeII ions, leading to the creation of large supramolecular prisms instead of helical or tetrahedral arrays.35 In contrast, the self-assembly of H with the ZnII/TREN or the CdII/TRPN systems resulted in formation of helicate Zn-9·SO4 or tetrahedron Cd-14·SO4 as the sole observed products, respectively. ESI-MS and NMR analyses reflected the anticipated solution structures. Their simple 1H NMR spectra, with one set of ligand resonances, are consistent with the formation of the fac coordination products; the 19F NMR spectra showed only one sharp signal for the perfluorinated phenyl ring in each case.
Treatment of an aqueous solution of Cd-14·SO4 with LiOTf induced precipitation of the acetonitrile-soluble Cd-14·OTf salt. ESI-MS and 1H NMR analyses indicated that this structure remained intact upon anion exchange (Fig. S68 and S69†). Slow vapour diffusion of Et2O into an acetonitrile solution of Cd-14·OTf containing KSbF6 allowed formation of suitable crystals for X-ray diffraction studies; Cd-14 structure crystallised in the cubic space group P213 (Fig. 3e).
The use of tritopic aldehyde D precluded Zn-TREN vertices from generating a helicate: the threefold connectivity of D requires the formation of a higher-nuclearity framework. The simplest structure possible is thus tetrahedral Zn-10·SO4 (Scheme 5), which was the only product observed following the reaction of TREN, D and ZnSO4. The analogous Cd-15·SO4 capsule (Scheme 5) was also observed to form from TRPN, D and CdSO4.
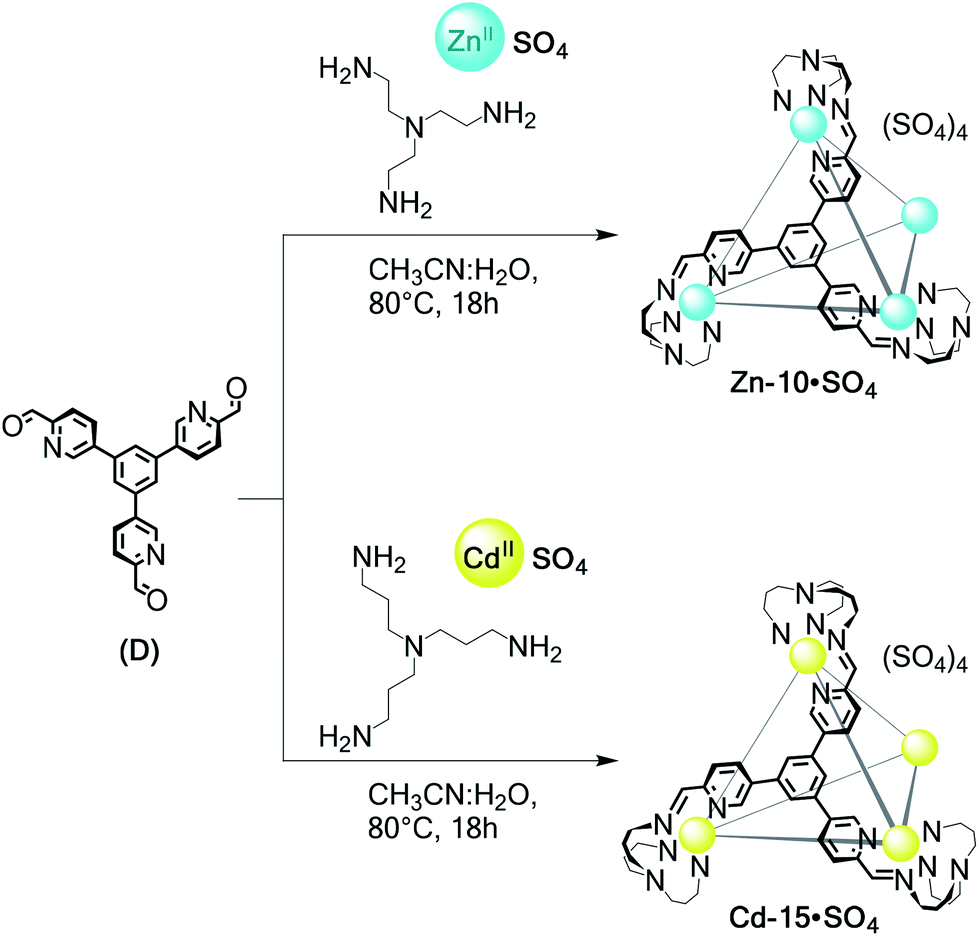 | ||
| Scheme 5 Self-assembly of water-soluble Zn-10·SO4 and Cd-15·SO4 from tritopic subcomponent D. | ||
NMR and ESI-MS analyses in water of Zn-10·SO4 and Cd-15·SO4 reflected their T-symmetric MII4L4 cage compositions (Fig. S70–S78†). 1H NMR showed only one set of ligand resonances for each assembly. DOSY measurements were consistent with species of comparable sizes to those observed for cages Cd-11·SO4 to Cd-14·SO4. These structures are stable for months at room temperature, but decomposition was observed after heating the cage D2O solutions above 80 °C. Anion exchange with LiOTf rendered these tetrahedra soluble in acetonitrile but water-insoluble (Fig. S78†). We have recently reported the crystal structure of Zn-10·NTf2, obtained as the water-insoluble triflimide salt, as part of a separate study.36 Its analytical data track with those obtained in the case of Zn-10·OTf prepared from the water-soluble sulfate salt.
Factors governing the stability of subcomponent self-assembled structures in water
The differences in behaviour observed for the architectures investigated throughout this study evidenced two principal factors associated with their kinetic stability in water: the strength of metal–ligand bonding for a given metal ion, and the density of connections between metals and ligands within a structure, considering both ligand topicity (tetratopic > tritopic > ditopic) and degree of chelation.The impact of metal ions on cage stability is evident from the comparison of the half lives (t1/2 at 20 °C) and conditions required for decomposition of the cubic structures M-1·SO4 in water (Fig. 4). While cubes constructed from ZnII and CdII survived only for minutes in aqueous solution, their NiII, FeII, and CoII congeners remained intact for months at room temperature.
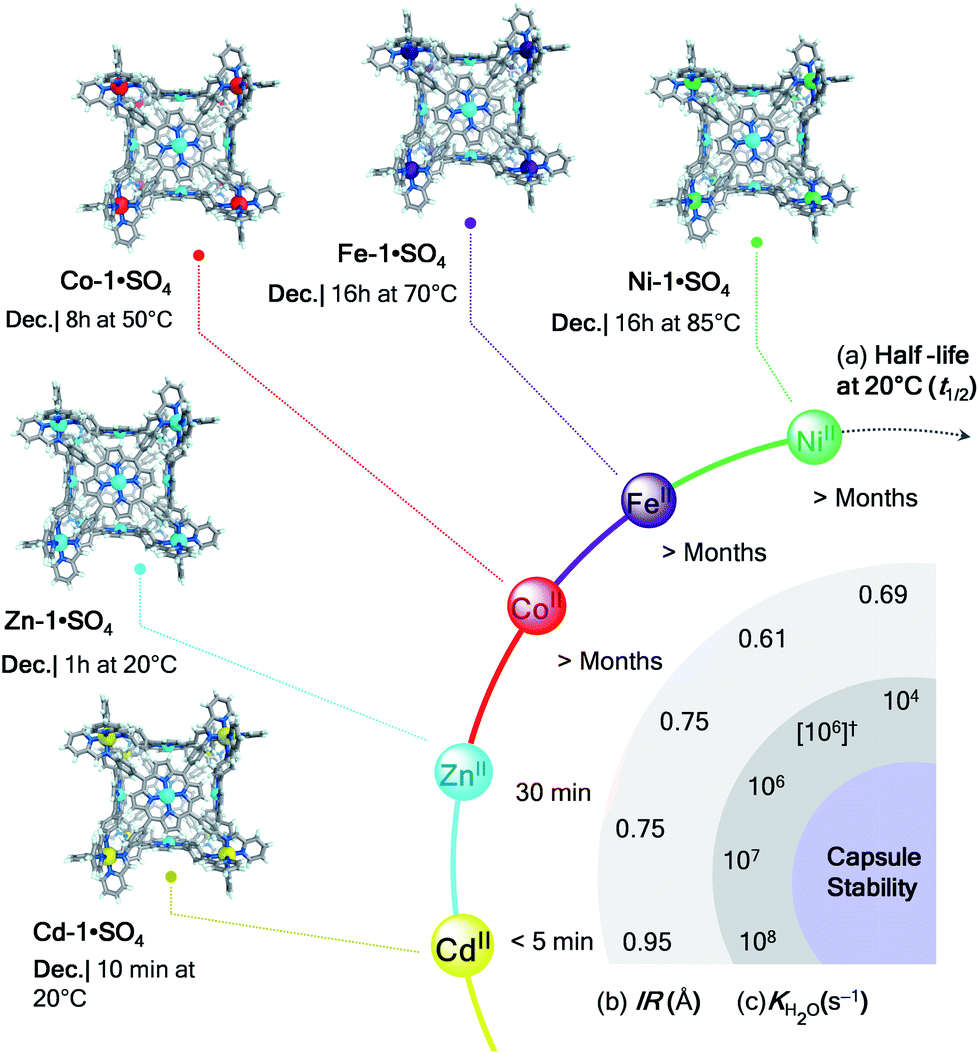 | ||
| Fig. 4 Relative stability of cubes M-1·SO4 in water. Crystal structures (Ni-1, Fe-1,22 and Co-1) and MM3 models (Zn-1 and Cd-1) of cubes with their decomposition conditions. (a) Half lives (t1/2 at 20 °C), (b) ionic radii (IR, Å) and (c) ligand-exchange rates for water (KH2O, s−1) for the different metal ions are displayed for comparison. (†) Although Fe-1 is low-spin, KH2O for high-spin FeII is given because [Fe(H2O)6]2+ is high-spin.19 | ||
Metal ions stabilise the structures into which they are incorporated following the series NiII > FeII > CoII > ZnII > CdII. Our observations mirror the trends in the stability constants found for mononuclear transition metal complexes of 2,2′-bipyridine and 1,10-phenanthroline.37 They are also consistent with the stabilities predicted by the Irving–Williams series,29 which gives an ordering of FeII < CoII < NiII > ZnII. The inversion of FeII and CoII is attributed to the low-spin character of FeII in our structures, whereas high-spin FeII, which forms weaker metal–ligand bonds,38 was considered in the original Irving–Williams series. These results collectively suggest that the ability of each metal to hold a supramolecular structure together may be gauged by examining the ionic radius (IR, Å)39 and, the ligand-exchange rate of aqua ligands (KH2O, s−1),19 and the relative energies of the metal–ligand bonds (Fig. 4). Slower exchange rates correlate well with capsule stabilities, as do shorter radii.
Whereas FeII (ref. 18) and CoII sulfate cages were only accessible through anion metathesis and exhibited limited stability in water, the ability of NiII to generate the most stable structures is reflected in the formation of Ni-2·SO4, Ni-3·SO4, and Ni-4·SO4 directly from NiSO4 in aqueous media. Cube Ni-1·SO4 also tolerated the highest temperature among the cube series before decomposing in water (Fig. 4).
Although NiII is known to form thermodynamically stable supramolecular40 and mononuclear41 complexes with chelating ligands such as 2,2′-bipyridine, NiII structures 1–4 may not be considered thermodynamically stable in water because their decomposition was observed when their aqueous solutions were heated (Ni-1·SO4 at 85 °C, Ni-2·SO4 at 75 °C, Ni-3·SO4 at 60 °C, and Ni-4·SO4 at 75 °C; Fig. 4) as evidenced by precipitation of water-insoluble subcomponents. We infer that the poor aqueous solubility of the subcomponents limits the reversibility of the dynamic covalent imine bonds and consequently the thermodynamic stability of the NiII assemblies.
An increase in aqueous stability as ligand topicity increases also emerges clearly from our study. The tetratopic ligands of 1 lend this cubic framework the greatest degree of stability as temperatures increase, and across the widest range of metal ions. Similarly, the tritopic ligands of tetrahedra 2 and 4 led to stability with a wider range of metal ions than was observed in the cases of their congeners 3 and 5, which incorporated ditopic ligands.
We infer the topicity effect upon stability to result from the higher degree of binding cooperativity20 imposed by these ligands. Dissociation of a single ligand arm from a tetratopic ligand of 1 would require three more arms to disengage in order to free the ligand to precipitate. In contrast, dissociation of one end of a ditopic ligand of 3 would only require the other end to come off for the ligand to be free. At room temperature, framework 3 thus disintegrated in water when prepared with FeII (ref. 18) and CoII; 3 was only stable in water when prepared with the most strongly binding metal, NiII.37,41
Cooperativity20 also plays a key role in the aqueous stabilisation of cryptate-like structures 6–15 (Fig. 3 and Scheme 5). These incorporate the most labile metals, ZnII and CdII, requiring the chelation imposed by TREN or TRPN. These dynamic cages must undergo imine hydrolysis steps in addition to delegation in order for their building blocks to become free to precipitate. A cage framework containing a single “defect”, a free TREN or TRPN amine arm in proximity to an aldehyde would thus experience a high effective molarity20 for imine condensation, whereas decomposition would require the two remaining imine arms to hydrolyse. We infer the tightly-knit, cooperative construction of these cages thus to underpin their aqueous stability. Our work thus points the way to the possible preparation of even more robust water-soluble cages through the use of more coordinatively-inert NiII or FeII to template42 the formation of cages containing TREN and TRPN vertices.
Conclusions
This study has demonstrated the versatility of the subcomponent self-assembly method in preparing a wide range of water-soluble and kinetically stable metal–organic architectures built from hydrophobic ligands, octahedral metal ions and sulfate counter anions. MII8L6 cubes, face-capped MII4L4 tetrahedra, edge-linked MII4L6 tetrahedra as well as MII2L3 triple helicates were obtained despite the hydrophobicity and minimal aqueous solubility of their organic subcomponents.These results may stimulate investigations involving the use of hydrophobic building blocks, which might not have been considered for use in aqueous solution, but the utility of which have been demonstrated herein. Such subcomponents show great promise for generating water-soluble architectures, when their assemblies are highly charged and paired with hydrophilic counterions.
The synthetic strategies developed herein provide a blueprint for the preparation of more intricate water-resistant metallosupramolecular structures, by employing the known concepts of using relatively inert metals and chelate cooperativity. Our implementation here takes advantage of the virtues of the subcomponent self-assembly method. Kinetic intermediates that may form at initial reaction stages were not trapped, allowing self-repair via rearrangement at the dynamic imine functions while maintaining a degree of structural integrity. We were thus able to prepare new classes of metal–organic capsules that had not previously been obtained as soluble and kinetically stable in aqueous solution.
Our study thus provides several means to control the lifetimes of capsules for prospective applications. Their disintegration in water under well-defined conditions may be advantageous for controlled drug delivery or cargo transport.43 For instance, ZnII-, FeII-, CoII-, and NiII-based cubes could be loaded with different cargoes, which can be released as the cages sequentially disassemble as the temperature is progressively increased. The cage components could be recovered and extracted for reassembly, as was demonstrated recently using a simple system involving a single capsule.10a We also anticipate the spectroscopic properties of the cubic capsules to be of interest in aqueous solution. These properties will be investigated in due course, in the context of aqueous luminescent sensing. Other applications, such as confined-space catalysis, thermal protection of reactive chemicals or guest transport could also be built upon the waterproof architectures accessible through the approaches developed herein.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council (695009) and the UK Engineering and Physical Sciences Research Council (EPSRC, EP/P027067/1). The authors thank the Diamond Light Source (UK) for synchrotron beamtime on I19 (MT15768) and the Cambridge University Chemistry NMR facility for performing some NMR experiments. E. G. P. acknowledges CONACYT-México for postdoctoral support. J. M. acknowledges postdoctoral fellowship support from Fundación Ramón Areces. A. J. P. acknowledges the Cambridge Trust (Vice Chancellor's Award). M. K. acknowledges the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 642192.Notes and references
- (a) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef CAS PubMed; (b) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (c) Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197 CrossRef CAS PubMed.
- (a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed; (b) V. M. Dong, D. Fiedler, B. Carl, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 14464 CrossRef CAS PubMed; (c) M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 4662 CrossRef CAS PubMed; (d) T. Sawada, M. Yoshizawa, S. Sato and M. Fujita, Nat. Chem., 2009, 1, 53 CrossRef CAS PubMed; (e) Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100 CrossRef CAS PubMed.
- (a) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231 CrossRef CAS PubMed; (b) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; (c) C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530 CrossRef CAS PubMed; (d) L. Catti, Q. Zhang and K. Tiefenbacher, Chem.–Eur. J., 2016, 22, 9060 CrossRef CAS PubMed.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS PubMed; (b) L. Shen, N. Cao, L. Tong, X. Zhang, G. Wu, T. Jiao, Q. Yin, J. Zhu, Y. Pan and H. Li, Angew. Chem., Int. Ed., 2018, 57, 16486 CrossRef CAS PubMed.
- (a) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744 RSC; (b) S. M. Biros, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 12094 CrossRef CAS PubMed; (c) M. Yamashina, M. Akita, T. Hasegawa, S. Hayashi and M. Yoshizawa, Sci. Adv., 2017, 3, e1701126 CrossRef PubMed; (d) C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408 CrossRef CAS; (e) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinsky, Chem. Rev., 2016, 116, 2414 CrossRef CAS; (f) F. Biedermann, M. N. Werner and H.-J. Schneider, Angew. Chem., Int. Ed., 2013, 53, 11158 CrossRef; (g) S. M. Biros and J. Rebek, Chem. Soc. Rev., 2007, 36, 93 RSC.
- (a) P. S. Cremer, A. H. Flood, B. C. Gibb and D. L. Mobley, Nat. Chem., 2018, 10, 8 CrossRef CAS PubMed; (b) P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952 CrossRef CAS PubMed; (c) J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893 CrossRef CAS PubMed; (d) K. Ono, J. K. Klosterman, M. Yoshizawa, K. Sekiguchi, T. Tahara and M. Fujita, J. Am. Chem. Soc., 2009, 131, 12526 CrossRef CAS PubMed; (e) B. Roy, A. K. Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916 CrossRef CAS PubMed; (f) F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754 CrossRef CAS PubMed.
- L. Taylor, I. Riddell and M. M. J. Smulders, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201806297.
- (a) G. Liu, Y. D. Yuan, J. Wang, Y. Cheng, S. B. Peh, Y. Wang, Y. Qian, J. Dong, D. Yuan and D. Zhao, J. Am. Chem. Soc., 2018, 140, 6231 CrossRef CAS PubMed; (b) Y.-P. He, L.-B. Yuan, G.-H. Chen, Q.-P. Lin, F. Wang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2017, 139, 16845 CrossRef CAS PubMed.
- (a) J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958 CrossRef CAS PubMed; (b) M. B. Hillyer, C. L. D. Gibb, P. Sokkalingam, J. H. Jordan, S. E. Ioup and B. C. Gibb, Org. Lett., 2016, 18, 4048 CrossRef CAS PubMed.
- (a) D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez and J. R. Nitschke, Angew. Chem., Int. Ed., 2018, 57, 3717 CrossRef CAS; (b) K. Yazaki, S. Yoshihisa, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2016, 22, 17557 CrossRef CAS PubMed; (c) B. Roy, E. Zangrando and P. S. Mukherjee, Chem. Commun., 2016, 52, 4489 RSC.
- (a) B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773 CrossRef CAS; (b) N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Chem.–Eur. J., 2011, 17, 9669 CrossRef CAS.
- (a) F. Ibukuro, T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 1998, 120, 8561 CrossRef CAS; (b) Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189 RSC.
- C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, J. A. Smith, F. R. Keene and C. Motti, Chem.–Eur. J., 2008, 14, 10535 CrossRef CAS PubMed.
- (a) J. A. Thomas, Chem. Soc. Rev., 2007, 36, 856 RSC; (b) P. R. Symmers, M. J. Burke, D. P. August, P. I. T. Thomson, G. S. Nichol, M. R. Warren, C. J. Campbell and P. J. Lusby, Chem. Sci., 2015, 6, 756 RSC.
- (a) N. Struch, C. Frömbgen, G. Schnakenburg and A. Lützen, Eur. J. Org. Chem., 2017, 4984 CrossRef CAS; (b) B. Sun, S. S. Nurttila and J. N. H. Reek, Chem.–Eur. J., 2018, 24, 14693 CrossRef CAS PubMed; (c) P. D. Frischmann, V. Kunz, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2015, 21, 2766 CrossRef CAS PubMed; (d) M. C. Young, A. M. Johnson, A. S. Gamboa and R. J. Hooley, Chem. Commun., 2013, 49, 1627 RSC; (e) P. F. Kuijpers, M. Otte, M. Dürr, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, ACS Catal., 2016, 6, 3106 CrossRef CAS; (f) D. Luo, X.-Z. Wang, C. Yang, X.-P. Zhou and D. Li, J. Am. Chem. Soc., 2018, 140, 118 CrossRef CAS PubMed; (g) D. Luo, M. Li, X.-P. Zhou and D. Li, Chem.–Eur. J., 2018, 24, 7108 CrossRef CAS PubMed; (h) H. Bunzen, Nonappa, E. Kalenius, S. Hietala and E. Kolehmainen, Chem.–Eur. J., 2013, 19, 12978 CrossRef CAS PubMed; (i) J. Roukala, J. Zhu, C. Giri, K. Rissanen, P. Lantto and V.-V. Telkki, J. Am. Chem. Soc., 2015, 137, 2464 CrossRef CAS PubMed; (j) K.-C. Sham, S.-M. Yiu and H.-L. Kwong, Inorg. Chem., 2013, 52, 5648 CrossRef CAS PubMed; (k) D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788 RSC; (l) S. Yi, V. Brega, B. Captain and A. E. Kaifer, Chem. Commun., 2012, 48, 10295 RSC.
- D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423 CrossRef CAS PubMed.
- (a) Y. Zhang, X. Zheng, N. Cao, C. Yang and H. Li, Org. Lett., 2018, 20, 2356 CrossRef CAS PubMed; (b) X.-Y. Hu, W.-S. Zhang, F. Rominger, I. Wacker, R. R. Schröder and M. Mastalerz, Chem. Commun., 2017, 53, 8616 RSC.
- E. G. Percástegui, J. Mosquera and J. R. Nitschke, Angew. Chem., Int. Ed., 2017, 56, 9136 CrossRef PubMed.
- M. Eigen, Pure Appl. Chem., 1963, 6, 97 CAS . The exchange rate constant of aqua ligands (KH2O) for low-spin FeII is not available; the KH2O for high-spin d6 FeII (106 s−1) is used for approximate comparison to KH2O for NiII (104 s−1).
- (a) L. K. S. von Krbek, C. S. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622 RSC; (b) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
- (a) C. G. P. Taylor, J. R. Piper and M. D. Ward, Chem. Commun., 2016, 52, 6225 RSC; (b) H.-K. Liu, W.-Y. Sun, D.-J. Ma, K.-B. Yu and W.-X. Tang, Chem. Commun., 2000, 591 RSC; (c) S. Aoki, S. Suzuki, M. Kitamura, T. Haino, M. Shiro, M. Zulkefeli and E. Kimura, Chem.–Asian J., 2012, 7, 944 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479 CrossRef CAS PubMed.
- R. A Bilbeisi, J. K. Clegg, N. Elgrishi, X. de Hatten, M. Devillard, B. Breiner, P. Mal and J. R. Nitschke, J. Am. Chem. Soc., 2012, 134, 5110 CrossRef PubMed.
- J. K. Clegg, J. Cremers, A. J. Hogben, B. Breiner, M. M. J. Smulders, J. D. Thoburn and J. R. Nitschke, Chem. Sci., 2013, 4, 68 RSC.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 751 CrossRef CAS PubMed.
- (a) K. Mahata, P. D. Frischmann and F. Würthner, J. Am. Chem. Soc., 2013, 135, 15656 CrossRef CAS PubMed; (b) A. M. Johnson, C. A. Wiley, M. C. Young, X. Zhang, Y. Lyon, R. R. Julian and R. J. Hooley, Angew. Chem., Int. Ed., 2015, 54, 5641 CrossRef CAS PubMed; (c) J. M. Dragna, G. Pescitelli, L. Tran, V. M. Lynch, E. V. Anslyn and L. Di Bari, J. Am. Chem. Soc., 2012, 134, 4398 CrossRef CAS PubMed; (d) N. Struch, J. G. Brandenburg, G. Schnakenburg, N. Wagner, J. Beck, S. Grimme and A. Lützen, Eur. J. Inorg. Chem., 2015, 5503 CrossRef CAS; (e) P. M. Bogie, L. R. Holloway, Y. Lyon, N. C. Onishi, G. J. O. Beran, R. R. Julian and R. J. Hooley, Inorg. Chem., 2018, 57, 4155 CrossRef CAS PubMed.
- (a) M. J. Burke, G. S. Nichol and P. J. Lusby, J. Am. Chem. Soc., 2016, 138, 9308 CrossRef CAS PubMed; (b) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (c) J. Ramírez, A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2007, 237 RSC.
- (a) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858 CrossRef CAS PubMed; (b) T. K. Ronson, C. Giri, N. K. Beyeh, A. Minkkinen, F. Topić, J. J. Holstein, K. Rissanen and J. R. Nitschke, Chem.–Eur. J., 2013, 19, 3374 CrossRef CAS PubMed; (c) I. A. Riddell, Y. R. Hristova, J. K. Clegg, C. S. Wood, B. Breiner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 2723 CrossRef CAS PubMed; (d) X.-P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062 CrossRef CAS PubMed; (e) D. Lewing, H. Koppetz and F. E. Hahn, Inorg. Chem., 2015, 54, 7653 CrossRef CAS PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192 RSC.
- I. Bertini, C. Luchinat and P. Giacomo, in Current Methods in Inorganic Chemistry, Elsevier, Amsterdam, 2001, vol. 2, pp. 187–189 Search PubMed.
- (a) S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704 CrossRef CAS PubMed; (b) M. G. Fisher, P. A. Gale, M. E. Light and S. J. Loeb, Chem. Commun., 2008, 5695 RSC; (c) H. Y. Lee, A. Olasz, M. Pink, H. Park and D. Lee, Chem. Commun., 2011, 47, 481 RSC.
- (a) M. Calvin and K. W. Wilson, J. Am. Chem. Soc., 1945, 67, 2003 CrossRef CAS; (b) A. E. Martell, R. D. Hancock and R. Motekaitis, Coord. Chem. Rev., 1994, 133, 39 CrossRef CAS; (c) A. W. Adamson, J. Am. Chem. Soc., 1953, 76, 1578 CrossRef.
- J. Mosquera, S. Zarra and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 1556 CrossRef CAS PubMed.
- (a) M.-P. Teulade-Fichou, J.-P. Vigneron and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1996, 2169 RSC; (b) J.-M. Lehn, R. Méric, J.-P. Vigneron, I. Bkouche-Waksman and C. Pascard, J. Chem. Soc., Chem. Commun., 1991, 62 RSC; (c) R.-C. Brachvogel, F. Hampel and M. von Delius, Nat. Commun., 2015, 6, 7129 CrossRef PubMed.
- M. Kieffer, B. S. Pilgrim, T. K. Ronson, D. A. Roberts, M. Aleksanyan and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 6813 CrossRef CAS PubMed.
- A. M. Castilla, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2016, 138, 2342 CrossRef CAS PubMed.
- H. Irving and D. H. Mellor, J. Chem. Soc., 1962, 5222 RSC.
- (a) S. Brooker, Chem. Soc. Rev., 2015, 44, 3192 RSC; (b) H. L. C. Feltham, A. S. Barltrop and S. Brooker, Coord. Chem. Rev., 2017, 344, 26 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- (a) R. Custelcean, P. V. Bonnesen, N. C. Duncan, X. Zhang, L. A. Watson, G. V. Berkel, W. B. Parson and B. P. Hay, J. Am. Chem. Soc., 2012, 134, 8525 CrossRef CAS PubMed; (b) R. Custelecean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025 CrossRef PubMed.
- For example: the association constants of MII complexes with 2,2′-bipyridine (bpy) [M(bpy)3]2+ reflect stronger binding to NiII; logK at 25 °C for FeII, CoII, NiII, ZnII, and CdII = 17.2, 16.0, 20.2, 13.2 and 10.4, respectively. From A. E. Martrell and R. M. Smith, Critical Stability Constants, vol. 5, Springer Science+Business, New York, 1982 Search PubMed.
- (a) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815 RSC; (b) H. L. Ozores, M. Amorín and J. R. Granja, J. Am. Chem. Soc., 2017, 139, 776 CrossRef CAS PubMed; (c) J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318 CrossRef CAS PubMed; (d) W. M. Bloch, J. J. Holstein, B. Dittrich, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 5534 CrossRef CAS PubMed; (e) S. Akine, M. Miyashita and T. Nabeshima, J. Am. Chem. Soc., 2017, 139, 4631 CrossRef CAS PubMed; (f) H. Löw, E. Mena-Osteritz and M. von Delius, Chem. Sci., 2018, 9, 4785 RSC; (g) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC; (h) E. Kim, D. Kim, H. Jung, J. Lee, S. Paul, N. Selvapalam, Y. Yang, N. Lim, C. G. Park and K. Kim, Angew. Chem., Int. Ed., 2010, 49, 4405 CrossRef CAS PubMed; (i) N. G. White and M. J. MacLachlan, Chem. Sci., 2015, 6, 6245 RSC; (j) N. Rodríguez-Vázquez, M. Amorín, I. Alfonso and J. R. Granja, Angew. Chem., Int. Ed., 2016, 55, 4504 CrossRef PubMed; (k) J. Yang, L. Shao, J. Yuan and F. Huang, Chem. Commun., 2016, 52, 12510 RSC; (l) G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233 CrossRef CAS PubMed; (m) H. Li, A. C. Fahrenbach, A. Coskun, Z. Zhu, G. Barin, Y.-L. Zhao, Y. Y. Botros, J.-P. Sauvage and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 6782 CrossRef CAS PubMed; (n) R. V. Mukhopadhyay, Y. Kim, J. Koo and K. Kim, Acc. Chem. Res., 2018, 51, 2730 CrossRef CAS PubMed.
- C. Bravin, E. Badetti, F. A. Scaramuzzo, G. Licini and C. Zonta, J. Am. Chem. Soc., 2017, 139, 6456 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and characterisation data. CCDC 1837541–1837547. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05085f |
| This journal is © The Royal Society of Chemistry 2019 |