
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed γ-arylation of γ,δ-unsaturated O-carbamates via an unusual haptotropic rearrangement†
Titouan
Royal
and
Olivier
Baudoin
 *
*
University of Basel, Department of Chemistry, St. Johanns-Ring 19, CH-4056 Basel, Switzerland. E-mail: olivier.baudoin@unibas.ch
First published on 26th December 2018
Abstract
An unusual γ-selectivity was observed in the arylation of γ,δ-unsaturated O-carbamates involving directed lithiation, transmetallation to zinc and Negishi coupling, when a specific combination of aryl electrophile and phosphine ligand is employed. Mechanistic studies indicate that an unusual, stereospecific haptotropic rearrangement of the palladium–diene intermediate is involved.
Introduction
The directed α-lithiation of N- and O-carbamates in the presence of a chiral diamine such as sparteine allows access to synthetically valuable enantioenriched amine and alcohol derivatives.1 In this context, we recently showed that α-zincated O-carbamates, obtained by sparteine-mediated α-lithiation and transmetallation to zinc, could be cross-coupled with aryl bromides to give highly enantioenriched α-arylated O-carbamates (Scheme 1a), which were further converted to secondary and tertiary alcohols.2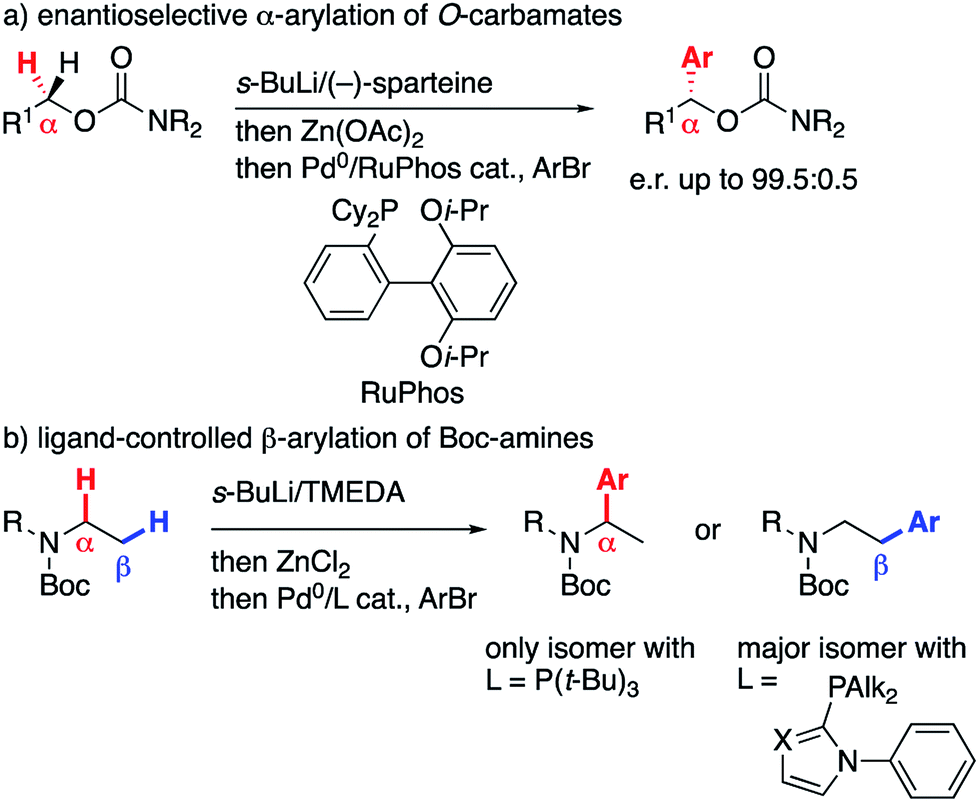 | ||
| Scheme 1 Previous work relevant to the current study. TMEDA = N,N,N′,N′-tetramethylethylenediamine. | ||
On the other hand, within a research program dedicated to the control of site-selectivity in the Pd-catalyzed cross-coupling of secondary nucleophiles,3 we reported that α-zincated acyclic Boc-amines undergo α- or β-selective arylation simply by switching the phosphine ligand (Scheme 1b),4 with the β-arylated product arising from a ‘chain walk’ mechanism.5 While we were trying to extend the migratory Negishi coupling from these N- to the above-mentioned O-carbamates, we uncovered a new reactivity of γ,δ-unsaturated substrates, which we wish to describe therein.
Results and discussion
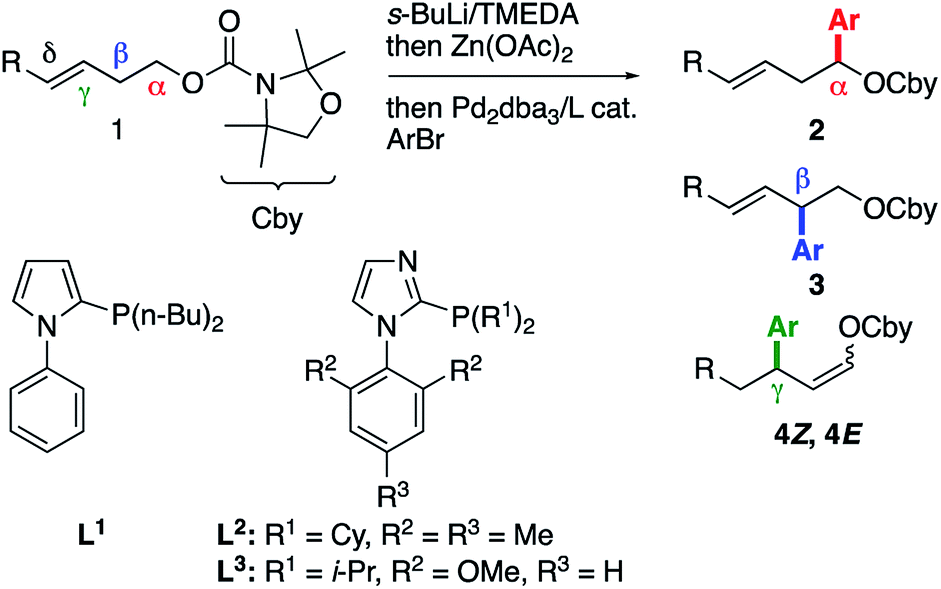
In our previous work, we showed that the α-lithiation/Li–Zn transmetallation/cross-coupling sequence6 applied to γ,δ-unsaturated carbamate 1a gave rise to α-arylated product 2a exclusively using bromobenzene as the electrophile and RuPhos7 as the ligand (Table 1, entry 1).2 When o-fluorobromobenzene, which was previously shown to favor Pd migration,8 was employed instead of PhBr, the same reaction outcome was observed (entry 2). Replacing RuPhos with the less bulky and more conformationally flexible phosphine L1, which was previously optimized to favor Pd migration,4,9 did not afford any cross-coupling product (entry 3). After a quick ligand screen,10 we found that imidazole-based phosphine11L2 furnished a mixture of α, β and γ-arylated products 2–4, with the latter being obtained as a Z/E mixture (entry 4). Interestingly, the proportion of these unexpected γ-arylated products increased when the reaction was conducted from the δ-substituted E-enecarbamate 1bE, which allowed isolation of the mixture of 4bZ and 4bE and confirmation of their structural identity (entry 5). Modifying the ligand structure impacted the yield and Z/E ratio.10 For instance, ligand L3 provided a lower yield of the γ-arylated products but a slightly enhanced Z/E ratio (entry 6). Despite significant experimentation, the modification of other reaction parameters did not lead to substantial improvements of the yield or the Z/E ratio of product 4b.10 Of note, the use of RuPhos (entry 7) or of a less electronically activated electrophile (entry 8) in the reaction of carbamate 1bE mainly furnished the α-arylated product, thereby highlighting the role of both the aryl electrophile and the phosphine ligand on the formation of the γ-arylated products 4. Moreover, the δ-arylated product was not observed in these experiments. Stimulated by the apparent lack of precedent for the formation of allylic products 4 from a homoallylic precursor such as 1,12 we decided to further study this transformation and elucidate its mechanism.|
|
|||||
|---|---|---|---|---|---|
| Entry | R | ArBr | Ligand | 2/3/4Z/4Eb | Yieldc (%) |
| a Reaction conditions: 1a–b (1.0 equiv.), s-BuLi (1.4 equiv.), TMEDA (1.4 equiv.), Et2O, −78 °C, 4 h, then Zn(OAc)2 (1.5 equiv.), −78 → 20 °C, 1 h, then evaporation of volatiles, then Pd2dba3 (1.75 mol%), ligand (3.5 mol%), ArBr (0.7 equiv.), toluene, 60 °C, 18 h. b Determined by GCMS analysis. c Combined NMR yield using trifluorotoluene as the internal standard. d Yield of the isolated product. e Yield of the isolated mixture of 4bZ and 4bE. | |||||
| 1 | H (1a) | PhBr | RuPhos | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:0:0 :0:0:0 |
60d |
| 2 | H (1a) | 2-F-C6H4Br | RuPhos | 100:0:0:0 |
54 |
| 3 | H (1a) | 2-F-C6H4Br | L1 | — | n.r. |
| 4 | H (1a) | 2-F-C6H4Br | L2 | 18:25:40:17 |
33 |
| 5 | Me (1bE) | 2-F-C6H4Br | L2 | 9:6:64:21 |
61 (60)e |
| 6 | Me (1bE) | 2-F-C6H4Br | L3 | 16:1:73:10 |
44 |
| 7 | Me (1bE) | 2-F-C6H4Br | RuPhos | 70:0:22:7 |
n.d. |
| 8 | Me (1bE) | 2-Me-C6H4Br | L2 | 96:0:1:3 |
n.d. |
First, we explored the effect of the aryl electrophile on the arylation of γ-enecarbamate 1bE (Scheme 2a). A range of 2-fluoroarenes bearing electron-withdrawing or -donating groups at the 4-position were found compatible (entries 2–6) and had a minor impact on the Z/E (65:35–77:23) and γ-selectivity (87–90%), hence further highlighting the main effect of the fluorine atom at the ortho position. Interestingly, 3-bromo-2-fluoropyridine had a stronger effect and provided the γ-arylated product 4h exclusively (entry 7). Other electron-withdrawing ortho substituents favored the γ-arylated product, but with lower Z/E selectivities (entries 8 and 9). A methoxy group afforded a lower γ-selectivity (entry 10), with an effect between a methyl (Table 1, entry 8) and electron-withdrawing groups. Next, other γ-enecarbamates were studied (Scheme 2b). The configuration of the alkene did not have a significant impact on the yield, Z/E or γ-selectivity (entry 11, compare with entry 1), hence showing that E and Z alkenes lead to the same reaction intermediate. Other δ-substituted γ-enecarbamates provided a similar outcome (entries 12–16), although the γ-selectivities could not be determined in all cases due to overlaps of the signals of the α, β and γ isomers in the GC analysis. Control experiments with δ-enecarbamate 1q and saturated enecarbamate 1r provided the α-arylated product exclusively, whereas the β-enecarbamate 1s provided traces of γ-arylated products. These results confirm the unique behavior of γ,δ-unsaturated carbamates, leading to reaction intermediates that are not accessible from other types of O-carbamates.
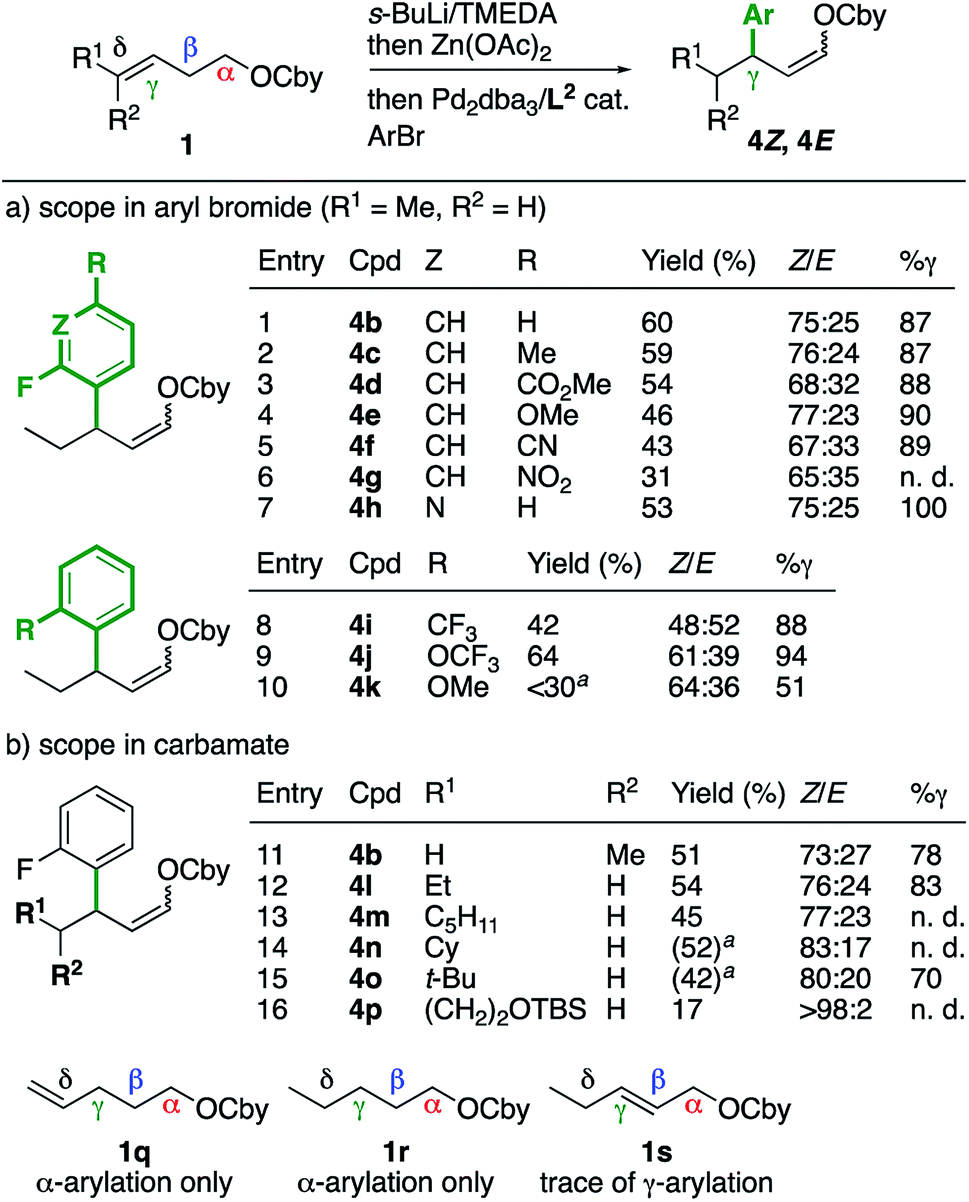 | ||
| Scheme 2 Scope of the γ-arylation of γ,δ-unsaturated O-carbamates. Yields refer to isolated mixtures of Z/E isomers unless otherwise noted. Z/E ratios were measured by 19F NMR. % γ refers to the percentage of γ-arylated product vs. other arylated products, as measured by GCMS. a Yield of the isolated mixture of inseparable arylated products. n. d. = could not be determined. | ||
A number of experiments were next performed to probe the reaction mechanism (Scheme 3). First, as an additional control experiment, lithiation of 1bE and quenching with water returned the reactant without isomerization of the alkene (Scheme 3a). This experiment complements the one reported in Table 1, entry 7, using RuPhos as the ligand, and shows that the double bond migration occurs after transmetallation to Pd and is ligand-induced. Performing the reaction under standard conditions from β-deuterated substrate 1m-D2 delivered a mixture of α-, β- and γ-arylated products as expected, which could be purified to isolate a small quantity of α- and γ-arylated products (Scheme 3b). The α-arylated product 2m-D2 retained a fully deuterated β position, whereas the γ-arylated products 4mZ-D2 and 4mE-D2 both showed a complete shift of one deuterium from the β to the δ position, consistent with a β-H elimination/insertion mechanism. Performing the lithiation with (+)-sparteine instead of TMEDA2 afforded other crucial pieces of information (Scheme 3c). Enecarbamates 1bE, 1bZ or a 1:1 mixture thereof led to the same 74:26 Z/E mixture of γ-arylated products 4bZ/4bE, with an e.r. of 98:2 for both Z and E isomers. Hence, the absolute configuration of 4bZ and 4bE does not depend on the geometry of the starting alkene. Hydrogenating this mixture provided saturated compound 5 with an e.r. of 75:25, showing that 4bZ and 4bE have opposite absolute configurations – if they had the same absolute configuration, the e.r. of 5 would be also 98:2. This experiment was repeated several times and was found to be reproducible. Similarly, an 87:13 mixture of 4bZ/4bE obtained using ligand L3 instead of L1 (see Table 1, entry 6) led to an 86:14 e.r. for hydrogenated product 5 (Scheme 3d). Determining the absolute configuration of the major enantiomer of 5 was key to elucidate the reaction mechanism (vide infra), however it also turned out to be a bigger challenge than expected. After significant experimentation, we found that the heavy p-nitrobiphenyl ester 7, obtained through cleavage of the Cby carbamate under Hoppe's conditions13 and esterification, was suitable for obtaining single crystals (Scheme 3d). First, a racemic sample of 7 was prepared using TMEDA instead of (+)-sparteine in the lithiation step, then the enantiomers were separated by semipreparative HPLC on a chiral column.10 Both enantiomers were crystallized and analyzed by X-ray diffraction, which allowed to ascribe their absolute configurations. The (S) and (R) enantiomers corresponded to the major and minor enantiomers, respectively, obtained through the (+)-sparteine-mediated sequence, and hence the absolute configuration of the major enantiomers of precursors 5 and 4bZ can be also ascribed as (S).
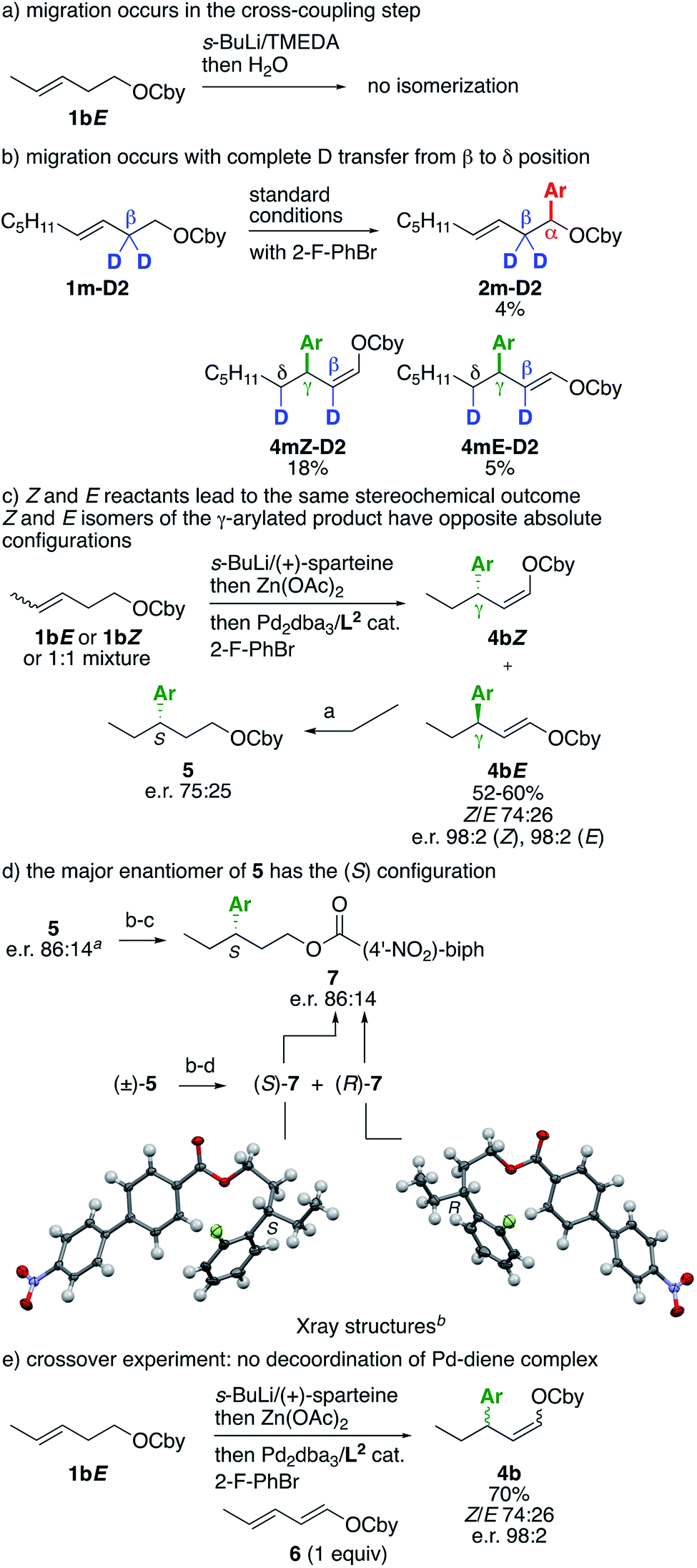 | ||
| Scheme 3 Mechanistic investigations. Reaction conditions: (a) H2 (50 bar), Pd/C, EtOH, 50 °C; (b) MeSO3H, MeOH, reflux, then Ba(OH)2, reflux; (c) ClC(O)(4′-NO2-biphenyl), Et3N, DMAP, CH2Cl2, 23 °C; (d) separation of enantiomers by semipreparative HPLC on a chiral stationary phase. Ar = 2-FC6H5. a Obtained from 1bE using L3 instead of L2 in the cross-coupling step. b Thermal ellipsoids at the 50% probability level. | ||
The ensemble of collected experimental information allows to propose the following mechanism (Scheme 4). As shown previously, the initial organopalladium A obtained by (+)-sparteine-mediated lithiation and stereoretentive Li–Zn and Zn–Pd transmetallations undergoes direct reductive elimination in the presence of usual ligands and electrophiles to give the α-arylated product 2 with the shown absolute configuration.2 Alternatively, ligand-enabled syn-stereospecific β-elimination of Ha or Hb provides π-complexes B1 or B2, respectively. From this point, two pathways are accessible: π-bond rotation and syn-insertion4,8a,14 to give complexes D1 and D2viaC1 and C2, and reductive elimination to give the β-arylated product 3 (the absolute configuration of which could not be determined). Alternatively, sterically favored migration of the Pd complex to the other double bond, known as haptotropic rearrangement,15 provides isomers E1–E2. Such a stereospecific haptotropic rearrangement along a conjugated polyene is extremely rare.16,17 Of note, π-complexes B1–B2 and E1–E2 are stable and do not undergo reversible decoordination–coordination, as indicated by the lack of observed racemization when the reaction was performed in the presence of dienecarbamate 6 (Scheme 3e). From E1 and E2, syn-insertion and reductive elimination lead to products 4E and 4Z, respectively, viaF1 and F2. The deuterium-labelling experiment shown in Scheme 3b is consistent with the pathway A → F1–F2. The opposite absolute configurations of the major enantiomers of 4bZ and 4bE deduced from Scheme 3c and d are also consistent with this pathway, wherein all elementary steps are stereospecific. This mechanistic proposal also explains the fact that both E and Z geometrical isomers of A furnish the same stereochemical outcome (Scheme 3c).
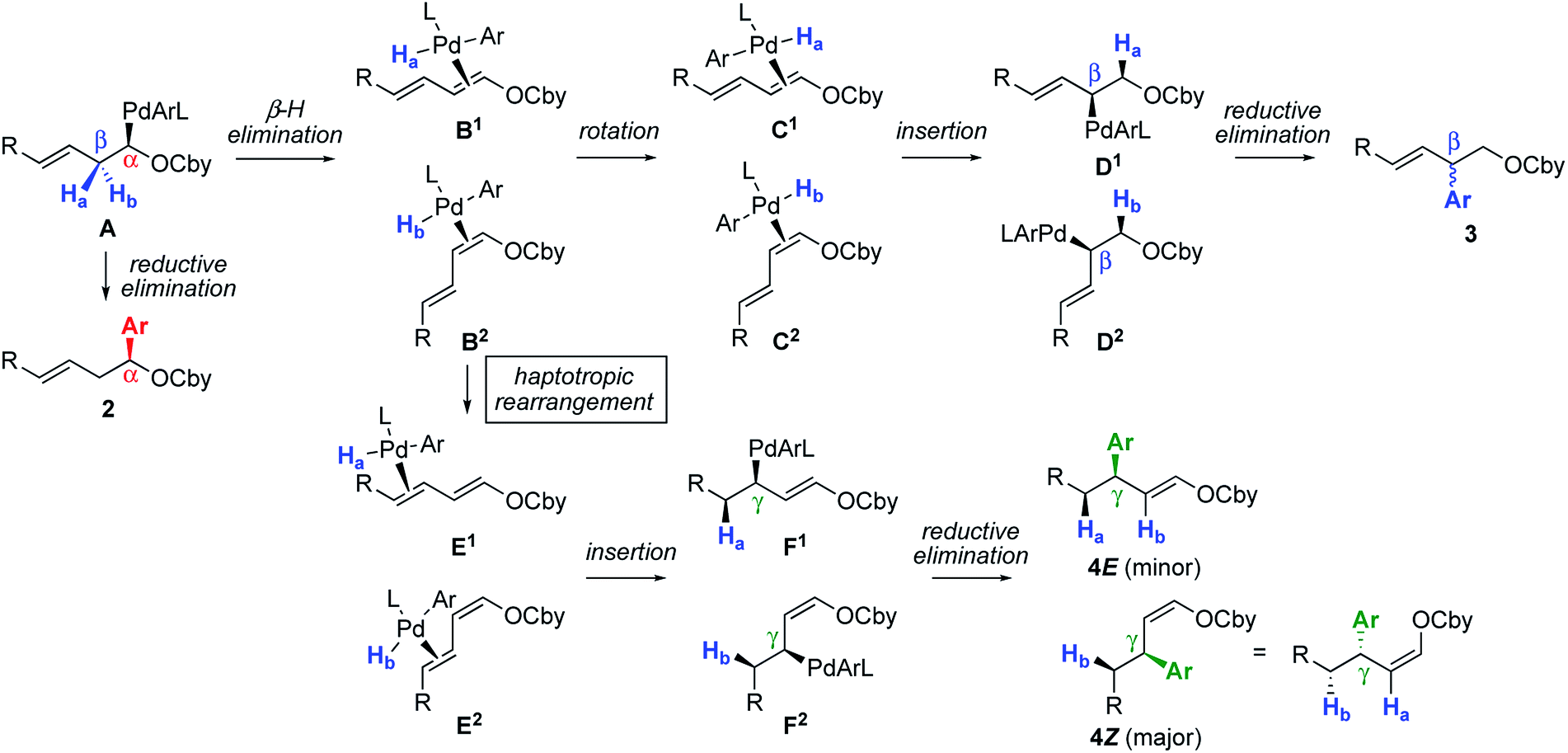 | ||
| Scheme 4 Proposed mechanism. | ||
Moreover, the absolute configuration of 4bZ and 4bE allows to specify that the haptotropic rearrangement occurs from the s-trans conformation for dienic intermediates B1 and B2. Haptotropic rearrangement from the s-cis conformer B′2 would lead to the main stereoisomer 4Z with the opposite, wrong absolute configuration (Scheme 5).
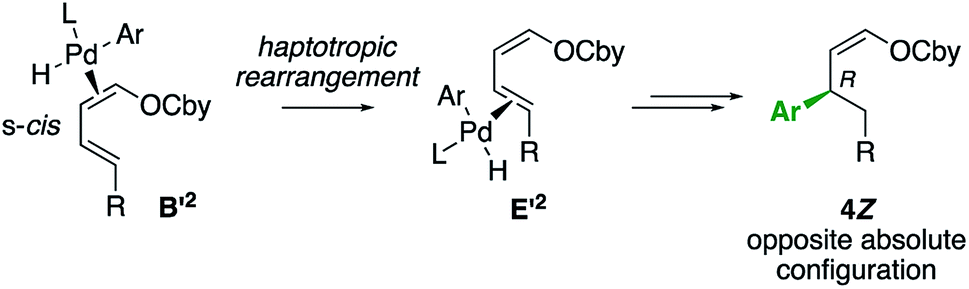 | ||
| Scheme 5 Haptotropic rearrangement from the s-cis conformation. | ||
Conclusions
The arylation of γ,δ-unsaturated O-carbamates via directed lithiation, transmetallation to zinc and Negishi coupling occurs with an unusual γ-selectivity when a specific combination of ortho-substituted aryl electrophile and phosphine ligand is employed. Mechanistic studies indicate that an unusual, stereospecific haptotropic rearrangement of the palladium–diene intermediate is involved.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the University of Basel. We thank Dr Alessandro Prescimone for X-ray diffraction analysis, Dr D. Häussinger for NMR experiments, S. Mittelheisser and Dr M. Pfeffer for MS analyses, and Dr J. Rotzler, Solvias AG, for a gift of cataCXium P ligands.Notes and references
- (a) P. Beak, A. Basu, D. J. Gallagher, Y. S. Park and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552 CrossRef CAS; (b) D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282 CrossRef CAS.
- T. Royal, Y. Baumgartner and O. Baudoin, Org. Lett., 2017, 19, 166 CrossRef CAS PubMed.
- O. Baudoin, Chimia, 2016, 70, 768 CrossRef CAS PubMed.
- A. Millet, D. Dailler, P. Larini and O. Baudoin, Angew. Chem., Int. Ed., 2014, 53, 2678 CrossRef CAS PubMed.
- (a) I. Franzoni and C. Mazet, Org. Biomol. Chem., 2014, 12, 233 RSC; (b) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209 CrossRef CAS PubMed; (c) H. Sommer, F. Juliá-Hernańdez, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153 CrossRef CAS PubMed.
- (a) K. R. Campos, A. Klapars, J. H. Waldman, P. G. Dormer and C.-y. Chen, J. Am. Chem. Soc., 2006, 128, 3538 CrossRef CAS PubMed; (b) I. Coldham and D. Leonori, Org. Lett., 2008, 10, 3923 CrossRef CAS PubMed.
- J. E. Milne and S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 13028 CrossRef CAS PubMed.
- (a) A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261 CrossRef CAS PubMed; (b) S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808 CrossRef CAS PubMed.
- S. Dupuy, K.-F. Zhang, A.-S. Goutierre and O. Baudoin, Angew. Chem., Int. Ed., 2016, 55, 14793 CrossRef CAS PubMed.
- See the ESI for details.†.
- S. Harkal, F. Rataboul, A. Zapf, C. Fuhrmann, T. Riermeier, A. Monsees and M. Beller, Adv. Synth. Catal., 2004, 346, 1742 CrossRef CAS.
- A. Millet and O. Baudoin, Org. Lett., 2014, 16, 3998 CrossRef CAS PubMed.
- D. Hoppe, F. Hintze and P. Tebben, Angew. Chem., 1990, 102, 1457 CrossRef CAS.
- R. J. DeLuca, B. J. Stokes and M. S. Sigman, Pure Appl. Chem., 2014, 86, 395 CAS.
- (a) J. Silvestre and T. A. Albright, J. Am. Chem. Soc., 1985, 107, 6829 CrossRef CAS; (b) I. D. Gridnev, Coord. Chem. Rev., 2008, 252, 1798 CrossRef CAS.
- For somewhat related cases: (a) Y. Takahashi, K. Tsutsumi, Y. Nakagai, T. Morimoto, K. Kakiuchi, S. Ogoshi and H. Kurosawa, Organometallics, 2008, 27, 276 CrossRef CAS; (b) Y.-L. Su, L.-L. Li, X.-L. Zhou, Z.-Y. Dai, P.-S. Wang and L.-Z. Gong, Org. Lett., 2018, 20, 2403 CrossRef CAS PubMed.
- For reviews on haptotropic rearrangements of Ni and Pd arene complexes in the context of catalyst-transfer polycondensation polymerization: (a) I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202 CrossRef CAS PubMed; (b) T. Yokozawa, Y. Nanashima and Y. Ohta, ACS Macro Lett., 2012, 1, 862 CrossRef CAS; (c) J. P. Lutz, M. D. Hannigan and A. J. McNeil, Coord. Chem. Rev., 2018, 376, 225 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full optimization tables, procedural, spectral and X-ray crystallographic (CIF) data. CCDC 1878132 and 1884922. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05058a |
| This journal is © The Royal Society of Chemistry 2019 |