
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient hydrogen bonding recognition in water using aryl-extended calix[4]pyrrole receptors†
G.
Peñuelas-Haro
 ab and
P.
Ballester
*ac
ab and
P.
Ballester
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans, 16, 43007-Tarragona, Spain. E-mail: pballester@iciq.es
bUniversitat Rovira i Virgili, Departament de Química Analítica i Química Orgànica, c/Marcel·li Domingo, 1, 43007-Tarragona, Spain
cICREA, Pg. Lluís Companys, 23, 08018-Barcelona, Spain
First published on 21st December 2018
Abstract
We describe the synthesis of four water-soluble aryl-extended calix[4]pyrrole receptors and report their binding properties with multiple neutral polar guests in water. The prepared receptors present different functionalization at their upper rims and have in common the placement of water solubilizing pyridinium groups at their lower rims. We investigate the interaction of the receptors with a guest series in water solution using 1H NMR titrations and ITC experiments. Despite the known competitive nature of water for hydrogen-bonding interactions, we demonstrate the formation of thermodynamically highly stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complexes stabilized by hydrogen-bonding interactions. We show that increasing the hydrogen-bond acceptor character of the guest has a positive impact on binding affinity. This result suggests that the receptor's cavity is indeed a better hydrogen-bond donor to interact with the guests than water molecules. We also assess the important contribution of the hydrophobic effect to binding by comparing the binding affinities of analogous inclusion complexes in water and chloroform solutions. The more polar guests are bound with similar affinities in the two solvents. We compare the binding properties of the different complexes in order to derive general trends.
:1 inclusion complexes stabilized by hydrogen-bonding interactions. We show that increasing the hydrogen-bond acceptor character of the guest has a positive impact on binding affinity. This result suggests that the receptor's cavity is indeed a better hydrogen-bond donor to interact with the guests than water molecules. We also assess the important contribution of the hydrophobic effect to binding by comparing the binding affinities of analogous inclusion complexes in water and chloroform solutions. The more polar guests are bound with similar affinities in the two solvents. We compare the binding properties of the different complexes in order to derive general trends.
Introduction
Molecular recognition of polar neutral molecules in water using synthetic receptors lacks simplicity.1–4 When the polar functional groups present in both binding partners (host and guest) are exposed to a bulk solvent, they become efficiently solvated by water molecules. Typically, the energetic cost required for their desolvation is not compensated by the polar interactions established upon complex formation. Thus, the free energy of binding becomes positive, leading to complex formation to a reduced extent. In contrast, biological receptors, like enzymes, bind efficiently polar neutral molecules containing a wide variety of functional groups that are susceptible to strong hydration. In this vein, it is worth mentioning that the avidine–biotine complex features the largest binding constant reported for a biological host–neutral guest complex in water (1015 M−1).5 The high binding constants and the selectivity exhibited by enzymes in the binding of polar neutral substrates in water is usually the result of a synergistic combination of polar interactions and the hydrophobic effect. To achieve high affinity and selectivity, enzyme binding takes place in hydrophobic cavities (low dielectric constant) featuring converging polar functional groups that are complementary to those of the substrate. In doing so, the polar groups of the enzyme's binding site and of the resulting enzyme–substrate complex are not easily accessible to bulk water molecules. Moreover, the water molecules that solvate the buried polar groups in the active site make energetically less favourable hydrogen bonding interactions than those in the bulk solution.Aiming at mimicking the binding behavior of enzymes, several aqueous and water soluble synthetic receptors possessing more or less open aromatic cavities with converging polar groups have been reported by the groups of Davis,6–8 Jiang,9–11 and Jabin,12,13 among others. The obtained binding results demonstrated that the combination of a functionalized aromatic cavity (binding site with a low dielectric constant) displaying multiple open portals (cages or tubes) with converging polar groups is a successful strategy for achieving effective binding of neutral polar molecules in aqueous media and water with synthetic receptors (Fig. 1). Most likely, the hydrophobic effect is key in enhancing affinity in aqueous media while the converging polar groups are responsible for endowing the receptors with binding selectivity beyond size and shape complementarity.
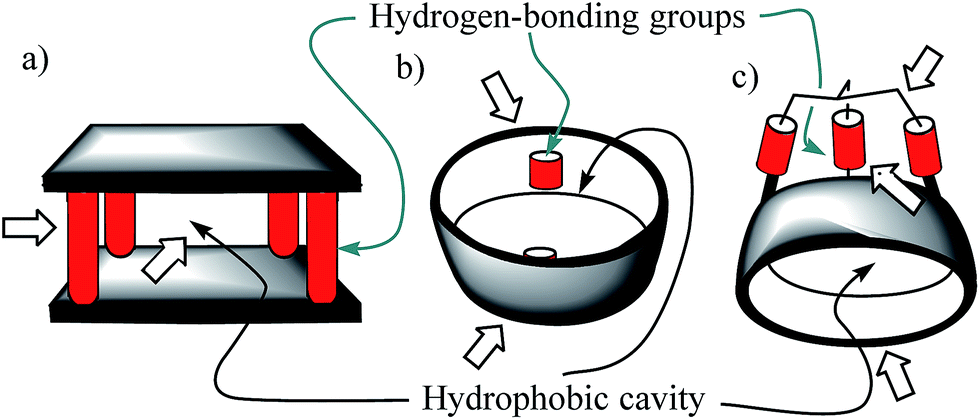 | ||
| Fig. 1 Schematic representations of the water-soluble receptors mentioned in the text: (a) Davis' “temple” architecture; (b) Jiang's “tubular” architecture; (c) Jabin's “cryptophane” architecture. Their aromatic cavities and polar converging groups are indicated by thin-arrows. Wide arrows highlight open portals suitable for guest inclusion and/or access of solvating water molecules of the polar groups. | ||
In 1999, Sessler14 and Floriani15 described almost simultaneously the synthesis of aryl extended calix[4]pyrroles bearing hydroxyl groups at the upper rim.
The compounds were obtained by the acid-catalysed cyclocondensation reaction of a hydroxyl-aryl-methyl ketone with pyrrole. The reaction produced the calix[4]pyrroles as a mixture of four configurational isomers, together with larger acyclic and cyclic oligomers. Remarkably, the cone conformation of the tetra-α-isomer of aryl-extended calix[4]pyrroles displays a polar aromatic cavity open only at one end (Fig. 2). This latter characteristic separates aryl extended calix[4]pyrroles from other water-soluble polar receptors described above. The solvation and concomitant dissociation of the deep inclusion complexes of calix[4]pyrroles requires a conformational change of the bound receptor (cone to alternate) owing to the blocking of their single open portal by the bound guest. These features suggested to us the potential application of tetra-α aryl extended calix[4]pyrrole isomers as scaffolds for the construction of effective synthetic receptors functioning in water solution. We produced tetra-α aryl extended calix[4]pyrroles that were water soluble by covalently attaching ionizing groups at either their upper (R1)16 or lower rims (R2).17 We evidenced that the prepared receptors formed 1:1 inclusion complexes in water with pyridine-N-oxide and its derivatives, which were highly stable kinetically and thermodynamically (Ka > 104 M−1). The incorporation of the water solubilizing groups at the lower rim of the calix[4]pyrrole receptor offered the advantage of further elaboration of the aromatic cavity through its upper rim aiming at further modulating its binding properties.
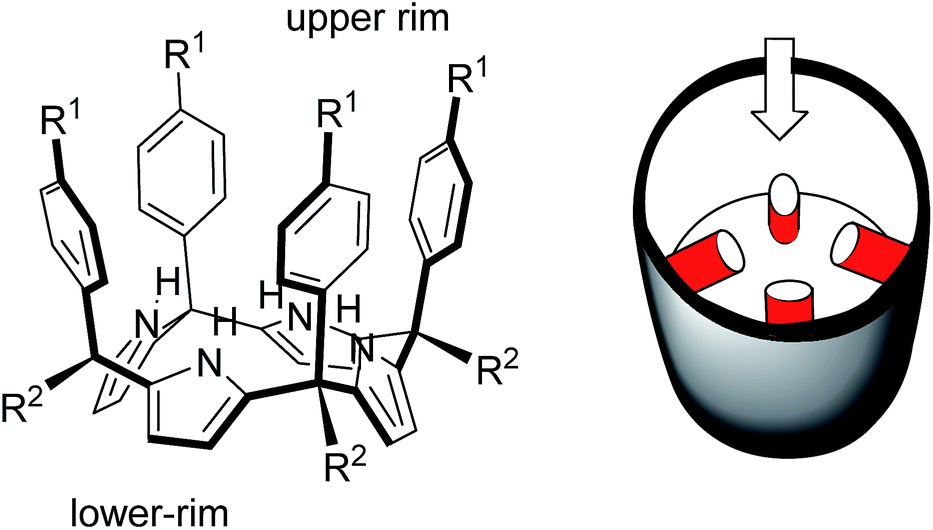 | ||
| Fig. 2 Molecular structure of a tetra-α aryl extended calix[4]pyrrole in cone conformation and its schematic representation highlighting the single open portal displayed by its cone conformation. | ||
This strategy was successfully implemented in calix[4]pyrrole receptors soluble in organic solvents.18 We reported aryl-extended calix[4]pyrrole cavitands soluble in organic solvents and bearing two bridging phosphonate groups at their upper rim.19,20
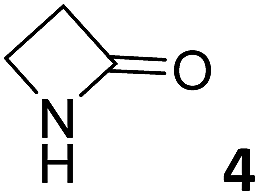
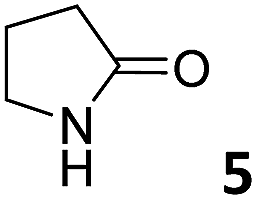
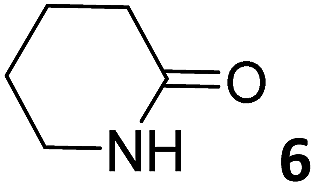
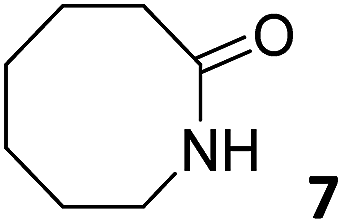
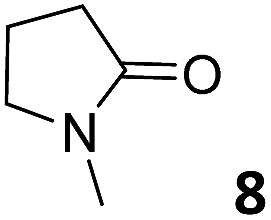
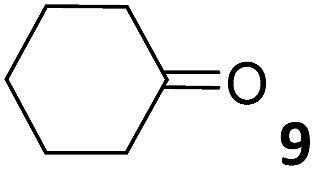
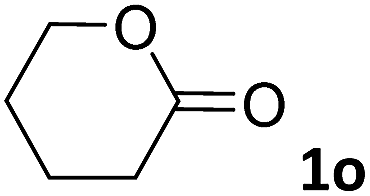
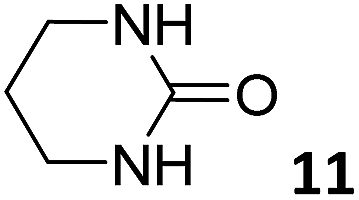
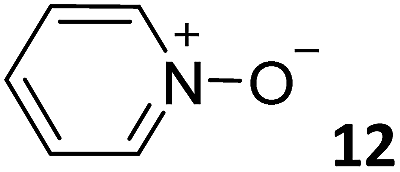
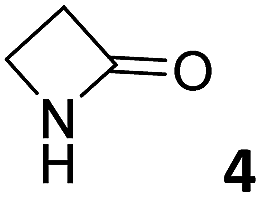
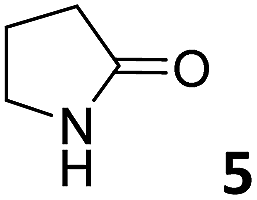
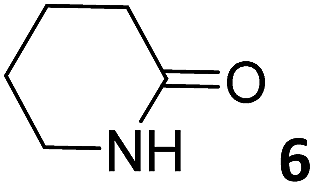
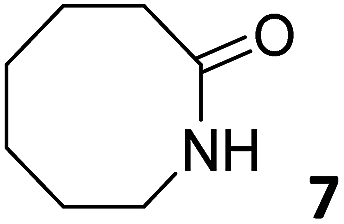
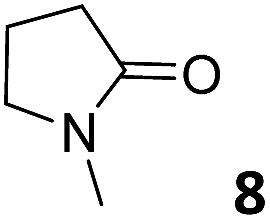
In this manuscript, we describe the synthesis of two unprecedented water-soluble calix[4]pyrrole cavitands also bearing two bridging phosphonate groups at their upper rims.21 The prepared receptors are diastereomeric displaying in–out (1io) and out–out (1oo) relative configurations of the two P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (Fig. 3). We also report the synthesis of water soluble bis-methylene bridged cavitand 2 and the conformationally more flexible tetra-α aryl extended calix[4]pyrrole 3 (Fig. 3). The incorporation of p-methyl substituents in the meso-phenyl groups of the receptors aimed at increasing the depth of their aromatic cavities. We employed pyridinium groups as water-solubilizing units, based on their performance for solubilizing related resorcinarene derived cavitands in water.11,13,22 We located the water solubilizing groups at the lower rim of the receptors, distal from the polar aromatic binding site. We also describe the binding properties of the synthesized receptors, in water, using: (a) a series of homologous lactams 4–7 (β, γ, δ, and ω); (b) N-methyl γ-lactam 8, which is used to probe the size selectivity of the receptor series; and (c) a range of monocyclic six-membered compounds 9–12 (Fig. 3). The later six-membered cyclic compounds contain a single polar group with different hydrogen-bond accepting properties at their oxygen atom (β).23 We analysed the thermodynamic constants derived from the titrations of the water-soluble 1oo receptor and the chloroform soluble analogue 17oo with the series of six membered cyclic guests to evaluate the relevance of the hydrophobic effect in water.
O groups (Fig. 3). We also report the synthesis of water soluble bis-methylene bridged cavitand 2 and the conformationally more flexible tetra-α aryl extended calix[4]pyrrole 3 (Fig. 3). The incorporation of p-methyl substituents in the meso-phenyl groups of the receptors aimed at increasing the depth of their aromatic cavities. We employed pyridinium groups as water-solubilizing units, based on their performance for solubilizing related resorcinarene derived cavitands in water.11,13,22 We located the water solubilizing groups at the lower rim of the receptors, distal from the polar aromatic binding site. We also describe the binding properties of the synthesized receptors, in water, using: (a) a series of homologous lactams 4–7 (β, γ, δ, and ω); (b) N-methyl γ-lactam 8, which is used to probe the size selectivity of the receptor series; and (c) a range of monocyclic six-membered compounds 9–12 (Fig. 3). The later six-membered cyclic compounds contain a single polar group with different hydrogen-bond accepting properties at their oxygen atom (β).23 We analysed the thermodynamic constants derived from the titrations of the water-soluble 1oo receptor and the chloroform soluble analogue 17oo with the series of six membered cyclic guests to evaluate the relevance of the hydrophobic effect in water.
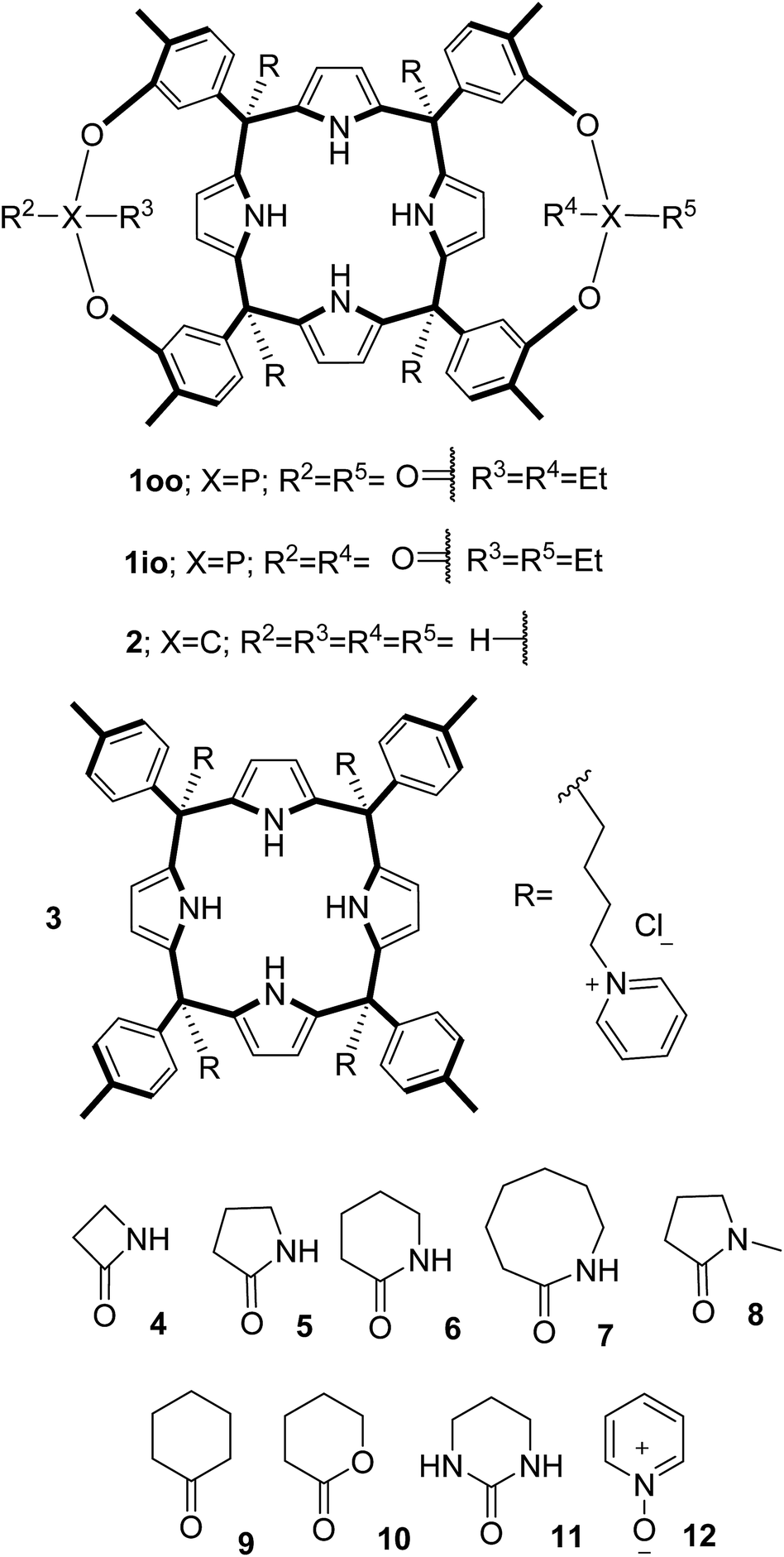 | ||
| Fig. 3 Molecular structures of the four water-soluble aryl-extended calix[4]pyrrole derivatives, 1oo, 1io, 2 and 3, and of the guests, 4–12, used in this study. | ||
Results and discussion
Synthesis
The aryl–alkyl ketones 14b and 15a were prepared uneventfully using standard reaction conditions and procedures described in the literature (Scheme 1, see the ESI† for details).24,25 A recently reported methodology for the stereo selective syntheses of tetra-α aryl extended calix[4]pyrrole isomers was used in the cyclocondensation reactions of the prepared ketones with pyrrole.26 This methodology consists in the incorporation of tributylmethylammonium chloride as a template under the standard acid mediated conditions (3 equiv. HCl (4 M dioxane) in dichloromethane solution). Remarkably, in the absence of the template the macrocyclization condensation reactions of 14b and 15a with pyrrole were not successful. The tetra-α-isomers of the aryl extended calix[4]pyrroles 16a and 16b were isolated, after column chromatography purification, in 10 and 4% yield, respectively (Scheme 1). The reaction of tetrol 16a with ethylphosphonic dichloride in THF solution using Et3N as the base produced a non-statistical mixture of three phosphonate stereoisomers 17ii, 17io and 17oo (Scheme 2).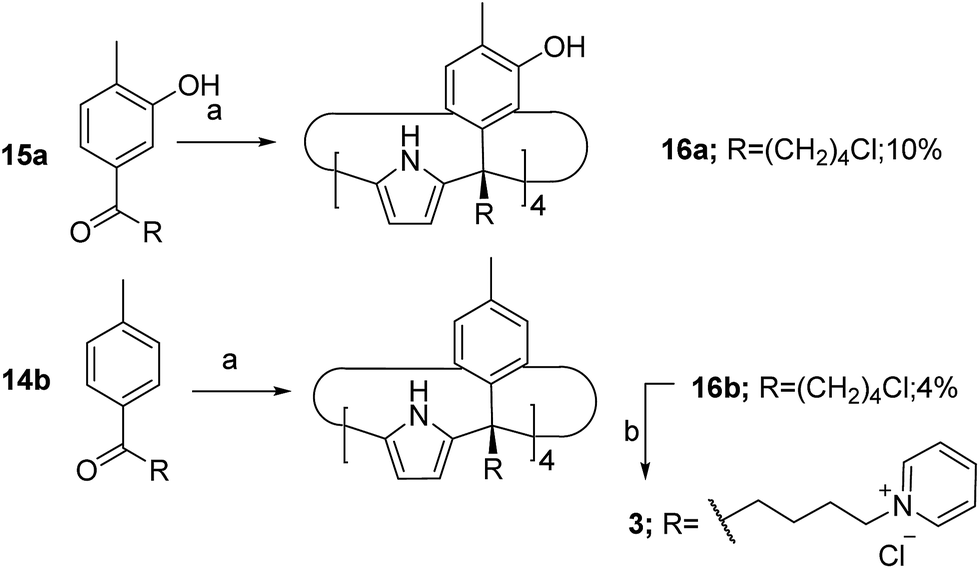 | ||
| Scheme 1 Synthetic schemes for the preparation of the calix[4]pyrroles 16a and 16b and the water soluble derivative 3. Reaction conditions: (a) MeNBu3Cl, HCl (4 M dioxane), pyrrole in DCM; (b) excess pyridine and heat. | ||
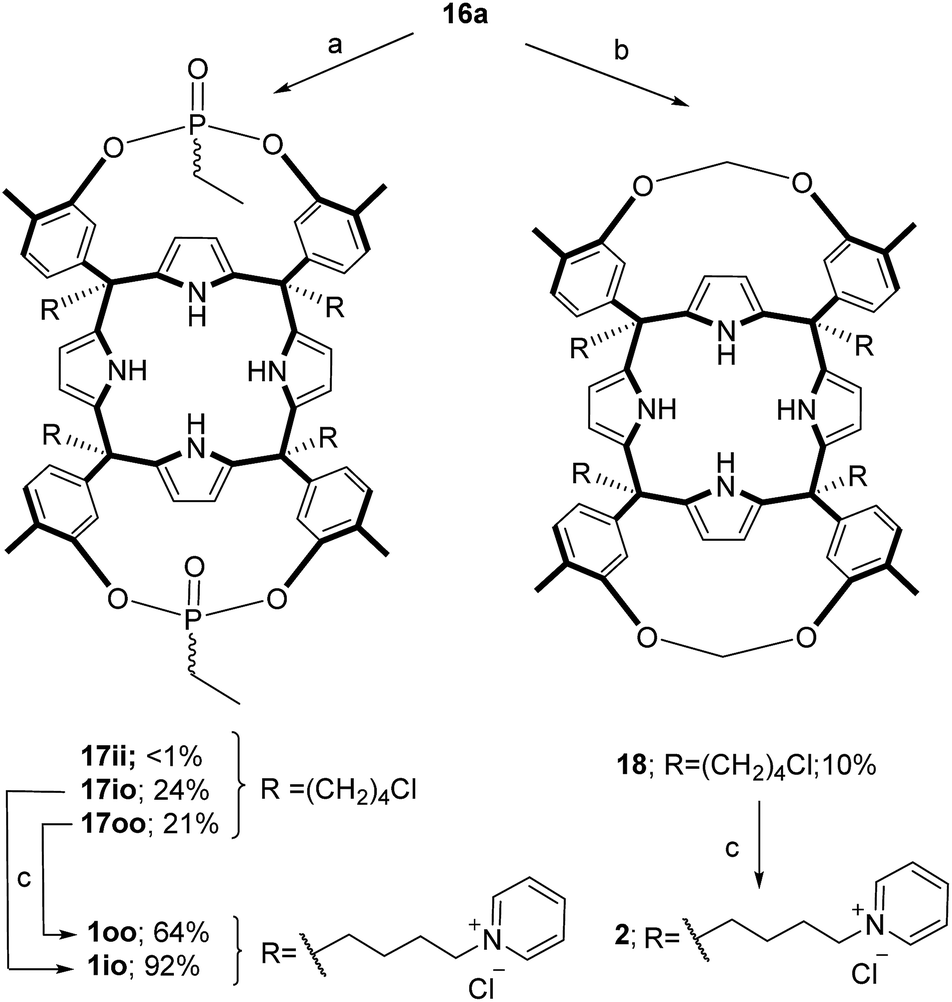 | ||
| Scheme 2 Synthetic schemes for the preparation of the cavitands 17 and 18 and their water soluble derivatives 1 and 2. aReaction conditions: (a) EtPOCl2, Et3N in THF; (b) K2CO3, MeBrCl in DMSO; (c) excess pyridine and heat. | ||
The three isomers were conveniently separated by column chromatography purification on silica gel of the crude reaction mixture. The configurational assignment of the isolated bisphosphonate isomers 17ii, 17io and 17oo was based on a combination of 1H NMR spectroscopy and X-ray crystallography (17ii and 17oo). The solid-state structure of the 17ii and 17oo stereoisomers is shown in Fig. 4. In both cases, the calix[4]pyrrole core adopts the cone conformation having one molecule of acetonitrile included in its aromatic cavity and hydrogen-bonded to the pyrrole NHs. Moreover, the 14-membered rings defined by the bridging phosphonato-group, two meso-phenyl groups, their corresponding meso-carbons and one pyrrole unit of each stereoisomer show conformations in which the ethyl substituents of the phosphorous atom are always equatorially oriented.
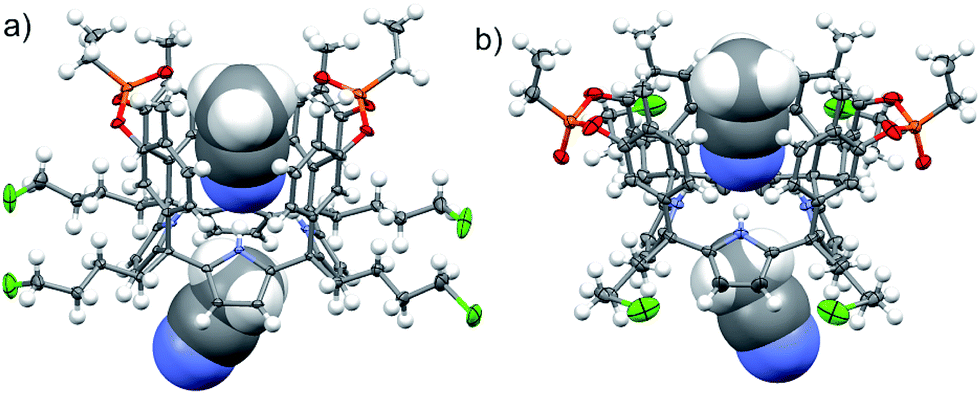 | ||
| Fig. 4 X-ray structures of the 17ii (a) and 17oo (b) isomers. Carbon, chloride and nitrogen atoms in 17ii and 17oo are shown as ellipsoids at the 50% probability level. Hydrogen atoms are shown as fixed-size spheres with a radius of 0.15 Å. Two bound solvent molecules (acetonitrile) are displayed as CPK models. Crystallographic disorder was removed for clarity. | ||
These conformations are analogous to the ones observed in previous studies for closely related tetramethyl bis-phenylphosphonate calix[4]pyrrole cavitands.27
Bis-methylene bridged cavitand 18 was synthesized by reacting calix[4]pyrrole 16a with bromochloro methane using potassium carbonate as a base in anhydrous DMSO (Scheme 2). Finally, we introduced the water solubilizing pyridinium groups in the terminal position of the meso aliphatic chain of the chloride derivatives 16b, 17io, 17oo and 18 by simply heating them overnight as separate solutions in dry pyridine. The reactions yielded the chloride salts of the tetrapyridinium receptors 3, 1io, 1oo and 2, as yellow solids, in good yields and high purities. Because only traces of the 17ii isomer were isolated, we did not attempt its conversion in the water-soluble version. The tetra-pyridynium salts of all receptors were highly soluble in D2O at mM concentration, allowing us to investigate their binding properties using 1H NMR titrations. All receptors were characterized by a full set of high-resolution spectra (NMR and MS).
Binding studies
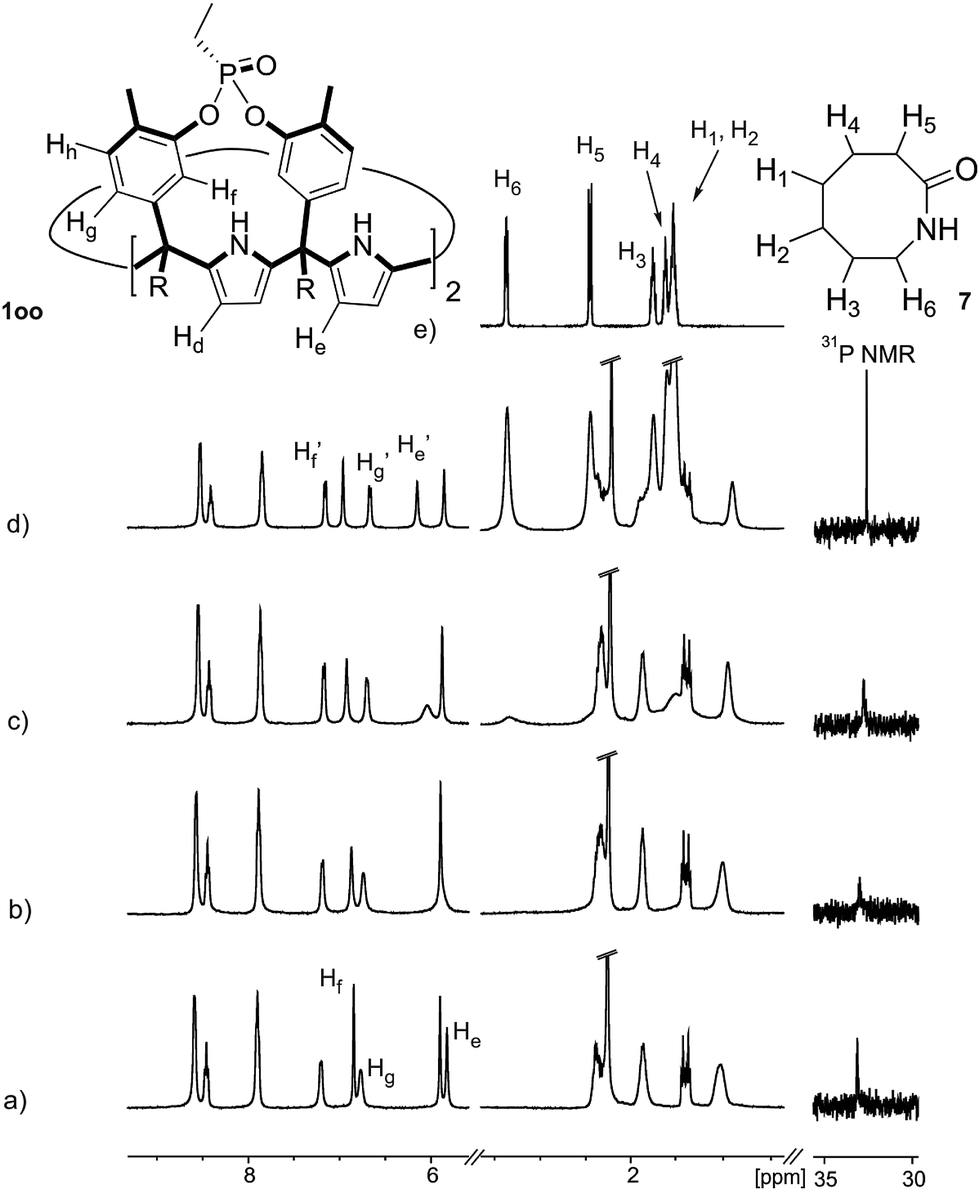
 | ||
| Fig. 5 Selected regions of the 1H NMR and 31P NMR spectra registered during the titration of a 1 mM solution of 1oo with incremental amounts of 6 in D2O solution. (a) Free 1oo; (b) 0.5 equiv. of 6 added; (c) 1.0 equiv. of 6 added; (d) 1.9 equiv. of 6 added; and (e) free 6. Primed labels correspond to the proton signals of 6 and 1oo in the 6⊂1oo inclusion complex. Proton assignment is shown in the molecular structures of the compounds. | ||
The addition of 1.9 equiv. of 6 did not produce noticeable changes in the signals assigned to protons of bound 1oo and included 6. However, new signals corresponding to the protons of free 6 emerged (Fig. 5d). Taken together, these results indicate that 1oo and 6 form a 1:1 complex, which experiences a slow chemical exchange on the chemical shift scale with the free counterparts and the binding constant value for the 6⊂1oo complex can be estimated to be larger than 104 M−1.
We performed a ROESY experiment using a solution containing an excess of guest (1.9 equiv., Fig. S51†). We observed the presence of cross peaks due to chemical exchange between the proton signals of free and bound 6. The presence of the cross peaks allowed the unequivocally assignment of the proton signals of bound 6 and the calculation of their corresponding complexation induced shifts (CIS) (ΔδH2 = −2.75 ppm; ΔδH1 = −0.9 ppm; ΔδH3 = −0.9; ΔδH4 = −0.6 ppm). The fact that all proton signals of bound 6 are significantly upfield shifted supports its inclusion in the aromatic cavity of the host where it experiences the shielding effect produced by the meso-aromatic substituents. Specifically, the proton alpha to the carbonyl group of 6 (H2) is not only the more upfield shifted one but also the one suffering the largest CIS. This result indicates that the guest is included in the host by burying the CO group in its aromatic cavity. Most likely, the inclusion process is driven by the hydrophobic effect and assisted with the formation of four hydrogen bonds between the pyrrole NHs of 1oo and the oxygen atom of 6, in combination with CH–π, and NH–π interactions. The relevance of hydrogen bonding in guest recognition will be addressed in more detail in the next section.
 | ||
| Fig. 6 Selected regions of the 1H NMR and 31P NMR spectra registered during the titration of a 1 mM solution of 1oo with incremental amounts of 7 in D2O solution. (a) Free 1oo; (b) 0.5 equiv. of 7 added; (c) 2.1 equiv. of 7 added; (d) 11.3 equiv. of 7 added; and (e) free 7. Primed labels correspond to the proton signals of 7 and 1oo in the 7⊂1oo inclusion complex. Proton assignment is indicated in the molecular structures of the compounds. | ||
Instead, some of the proton signals of the 1oo receptor experienced noticeable chemical shift changes, which required the addition of more than 11 equivalents of 7 to reach saturation. In particular, one of the signals of the β-pyrrole protons (He) moved downfield (CIS = 0.3 ppm) and sharpened in the course of the incremental addition of 7. Similarly, the singlet of the aromatic proton Hf of the meso-aryl substituent also shifted downfield (CIS = 0.1 ppm). The calculated CIS values for these two proton signals are in excellent agreement with those observed for the same protons in complexes featuring slow chemical exchange on the NMR time scale i.e.6⊂1oo. The signals of the protons of 7 broadened beyond detection in the initial phases of the titration emerging again, as significantly broadened signals, when more than 2 equiv. of 7 were added. The addition of a larger excess of 7 produced small changes in the chemical shift values of the mentioned protons but an evident sharpening. These findings indicate that the binding equilibrium of 1oo with 7 is fast on the chemical shift timescale and that the binding constant of the resulting 7⊂1oo complex can be estimated to be smaller than 104 M−1. The fast kinetics of the chemical exchange between free and bound 7 together with the large difference expected in chemical shifts for these protons in the free and bound states explain their broadening beyond detection in the initial phase of the titration.
In summary, the increase of two methylene units in the lactam ring provoked a significant reduction in the binding affinity and kinetic stability of the corresponding 7⊂1oo complex.
Theoretical and experimental studies on the binding geometry of the inclusion complexes
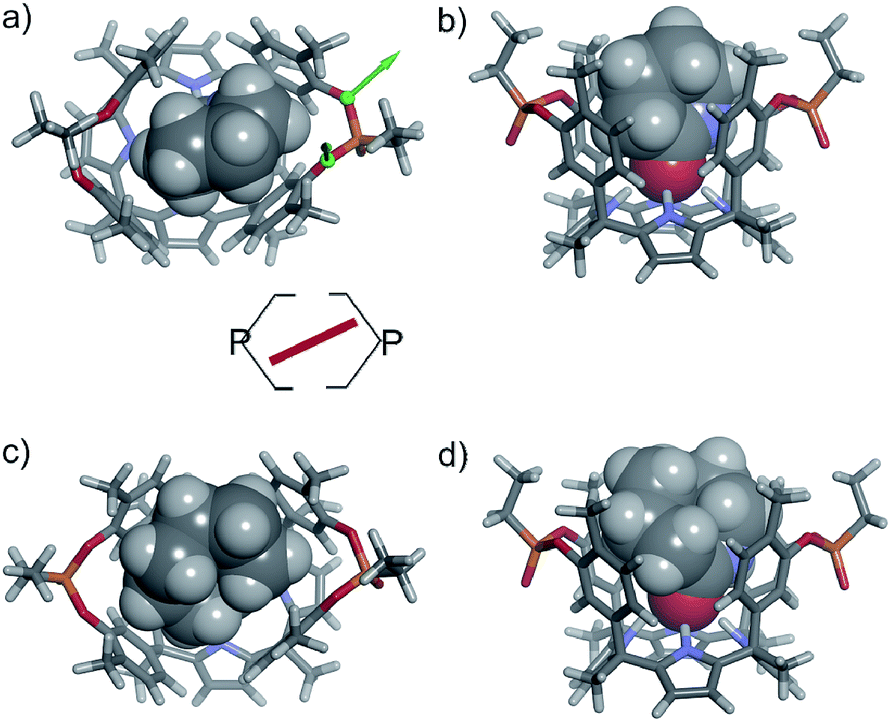 | ||
| Fig. 7 Top views and schematic representation of the inclusion complexes 6⊂1oo and 7⊂1oo (a and c panels). The corresponding side views are shown in the (b and d) panels. The structures are energy minima at the PB86-D3-def2-SVP level of theory. The host is shown in stick representation and the included guests as a CPK model. The meso-alkyl chains of 1oo were pruned to methyl groups for clarity. | ||
In the energy-minimized complexes, the calix[4]pyrrole unit adopts the cone conformation and forms four hydrogen bonds with the bound lactam. The establishment of multiple CH and NH–π-interactions can be inferred from the excellent complementarity in functionality, shape and size featured by the complex components (Fig. 7). According to the modelling, bound 1oo adopts a geometry resembling a hexagonal prismatic box, closed at one base by the calix[4]pyrrole core and open at the opposite one. The hexagon is defined by the four bridged aromatic walls, the bridging phosphonate groups, that reside on the vertex of the long diagonal, and two additional parallel faces of the hexagon that remain partially open. The oxygen atoms of the bridging phosphonate groups are not symmetric with respect to the orientation of their electron lone pairs (green arrows in Fig. 7). The conformation adopted by the outwardly directed phosphonate groups in 1oo coincides with the one observed in the X-ray of related calix[4]pyrrole bis-phenylphosphonate cavitands soluble in organic solvents, as well as their complexes with alkylammonium chloride salts.27 However, it is slightly different to the one observed for the acetonitrile solvate in the X-ray structure of 17oo (Fig. 4). In turn, the bound lactams seem to prefer an orientation that maximizes edge-to-face, CH–π and NH–π interactions, between perpendicular rings of the binding partners and CH–π interactions between parallel ring surfaces.
The observation of a single set of proton signals in the NMR spectra of all inclusion complexes reflects the existence of a time averaged binding geometry, which results from a fast chemical exchange between different binding geometries.
In short, at room temperature, the bound guests spin rapidly around the axial axis of the inclusion complexes. In the specific case of the 6⊂1oo complex, we computed an energy difference of ca. 4.5 kcal mol−1 in favour of the geometry in which the guest bisects the large diagonal of the hexagonal-like host rather than being oriented with it (see schematic representation in Fig. 7). The computed value provides an estimate for the energy barrier of the spinning process of the bound guest.
Aiming at experimentally evidencing the implication of the pyrrole NHs of receptor 1oo in hydrogen-bonding interactions with the included lactam, we acquired the 1H NMR spectra of the free receptor and the 6⊂1oo complex in a 9:1 H2O:D2O solution mixture employing the W5 pulse sequence to suppress the large water signal (Fig. S100†).33 Unfortunately, the 1H NMR spectra of the free receptor did not show any signal that could be assigned to the free pyrrole NHs. Most likely, the NH protons are involved in a fast/intermediate chemical exchange with the hydrogen atoms of the water molecules. In contrast, the water suppressed 1H NMR spectrum of a 1 mM equimolar mixture of 1oo and 6 showed two broad singlets resonating at δ = 9.37 and 9.11 ppm, respectively. We assigned these signals to the pyrrole NHs of the 6⊂1oo complex, which is formed quantitatively under the abovementioned conditions. The measured chemical shift values are in complete agreement with those observed for the NHs in related hydrogen-bonded complexes of water-soluble four-wall aryl-extended calix[4]pyrroles with pyridyl-N-oxides.16 The obtained result supports the involvement of the pyrrole NHs in hydrogen-bonding interactions in the formation of the 6⊂1oo complex. A 2D-ROESY spectrum of the 6⊂1oo complex (Fig. S101†) showed multiple cross peaks due to intramolecular close contacts, which are in complete agreement with the proposed structure (Fig. 7a and b). For instance, the singlet of the ortho aromatic proton (Hf) in the meso-aryl substituents showed a selective cross peak with the β-pyrrole protons of the unit involved in the 14-membered ring of the receptor (Hd). On the other hand, the doublet ortho aromatic proton (Hg) of the meso-aryl substituent showed close-contact cross peaks exclusively with the β-pyrrole protons of the other pyrrole unit (He). All these observations indicate that bound 1oo adopts the cone conformation. Moreover, cross peaks due to intermolecular close-contacts between the methyl and the meso-aryl protons of the receptor with the methylene protons of the included δ-lactam 6 were also detected. (See the ESI† for theoretical studies on the geometry of free 1oo in water solution).
Dissecting hydrogen-bonding interactions and the hydrophobic effect in the binding of 1oo
For the aliphatic cyclic lactams 4–6 (C2–C4), the determined ΔG values for binding to 1oo assign a gain of 0.8–0.5 kcal mol−1 to each additional hydrophobic methylene group (Table 1). The magnitude of this estimate is smaller than the 1.2 kcal mol−1 value derived from their encapsulation in the cavity of an octahedral coordination cage.34,35 Most likely, this difference is due to the partial desolvation of the guest required for inclusion in 1oo (Fig. 7) compared to the full hydrophobic desolvation compulsory for encapsulation in the coordination cage. Remarkably, the binding affinity of 1oo for the larger and more hydrophobic 1-aza-2-cyclooctaone 7 is reduced by 2.9 kcal mol−1 compared to 6. The large lactam 7 is no longer size and shape complementary to the polar cavity of 1oo. Likewise, the N-methyl-γ-lactam 8, although being more hydrophobic than the parent compound 5, experiences a drop of two orders of magnitude in affinity for the cavity 1oo owing to size mismatch. Clearly, the 6-membered ring lactam 6 provides the best fit for the cavity of 1oo. Consequently, we undertook the evaluation of the hydrophobic and hydrogen-bonding contributions to the binding of 1oo using the thermodynamic data of the binding processes of five 6-membered cyclic guests, 6, 9–12. The members of this series have in common a hydrogen-bonding accepting oxygen atom and differ in their hydrophobicity and hydrogen-bond acceptor abilities of their polar groups: ketone 9, lactone 10, lactam 6, urea 11 and N-oxide 12 (see Table 1). Several parameters have been used to quantify the hydrophobic (lipophilic) nature of organic molecules. We opted for the use of the logP value, partition coefficient between octanol/water, calculated using ChemDraw 16. The software assign the following logP values to the selected guests for this study: 1.43 (ketone 9), 0.52 (lactone 10), −0.16 (lactam 6), −0.95 (urea 11), and −1.11 (N-oxide 12). The more positive the logP value the more lipophilic is the compound. It is accepted that in water solution and using synthetic H-bonding receptors an increase in binding should be directly related to a decrease in guest polarity.35–37
:1 complexes of the 1oo receptor in H2O or D2O and 17oo in CHCl3 or CDCl3 [association constant Ka (M−1), free Gibbs energy ΔG (kcal mol−1), ΔH enthalpy (kcal mol−1) and entropy TΔS (kcal mol−1)]
| Guest | Receptor | |||||||
|---|---|---|---|---|---|---|---|---|
| 1oo | 17oo | |||||||
| K a × 10−4 | ΔG | ΔH | TΔS | K a × 10−4 | ΔG | ΔH | TΔS | |
|
a ITC titration experiments were repeated at least twice, and the reported Ka value is the average of the values obtained from the fit of the integrated heat data to a 1:1 binding model.
b
1H NMR titration experiments were also repeated at least twice, and the reported Ka is the average value returned from the fit of the chemical shift changes observed for the diagnostic proton signals to a 1:1 binding isotherm. Error values for Ka are reported as standard deviations. Errors values for ΔG, ΔH and TΔS are determined to be less than 10%. nd not determined. See the ESI for experimental details of the titrations.
|
||||||||
|
2.1 ± 0.1a | −5.9 | −6.1 | 0.2 | ||||
|
10.8 ± 0.2a | −6.8 | −6.8 | 0.0 | ||||
|
22.6 ± 1.0a | −7.3 | −6.8 | 0.5 | 6.7 ± 0.1a | −6.6 | −9.9 | −3.3 |
|
0.08 ± 0.01b | −3.9 | nd | nd | ||||
|
0.04 ± 0.01b | −3.6 | nd | nd | ||||
|
0.14 ± 0.02b | −4.3 | nd | nd | 0.0018 ± 0.0001b | −1.7 | nd | nd |
|
0.95 ± 0.04a | −5.4 | −6.2 | −0.8 | 0.0060 ± 0.0002b | −2.4 | nd | nd |
|
68.5 ± 1.0a | −8.0 | −8.4 | −0.4 | 100.0 ± 30.0a | −8.2 | −12.2 | −4.0 |
|
685.0 ± 20.0a | −9.1 | −11.4 | −2.2 | 120.0 ± 10.0a | −8.3 | −14.3 | −6.0 |
The idea behind this relies on the common believe that in water more polar guests are more difficult to desolvate and therefore will bind weakly through hydrogen-bonding interactions.
In striking contrast to the idea above, the absolute ΔG values determined for the binding of the five 6-membered cyclic guests with 1oo in water increased as the guest hydrophilicity increased (Table 1). This finding suggested that the binding of the guest series by 1oo in water was not dominated by their hydrophobicity.
Moreover, the plot of the ΔG values versus the H-bonding accepting capabilities of the guest's oxygen atom, as condensed in its β-value,23 produced a linear relationship with a negative slope (dashed line in Fig. 8). This result reflects that the H-bonding contribution to guest binding seems to get stronger as the H-bonding accepting value (β) of the guest's polar group increases. This is the expected result in binding experiments performed in relatively non-competitive hydrogen bonding solvents (vide infra) but, to the best of our knowledge, it is unprecedented in water, the most competitive hydrogen-bonding solvent. In short, we discovered that for the employed guest series the polar cavity of 1oo seems to provide a better environment as a hydrogen-bond donor than the water molecules.
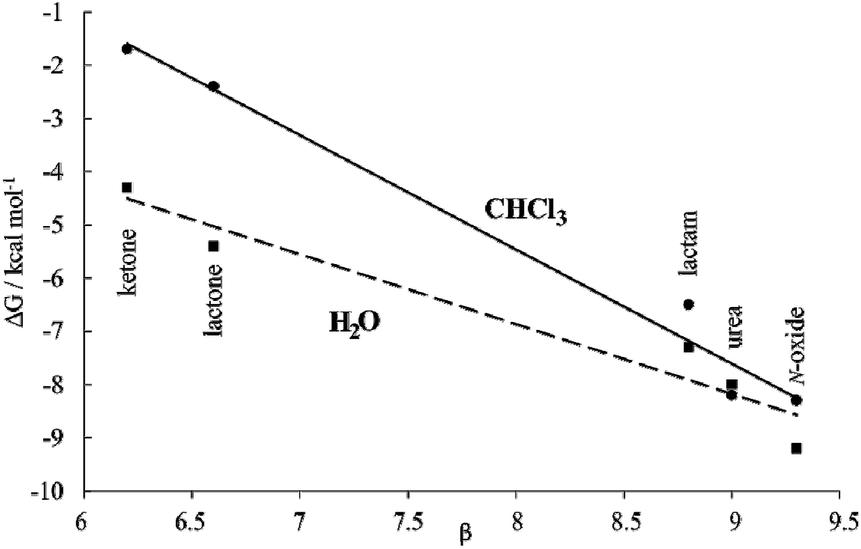 | ||
| Fig. 8 Plot of ΔG vs. H-bonding acceptor capabilities (β) for the inclusion complexes of the five 6-membered cyclic guest with 1oo (squares, H2O dashed-line) and 17oo (circles, CHCl3 solid-line) receptors. This graphical analysis serves to illustrate the hydrophobic effect contribution to binding. | ||
We became interested in determining the ΔG values of the analogous inclusion complexes formed in a non-competitive hydrogen-bonding solvent. For this reason, we used the isostructural bis-phosphonate tetrachloride derivative 17oo as a host to perform binding experiments with the guest series in chloroform solution (Table 1). In this solvent, the binding event should be mainly driven by polar interactions (hydrogen-bonding, CH–π, π–π, NH–π) and van der Waals forces. As expected, the slope of the line defined by the ΔG values in chloroform is also negative reflecting the substantial contribution of hydrogen-bonding to guest binding in the non-polar and non-protic solvent. The two lines have a marked separation in the region defined by the guests with poor hydrogen-bonding accepting abilities.
The separation between them could be used to quantify the contribution of the hydrophobic effect to binding as ∼2.5–3.0 kcal mol−1 for guests with β < 7. In contrast, the hydrophobic effect seems to have a reduced contribution to the binding for guests with β ∼ 9 (0.5 kcal mol−1) and the two lines tend to intersect. However, a detailed analysis of the ITC results for guests with β ∼ 9 paints a different picture (Table 1). Significant negative entropy values are measured in chloroform solution, where the hydrophobic driving force is not in play (−3.3 to −6.0 kcal mol−1 at 298 K). In contrast, in water, where the hydrophobic driving force is turned on the entropy values become close to zero or slightly negative. Overall, an average of 3.7 kcal mol−1 is added to the entropy values in water compared to those in chloroform, which is an important factor to understand why the free energy values are almost identical in both solvents. An enthalpy–entropy compensation effect hides the beneficial impetus of the hydrophobic effect (2.5–3.7 kcal mol−1) to the binding of the more polar guests if we only consider the free energy values. Unfortunately, the lack of entropy values for the binding of the less polar guests, 9 and 10, does not allow us to provide the corresponding quantification of the hydrophobic effect. Taken together, the obtained results indicate that water and the hydrophobic effect are very important in enhancing the binding affinity of the polar six-membered cyclic guests for 1oo in comparison to 17oo in CHCl3 solution. Jiang et al. proposed the release, upon complex formation, of weakly hydrogen-bonded water molecules present in the cavity of the receptor as an important driving force for the efficient binding of polar guests in water using synthetic receptors.10,11 We surmise that this process is also operative in the complexation of 1oo with the polar guest series.
In the case at hand, we suggest that the plots of the free energy values vs. β are better evaluated considering that the slopes of the lines represent a composite of the hydrogen-bond donor properties of the host relative to that of the solvent (αHost–αsolvent).§38 The slope is larger for chloroform because water is a better hydrogen-bond donor. Hydrogen bonding interactions are better in chloroform because the solvent is less competitive. Nevertheless, the negative slope of the line defined by the ΔG values in water indicates the better hydrogen-bond donor properties of the receptor compared to the solvent.
Having achieved a ΔG value of −9.2 kcal mol−1 for the binding of pyridine N-oxide 12 by the synthetic receptor 1oo in water is highly remarkable owing to the reduced size and surface of the guest. The hydrophobic effect is known to be proportional to the hydrophobic surface contact of the bound guest.39 Our previous attempts to bind 12 with water soluble calix[4]pyrrole receptors being conformationally more flexible and having a shallower aromatic cavity provided ΔG = −6.2 kcal mol−1.17
Comparison of the binding properties of 1oo with the reference receptors 1io, 2 and 3
In order to evaluate which structural features of 1oo were responsible for its excellent binding properties, we assessed the thermodynamic constant of the reference calix[4]pyrrole receptors 1io, 2 and 3 with the lactam series (Table 2). Receptor 1io displays a conformational restriction analogous to 1oo but features one PO group inwardly directed. Calix[4]pyrrole 2 is also a cavitand featuring the replacement of the PO bridging units of 1 by methylene groups. Finally, receptor 3 is not cavitated and thus, conformationally more flexible. The direct comparison of the thermodynamic binding constants of different receptors is not exempt of shortcomings owing to dissimilar solvation/desolvation processes. Nevertheless, the direct comparison of thermodynamic constants is expected to provide general guidelines explaining the superior binding properties of 1oo. These guidelines may be transferred to the design of other effective hydrogen-bonding receptors working in water solution.:1 complexes of the receptors 1io, 2 and 3 in H2O or D2O [association constant Ka (M−1), free Gibbs energy ΔG (kcal mol−1), ΔH enthalpy (kcal mol−1) and entropy TΔS (kcal mol−1)]
| Guest | Receptor | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1io | 2 | 3 | ||||||||||
| K a × 10−4 | ΔG | ΔH | TΔS | K a × 10−4 | ΔG | ΔH | TΔS | K a × 10−4 | ΔG | ΔH | TΔS | |
|
a ITC titration experiments were repeated at least twice, and the reported Ka value is the average of the values obtained from the fit of the integrated heat data to a 1:1 binding model.
b
1H NMR titration experiments were also repeated at least twice, and the reported Ka is the average value returned from the fit of the chemical shift changes observed for the diagnostic proton signals to a 1:1 binding isotherm. Error values are reported as standard deviations. Error values for Ka are reported as standard deviations. Errors values for ΔG, ΔH and TΔS are determined to be less than 10%. nd not determined. See the ESI for experimental details of the titrations.
|
||||||||||||
|
0.35 ± 0.01a | −4.8 | −6.5 | −1.7 | 1.24 ± 0.04a | −5.5 | −9.1 | −3.6 | 0.55 ± 0.11a | −5.1 | −7.4 | −2.3 |
|
0.94 ± 0.04a | −5.4 | −6.7 | −1.3 | 5.94 ± 0.13a | −6.4 | −8.7 | −2.3 | 4.13 ± 0.07a | −6.2 | −7.2 | −1.0 |
|
2.82 ± 0.01a | −6.1 | −8.0 | −1.9 | 7.05 ± 0.22a | −6.5 | −8.2 | −1.7 | 7.10 ± 0.40a | −6.5 | −5.9 | 0.6 |
|
0.009 ± 0.002b | −2.7 | nd | nd | 0.02 ± 0.01b | −3.1 | nd | nd | 0.05 ± 0.01b | −3.7 | nd | nd |
|
0.005 ± 0.002b | −2.3 | nd | nd | 0.008 ± 0.001b | −2.6 | nd | nd | 0.08 ± 0.01b | −3.9 | nd | nd |
Consistently, the 1oo isomer delivers a thermodynamic advantage of ca. 1.2 kcal mol−1 in the binding of the cyclic lactam series in water with respect to the 1io counterpart (Tables 1 and 2).
We assign this difference to the need for partial desolvation of the inwardly directed PO group upon complex formation in the case of 1io. That is, in the inclusion complexes of the 1io receptor the inwardly directed PO group is partially shielded from solvation owing to its proximity to the hydrophobic section of the included guest (Fig. 9). In contrast, the outwardly directed PO groups of the 1oo receptor are not strongly affected by solvation processes upon complex formation. In short, receptor's polar groups experiencing desolvation upon complex formation and not being compensated by establishing polar interactions with the guest are detrimental to binding affinity.
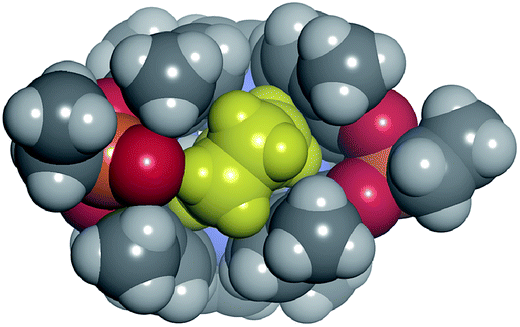 | ||
| Fig. 9 Top view of the energy minimized 6⊂1io complex depicted as a CPK model. The γ-butyrolactam 6 is shown in yellow. The van der Waals contact between the methylene of the bound guest and the oxygen atom of the inwardly directed PO group of the receptor demands its partial desolvation. | ||
Receptor 1oo shows a thermodynamic advantage of ca. 0.2–0.7 kcal mol−1 in the binding of the lactam series compared to 2 and 3. Most likely, these energetic differences are related to different desolvation/solvation processes experienced by the receptors and their complexes. Unfortunately, these processes are difficult to rationalize and to take into account during receptor's design.
The similar binding constant values measured for the complexes of the lactam series with receptors 2 and 3 demonstrate that the bridging of the meso-phenyl rings in order to reduce the conformational flexibility of the receptor has a limited impact on binding affinity. In water, solvation/desolvation processes seem to be more important to define the entropy term of binding than the receptor's preorganization.
General considerations of this study
In water, the formation of the studied 1:1 inclusion complexes is highly enthalpy driven. The entropy term represents a small positive contribution to binding for the complexes of the 1oo receptor. On the other hand, it opposes binding, although to a reduced extent, in most of the complexes of receptors 1io, 2 and 3. The significant enthalpic contribution to binding results from a combination of host–guest interactions, including hydrogen bonding, CH–π, and/or NH–π interactions. In water, the hydrogen bonding interactions of the 1:1 inclusion complexes of four-wall aryl-extended calix[4]pyrrole receptors are very efficient because they are shielded from bulk water molecules. This shielding is provided by the well-defined aromatic cavity of the bound receptors that is closed at one end. The only open end of the cavity is blocked to the passage of water molecules due to the bound guest. The dissociation of the complex by water solvation requires a conformational change of the receptor.
The hydrophobic effect plays an important enthalpic and entropic role in promoting the formation of the 1:1 inclusion complexes of aryl-extended calix[4]pyrrole receptors. Enthalpically, through the release of weakly hydrogen-bonded solvating water molecules that reorganize in the bulk forming stronger hydrogen-bonding interactions and also by increasing dispersion interactions in the complex in comparison to the solvated solutes. Entropically, because the solvating water molecules are highly organized and their release to the bulk solution increases disorder. The 1:1 inclusion complexes are structurally very well-defined and their formation requires extensive desolvation of large hydrophobic surfaces and polar groups of both the host and guest.40
It is worth noting that some of the polar compounds used in this study were investigated previously as guests with other synthetic receptors working in water and aqueous solutions.9,11,12,38 Without exception the affinity values displayed by 1oo for these guests are several orders of magnitude larger than the ones reported in the literature.
Conclusions
We report the synthesis of unprecedented water-soluble aryl-extended calix[4]pyrrole derivatives. The synthetic strategy allowed the functionalization of both upper and lower rims of the receptors. In particular, the upper rim functionalization leading to the bis-phosphonate cavitand with two outwardly directed PO groups, 1oo, has a positive impact on its binding properties compared to the in–out stereoisomer, 1io, the methylene bridged analogue, 2, and the calix[4]pyrrole counterpart 3. The results of the binding studies of the receptors with a library of neutral polar guests performed in water, using 1H NMR titrations and ITC experiments, demonstrated the formation of thermodynamically and kinetically stable 1:1 inclusion complexes. Despite the well-known competitive nature of water for hydrogen bonding, some guests were bound with affinity constants larger than 105 M−1. We assessed the hydrogen-bonding and hydrophobic contributions to binding by comparing the thermodynamic signatures of the processes in water and chloroform solutions. Both contributions turned out to be highly relevant. Remarkably, the more hydrophilic guests were bound more strongly in both water and chloroform solutions. Surprisingly, the polar aromatic cavity of the calix[4]pyrrole 1oo receptor exhibited better hydrogen bond donor properties than bulk water. The reason why these receptors work well in water is probably related to the difficult access of the water molecules to the polar groups in the complexes and its reduced energetic cost of solvation compared to the free counterparts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Gobierno de España MINECO and FEDER (CTQ2017-84319-P), and the CERCA Programme/Generalitat de Catalunya and AGAUR (2017 SGR 1123) for funding. G. P. thanks MINECO for a FPI fellowship. We also thank Dr Eduardo C. Escudero-Adán, X-ray Diffraction Unit of ICIQ, for help with the analysis of the X-ray crystallographic data and Dr Daniel Hernández-Alonso for experimental assistance and helpful discussions in the initial phases of the project.Notes and references
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- E. A. Kataev and C. Müller, Tetrahedron, 2014, 70, 137–167 CrossRef CAS.
- Y. Kato, M. M. Conn and J. Rebek, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1208–1212 CrossRef CAS.
- F. Diederich, Angew. Chem., Int. Ed., 1988, 27, 362–386 CrossRef.
- N. M. Green, Biochem. J., 1963, 89, 599–609 CrossRef CAS PubMed.
- A. P. Davis, Org. Biomol. Chem., 2009, 7, 3629–3638 RSC.
- B. Sookcharoenpinyo, E. Klein, Y. Ferrand, D. B. Walker, P. R. Brotherhood, C. Ke, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2012, 51, 4586–4590 CrossRef CAS PubMed.
- C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718 CrossRef CAS PubMed.
- G.-B. Huang, S.-H. Wang, H. Ke, L.-P. Yang and W. Jiang, J. Am. Chem. Soc., 2016, 138, 14550–14553 CrossRef CAS PubMed.
- L.-L. Wang, Z. Chen, W.-E. Liu, H. Ke, S.-H. Wang and W. Jiang, J. Am. Chem. Soc., 2017, 139, 8436–8439 CrossRef CAS PubMed.
- H. Yao, H. Ke, X. Zhang, S.-J. Pan, M.-S. Li, L.-P. Yang, G. Schreckenbach and W. Jiang, J. Am. Chem. Soc., 2018, 140, 13466–13477 CrossRef CAS PubMed.
- A. Lascaux, G. De Leener, L. Fusaro, F. Topic, K. Rissanen, M. Luhmer and I. Jabin, Org. Biomol. Chem., 2016, 14, 738–746 RSC.
- A. Lascaux, G. Delahousse, J. Ghostin, J. P. Bouillon and I. Jabin, Eur. J. Org. Chem., 2011, 5272–5278 CrossRef CAS.
- P. Anzenbacher, K. Jursíková, V. M. Lynch, P. A. Gale and J. L. Sessler, J. Am. Chem. Soc., 1999, 121, 11020–11021 CrossRef CAS.
- L. Bonomo, E. Solari, G. Toraman, R. Scopelliti, C. Floriani and M. Latronico, Chem. Commun., 1999, 2413–2414 RSC.
- B. Verdejo, G. Gil-Ramírez and P. Ballester, J. Am. Chem. Soc., 2009, 131, 3178–3179 CrossRef CAS PubMed.
- D. Hernandez-Alonso, S. Zankowski, L. Adriaenssens and P. Ballester, Org. Biomol. Chem., 2015, 13, 1022–1029 RSC.
- L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef CAS PubMed.
- M. Ciardi, A. Galán and P. Ballester, J. Am. Chem. Soc., 2015, 137, 2047–2055 CrossRef CAS PubMed.
- T. Guinovart, D. Hernández-Alonso, L. Adriaenssens, P. Blondeau, M. Martínez-Belmonte, F. X. Rius, F. J. Andrade and P. Ballester, Angew. Chem., Int. Ed., 2016, 55, 2435–2440 CrossRef CAS PubMed.
- B. Cantadori, P. Betti, F. Boccini, C. Massera and E. Dalcanale, Supramol. Chem., 2008, 20, 29–34 CrossRef CAS.
- R. Pinalli, G. Brancatelli, A. Pedrini, D. Menozzi, D. Hernández, P. Ballester, S. Geremia and E. Dalcanale, J. Am. Chem. Soc., 2016, 138, 8569–8580 CrossRef CAS PubMed.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- K. Ritter and M. Hanack, Tetrahedron Lett., 1985, 26, 1285–1288 CrossRef CAS.
- V. Fiandanese, G. Marchese, V. Martina and L. Ronzini, Tetrahedron Lett., 1984, 25, 4805–4808 CrossRef CAS.
- A. Díaz-Moscoso, D. Hernández-Alonso, L. Escobar, F. A. Arroyave and P. Ballester, Org. Lett., 2017, 19, 226–229 CrossRef PubMed.
- M. Ciardi, F. Tancini, G. Gil-Ramírez, E. C. Escudero Adán, C. Massera, E. Dalcanale and P. Ballester, J. Am. Chem. Soc., 2012, 134, 13121–13132 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- M. Liu, X.-a. Mao, C. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125–129 CrossRef CAS.
- S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475–8483 CrossRef CAS PubMed.
- A. J. Metherell, W. Cullen, N. H. Williams and M. D. Ward, Chem.–Eur. J., 2018, 24, 1554–1560 CrossRef CAS PubMed.
- C. Allott, H. Adams, P. L. Bernad, C. A. Hunter, C. Rotger and J. A. Thomas, Chem. Commun., 1998, 2449–2450 RSC.
- M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Chem. Sci., 2013, 4, 2744–2751 RSC.
- S. Turega, M. Whitehead, B. R. Hall, A. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122–1132 CrossRef CAS PubMed.
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS.
- F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318–15323 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterisation data, and binding studies by NMR and ITC. CCDC 1877620 and 1878126. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05034a |
| ‡ TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com. |
| § As described by Hunter and Ward,38 we used the slope of the correlation in water for guests 6, 9–12 to estimate an effective H-bond donor parameter for 1oo. The slope value is −1.3, which corresponds to −(α1oo–αH2O), because αH2O = 2.8, we calculated α1oo = 4.1. Thus, the binding pocket of 1oo provides an effective H-bond donor interaction that is comparable to a strong H-bond donor i.e. phenol. The slope (−2.1) of the free-energy correlation for the same guests in CHCl3 solution assigned a H-bond donor parameter for 17oo of α17oo = 4.3, as αCHCl3 = 2.2. Remarkably, the results of the estimated H-bond donor abilities for an aryl-extended calix[4]pyrrole core are quite consistent for the two solvents. |
| This journal is © The Royal Society of Chemistry 2019 |