
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA catalytic antioxidant for limiting amyloid-beta peptide aggregation and reactive oxygen species generation†
Luiza M. F.
Gomes
a,
Atif
Mahammed
b,
Kathleen E.
Prosser
a,
Jason R.
Smith
a,
Michael A.
Silverman
 c,
Charles J.
Walsby
a,
Zeev
Gross
*b and
Tim
Storr
*a
c,
Charles J.
Walsby
a,
Zeev
Gross
*b and
Tim
Storr
*a
aDepartment of Chemistry, Simon Fraser University, V5A-1S6, Burnaby, BC, Canada. E-mail: tim_storr@sfu.ca
bSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa, 32000, Israel. E-mail: chr10zg@technion.ac.il
cDepartment of Biological Sciences, Simon Fraser University, V5A-1S6, Burnaby, BC, Canada
First published on 3rd December 2018
Abstract
Alzheimer's disease (AD) is a multifaceted disease that is characterized by increased oxidative stress, metal-ion dysregulation, and the formation of intracellular neurofibrillary tangles and extracellular amyloid-β (Aβ) aggregates. In this work we report the large affinity binding of the iron(III) 2,17-bis-sulfonato-5,10,15-tris(pentafluorophenyl)corrole complex FeL1 to the Aβ peptide (Kd ∼ 10−7) and the ability of the bound FeL1 to act as a catalytic antioxidant in both the presence and absence of Cu(II) ions. Specific findings are that: (a) an Aβ histidine residue binds axially to FeL1; (b) that the resulting adduct is an efficient catalase; (c) this interaction restricts the formation of high molecular weight peptide aggregates. UV-Vis and electron paramagnetic resonance (EPR) studies show that although the binding of FeL1 does not influence the Aβ–Cu(II) interaction (Kd ∼ 10−10), bound FeL1 still acts as an antioxidant thereby significantly limiting reactive oxygen species (ROS) generation from Aβ-Cu. Overall, FeL1 is shown to bind to the Aβ peptide, and modulate peptide aggregation. In addition, FeL1 forms a ternary species with Aβ–Cu(II) and impedes ROS generation, thus showing the promise of discrete metal complexes to limit the toxicity pathways of the Aβ peptide.
Introduction
Alzheimer's disease (AD) is the most common form of dementia, representing between 50–75% of all cases.1 In 2017, an estimated 50 million people worldwide suffered from dementia, and this number is projected to grow sharply due to increased life expectancy.2 The lack of effective treatment strategies for AD, coupled with increased incidence, has stimulated extensive research efforts in this important field.3Clinical diagnosis of AD is currently based on progressive loss of memory and impairment in cognition,4 with final diagnosis requiring post-mortem examination of the brain to determine the severity of two neuropathological hallmarks; amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs). It is still unclear whether Aβ-plaques, NFTs, or both, are a cause or an effect of the neurodegeneration in AD.5 NFTs are intracellular aggregates of oxidatively-modified and hyperphosphorylated microtubule-associated protein tau,6 while Aβ plaques are extracellular and contain the Aβ peptide as the major constituent. The Aβ peptide is a product of the amyloid precursor protein (APP), and a series of cleavage events by α-, β-, γ-secretases,7 afford the Aβ peptide as predominantly Aβ1–40 or Aβ1–42 (a 40- or 42-residue peptide).8 In addition, truncation at the N-terminus results in Aβ3(p)–n, Aβ4–n, and Aβ11(p)–n (where p refers to pyroglutamate) peptides that are also significant components of amyloid deposits.9 Aβ can be found in three general forms in the brain: membrane associated, aggregated, and soluble. Most of Aβ is membrane-associated in healthy individuals, but in individuals with AD the aggregated and soluble fractions increase considerably.8,10
Early neuronal and pathological changes show indications of oxidative damage, indicating oxidative stress is involved in AD.11 The cause of oxidative stress in AD has been attributed to a number of factors, including impaired cellular energy metabolism and/or Fenton-type processes involving redox-active metal-ions (Fe, Cu), and metal-containing aggregates.12 Metal-ions, such as Zn, Cu and Fe, are essential for healthy organisms and brain function, and are tightly regulated under normal circumstances.13 However, a change in metal-ion homeostasis in the brain has been associated with protein aggregation, and the generation of reactive oxygen species (ROS) in neurodegenerative diseases such as AD.14 Metal-ions are present in increased concentrations in Aβ plaques in comparison to normal brain tissue, with concentrations of ca. 0.4 mM for Cu, 1 mM for Zn, and 0.9 mM for Fe.13a,13b,15 Metal-ion binding can modify the aggregation pattern of Aβ, disrupt normal metalloenzyme activity, and facilitate the production of ROS.10b,14a,14c,16 Recent studies have shown that the Aβ–Cu(II) complex exhibits detrimental catalytic ROS generation, particularly so in the presence of cholesterol and vitamin C, and is able to reduce O2 generating the superoxide anion (O2˙−), hydrogen peroxide (H2O2), and hydroxyl radical (·OH).17
As a result of the possible role of metal-ion dyshomeostasis in AD, the development of multifunctional metal binding molecules as therapeutics has been actively explored.18 We, and others, have developed metal-binding agents with additional properties such as radical scavenging, peptide binding and aggregation inhibition, and acetylcholine esterase (AChE) activity.19 In addition, a number of groups have explored the use of discrete metal complexes for the diagnosis and treatment of AD.20 In terms of therapeutics, Pt,21 Ru,22 Ir,23 Co,10b,24 Re,25 Rh,26 Mn,27 and V28 complexes have been investigated for their ability to modify the aggregation of the Aβ peptide, and certain compounds have shown promising results in disease models. For example, an orally-available Pt(IV) complex (Scheme 1) was shown to cross the blood–brain barrier (BBB), reduce plaque burden, and reduce Aβ peptide levels in a APP/Ps1 mouse model.29 Therefore, a metal complex that can bind to the Aβ peptide, modulate aggregation, and reduce ROS production is a promising therapeutic for AD.
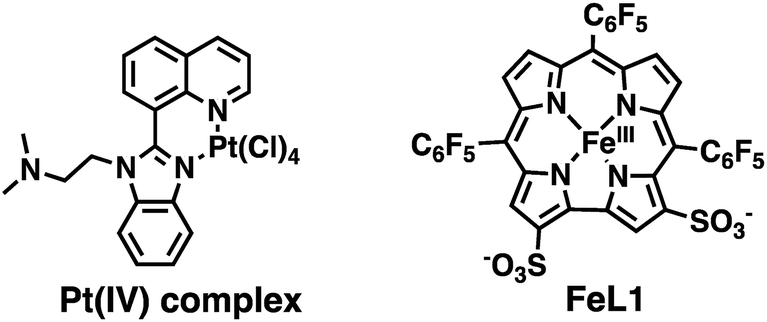 | ||
| Scheme 1 Structures of Pt(IV) complex, and Fe(III) corrole (FeL1) complex. | ||
Corrole ligands are known to bind to metal ions, such as Al, Cu, Fe, Ga, and Au, and the corresponding metal complexes display outstandingly high hydrolytic stability.30 The iron(III) complex (FeL1, Scheme 1) displays excellent catalase activity,31 superoxide dismutase (SOD) activity,30a and catalytic activity for the decomposition of peroxynitrite (PN, ONOO−).30d,32 Additionally, FeL1 binds to and protects the cholesterol-carrying lipoproteins from oxidative stress; and oral administration of FeL1 to a mouse model of atherosclerosis leads to a decrease in atherosclerotic lesions.30c,33 We were thus interested to investigate the interaction of FeL1 with the Aβ peptide, and how this would modulate peptide aggregation and ROS generation. Strong inspiration came from reports by Dey et al., who have shown that heme binds to the Aβ peptide, that one of the three histidine residues (His6) of Aβ is ligated to the heme's iron, and that the heme-Aβ adduct induces ROS formation.17a,34 Furthermore, a study that focused on the uptake of iron complexes by macrophages, which are a major source of ROS, revealed that heme is cytotoxic while FeL1 is cytoprotective.35 Additionally, FeL1 was reported to have low cytotoxic activity while maintaining cell cycle distribution similar to untreated cancer cells.36 We report the interaction of FeL1 with the Aβ peptide, how it affects peptide aggregation, and the radical scavenging ability of the FeL1–Aβ adduct, in both the presence and absence of Cu(II) ions.
Results and discussion
Binding of Aβ His residues to FeL1
The Fe(III) complex of the amphipolar 2,17-bis-sulfonato-5,10,15-tris(pentafluorophenyl)corrole (FeL1) has very strong affinity to human serum albumin (HSA) and lipoproteins, which is in part due to binding of histidine (His) residues to the metal ion.30c,37 The His ligation causes a shift in the Soret band of FeL1 from 390 to 410 nm, as well as the formation of a new band at 620 nm, and the intensity of the latter band is associated with the binding of either one or two axial His residues.30c,31,37 There are three Aβ His residues (His6, His13, and His14) and they play an important role in metal-ion binding (Scheme 2), with dissociation constants (Kd) of ∼10−10 M for Cu(II) and ∼10−5 M for Zn(II).10b,14a,38 In addition, Aβ His residues have been reported to bind to discrete metal complexes such as heme,34a Ru complexes,22a,23a,39 and Co complexes.10b,24 In addition to His binding, residues Asp1, Tyr10, and Glu11 play a role in the coordination of Aβ to Cu(II) and Zn(II).40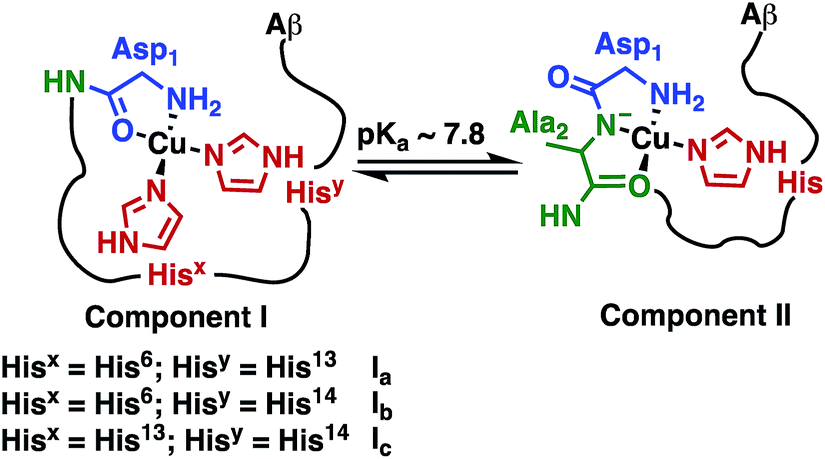 | ||
| Scheme 2 Representation of Component I (Ia, Ib, Ic) and Component II, the two major pH-dependent Aβ–Cu(II) binding modes (modified from Borghesani et al., 2018).9c | ||
Prior to investigating the interaction(s) of FeL1 with the Aβ peptide by UV-Vis spectroscopy, its binding to 1-methylimidazole (1-MeIm) was examined as to determine the spectral features and binding affinity associated with exogenous imidazole as the axial ligand. Gradual addition of up to 150 equiv. of 1-MeIm led to a shift in the near UV (Soret) band, a decrease in the band at 533 nm, and the formation of a new band at 620 nm (Fig. 1). The spectral changes matched those for histidine binding (Fig. S1†),30c however in both cases a large excess of ligand is required (150 and 700 equiv. respectively) to observe spectral endpoints. A variable pH UV-Vis titration (Fig. S2†), at a concentration ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 FeL1:1-MeIm, together with subsequent data fitting using Hypspec and HySS,41,45 provides binding constants of logM(1-MeIm) = 5.81 ± 0.01 (where M = FeL1) and a much smaller logM(1-MeIm)2 = 2.57 ± 0.02. Our results are in accord with the higher stability of 5-coordinate mono-axial ligated Fe(III) corroles in comparison to 6-coordinate bis-axial ligated Fe(III) corroles,42 which is opposite to that reported for Fe(III) porphyrins.43 The main reason for this difference is that upon bis-axial ligation Fe(III) porphyrins gain more crystal field stabilization energy (CFSE) as they transform from high spin (HS) to low spin (LS), while Fe(III) corroles only transform from intermediate spin (IS) to LS.42b,42c
:2 FeL1:1-MeIm, together with subsequent data fitting using Hypspec and HySS,41,45 provides binding constants of logM(1-MeIm) = 5.81 ± 0.01 (where M = FeL1) and a much smaller logM(1-MeIm)2 = 2.57 ± 0.02. Our results are in accord with the higher stability of 5-coordinate mono-axial ligated Fe(III) corroles in comparison to 6-coordinate bis-axial ligated Fe(III) corroles,42 which is opposite to that reported for Fe(III) porphyrins.43 The main reason for this difference is that upon bis-axial ligation Fe(III) porphyrins gain more crystal field stabilization energy (CFSE) as they transform from high spin (HS) to low spin (LS), while Fe(III) corroles only transform from intermediate spin (IS) to LS.42b,42c
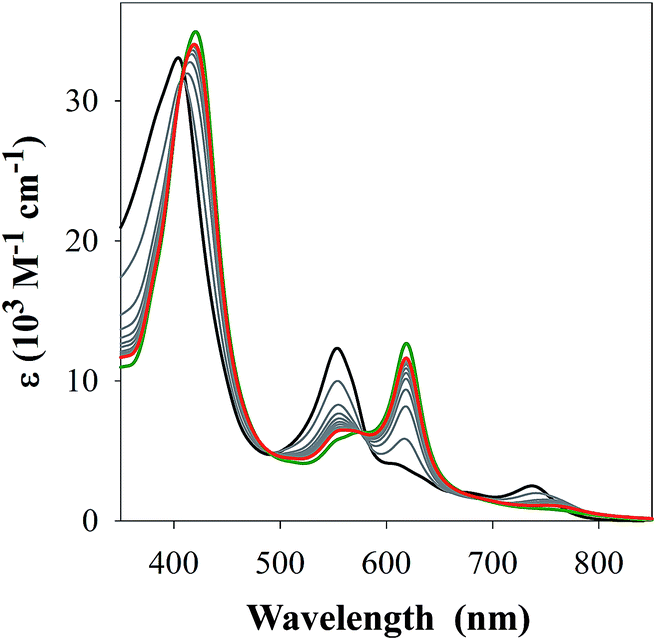 | ||
| Fig. 1 UV-Vis spectra of 1-MeIm additions to FeL1 (30 μM, black) in PBS buffer (0.01 M, pH 7.4). Grey lines represent additions of 5 equiv. of 1-MeIm up to 50 equiv. (red) with a maximum of absorption at 620 nm for 150 equiv. shown in green. | ||
The studies with 1-MeIm provided critical information for examining the interaction of FeL1 with the full length Aβ1–42 and two truncated peptides: Aβ1–16 that contains the metal binding N-terminus (His6, His13, His14), and Aβ17–40 with the hydrophobic portion of the peptide lacking any His. Addition of Aβ17–40 to FeL1 did not induce any significant spectral changes, while even a single equivalent of either Aβ1–42 or Aβ1–16 led to a red shift and intensity-increase of the Soret band, accompanied by the appearance of a λmax = 620 nm band (Fig. 2). While this experiment clearly proves the importance of His–Fe binding, the comparison of Fig. 2 and 1 exposes major differences. Importantly, the binding of the protein-provided histidine must be much stronger than that of 1-MeIm as full spectra changes are achieved with 1 vs. >100 equivalents, respectively. In the former case, the observed spectral changes occur immediately upon mixing, with no further spectral changes apparent after monitoring for 1 h, and addition of excess Aβ1–16 (up to 16 equivalents, Fig. S3†) did not induce further spectral changes. The last result is also highly relevant to the other spectral difference: while the 533 nm band disappears in the presence of a large excess of 1-MeIm (Fig. 1), the bands at 533 and 620 nm remain of essentially equal intensity starting from a 1:1 (Fig. 2) to a 1:16 (Fig. S3†) ratio of FeL1:Aβ1–16. Taken together, the results show that FeL1 and Aβ form a 1:1 adduct that relies on only one of the His residues in Aβ. The other two His residues are either too far away to approach the metal center and/or are unable to bind due to steric interference. Previous reports agree with our findings in that axial ligand binding to Fe(III) corroles shows that the 5-coordinate species is stabilized in comparison to the 6-coordinate bis-axial ligated species.42a,42b,44
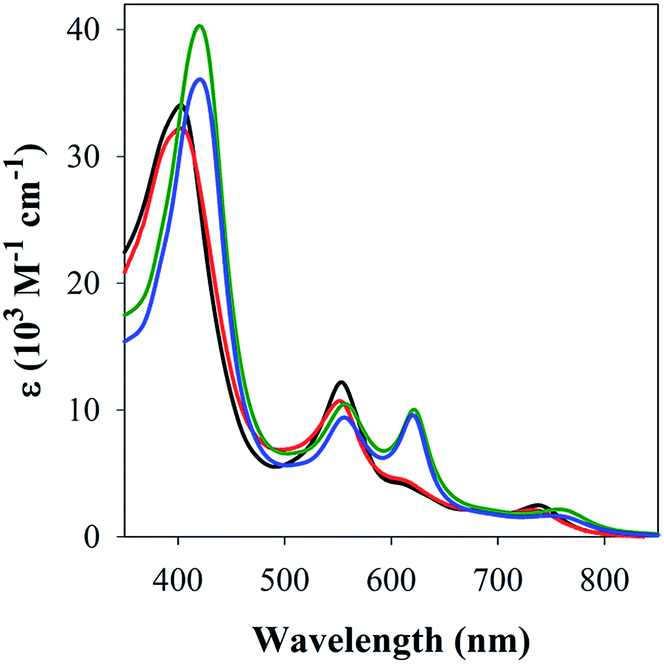 | ||
| Fig. 2 UV-Vis spectra of FeL1 (30 μM, black) in the presence of 1 equiv. of Aβ17–40 (red), Aβ1–16 (green) and Aβ1–42 (blue) in PBS buffer (0.01 M, pH 7.4). | ||
The stability of the 1:1 FeL1:Aβ1–16 adduct was determined via a variable pH titration (Fig. 3), which together with subsequent data fitting using Hypspec and HySS,41,45 provided binding constants of logM(Aβ1–16) = 11.90 ± 0.01, and logM(Aβ1–16)(H) = 4.90 ± 0.02 (where M = FeL1, and (H) indicates a mono-protonated peptide species). This experiment demonstrates the much higher affinity of FeL1 for the Aβ peptide in comparison to 1-MeIm (see below). At higher pH values (>9.5) a metal hydrolysis species is evident (modelled as logM(Aβ1–16)(OH), presumably due to deprotonation of a bound H2O ligand). As indicated from the speciation diagram (Fig. 3), the interaction of FeL1 with the Aβ peptide coincides with His deprotonation (reported pKa values of 5.72, 6.5, and 6.95).46 Further analysis of the speciation diagram of FeL1 with Aβ1–16 provides the binding affinity at physiological pH. The concentration of free FeL1 present in solution at a given pH, referred to as pM (p(FeL1) = −log[(FeL1)unchelated]), is a direct estimate of metal–ligand affinity when all species in solution are considered.47 The calculated value for p(FeL1) is 6.6 ([FeL1] = [Aβ1–16] = 30 μM), which affords a Kd value of ∼10−7 M.18a,48 This value shows that the affinity of Aβ for FeL1 is lower than for Cu(II) but larger than for Zn(II).
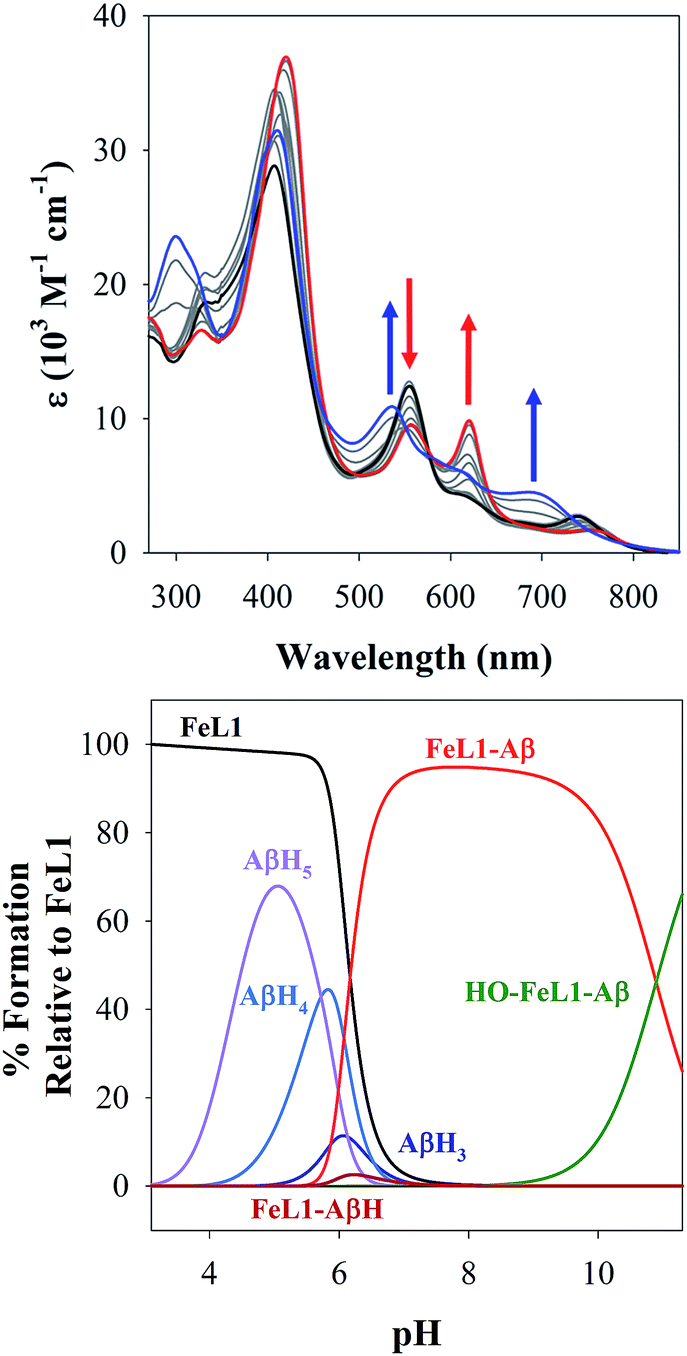 | ||
| Fig. 3 (Top) variable pH UV-Vis titration of FeL1 (30 μM) and Aβ1–16 (30 μM) from pH 3.1 (black) to pH 11.5 (blue). The red spectrum represents the maximum absorbance for the FeL1–Aβ complex at pH 8.2. (Bottom) using HypSpec and HySS,41 the variable pH data were fitted to a model including FeL1–Aβ, FeL1–Aβ(H), and a HO-FeL1–Aβ component at high pH. At pH 7.4 the majority of FeL1 is bound to Aβ1–16 (>99%). | ||
To gain more insight into the binding event, both 1H NMR and ESI-MS studies were performed. The MS spectrum of a 1:1 FeL1:Aβ1–16 adduct showed multiple m/z peaks consistent with FeL1 binding to Aβ1–16, with the most intense adduct peak corresponding to [FeL1–Aβ1–16]2+ (Fig. S5†). FeL1 has been reported to bind to human serum albumin (HSA),31 and in addition the Aβ peptide shows a specific interaction with HSA.49 Based on these reports we investigated the binding of FeL1 to Aβ in the presence of HSA, and under these conditions observed the [FeL1–Aβ1–16]2+ adduct (Fig. S6†). The 1H NMR of Aβ1–16 was recorded in the presence of 0.10 and 0.25 equivalents of the paramagnetic FeL1.10b,50 Initially, the signals from the three histidines and the tyrosine were quite sharp and well resolved (Fig. 4, bottom trace). Addition of FeL1 induced broadening of all signals attributed to the histidines (7.95, 7.05, 7.00 ppm), while those of Tyr10 (7.10 and 6.82 ppm) were not affected (Fig. 4, mid and top traces). Overall, the data are consistent with binding of an Aβ His residue to FeL1, and there is likely no preference for any of the available peptide His residues (His6, His13, His14).
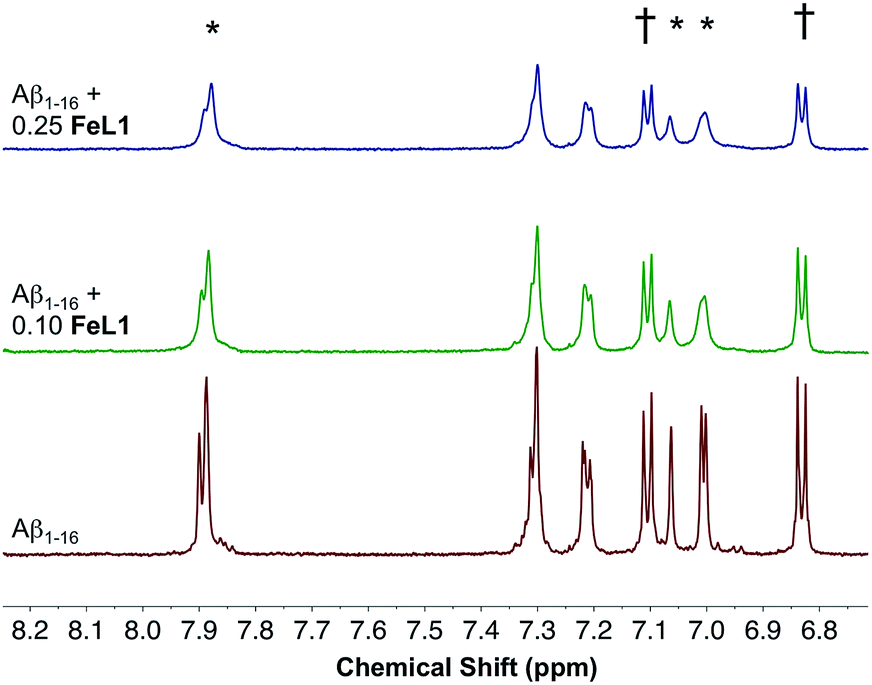 | ||
| Fig. 4 Changes in the 1H NMR spectra of Aβ1–16 in the presence of FeL1. Shown are spectra obtained at 210 μM Aβ1–16, in PBS buffer prepared in D2O pH 7.4 at 25 °C (red), with addition of 0.10 (green) and 0.25 equivalents (blue) of FeL1. *His6, His13, and His14. †Tyr10. | ||
FeL1 binding to Aβ in the presence of Cu(II)
:1:1 FeL1:Aβ1–16:Cu adduct) was further confirmed by ESI-MS, with m/z peaks corresponding to [FeL1–Aβ–Cu(II)]2+ and sodium adducts (Fig. S5†).
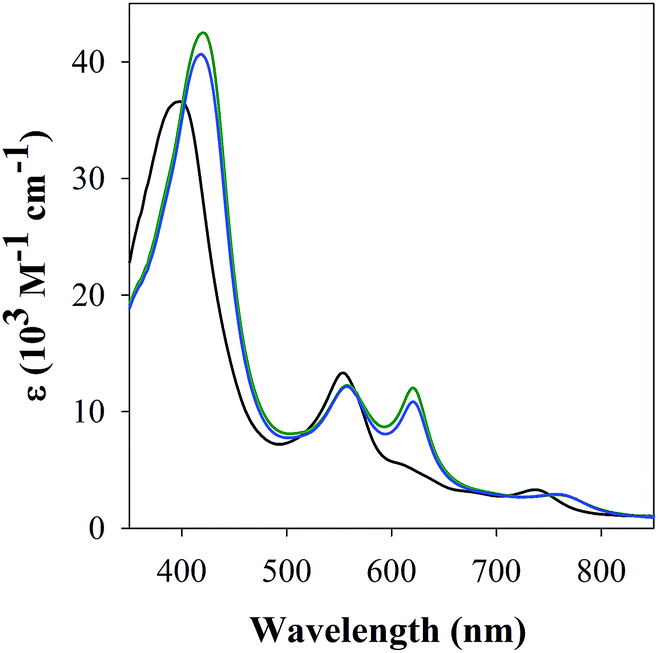 | ||
| Fig. 5 UV-Vis spectra of binding of FeL1 (30 μM, black) in PBS buffer (0.01 M, pH 7.4) to 1 equiv. of Aβ1–16 with (blue) or without (green) Cu(II) (0.9 equiv.). | ||
:0.4 for Component I:Component II at pH 7.4 is in agreement with the measured pKa value of 7.8 ± 0.5 (Scheme 2) via the Henderson–Hasselbalch equation.12b,50
| Aβ–Cu(II) – Component I | Aβ–Cu(II) – Component II | FeL1 (S = 3/2) | Fe bound (S = 1/2) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g ∥ | g ⊥ | A Cu∥ | χ | g ∥ | g ⊥ | A Cu∥ | χ | g ∥ | g ⊥ | g 1 | g 2 | g 3 | |
| a See experimental section for details. b Component relative abundance. | |||||||||||||
| Aβ–Cu(II) | 2.26 | 2.05 | 186 | 0.6 | 2.22 | 2.05 | 170 | 0.4 | — | — | — | — | — |
| FeL1 | — | — | — | — | — | — | — | — | 2.0 | 3.9 | — | — | — |
| FeL1 MeIm | — | — | — | — | — | — | — | — | 2.0 | 3.9 | 2.70 | 2.2 | 1.8 |
| FeL1–Aβ | — | — | — | — | — | — | — | — | 2.0 | 3.9 | 2.75 | 2.2 | 1.7 |
| FeL1–Aβ–Cu(II) | 2.26 | 2.05 | 186 | 0.7 | 2.22 | 2.05 | 170 | 0.3 | 2.0 | 3.9 | 2.75 | 2.2 | 1.7 |
FeL1 in buffer displays a weak intermediate-spin Fe(III) signal at g⊥ = 3.9 and g∥ = 2.0,42b,53 whereas FeL1–Aβ1–16 shows, in addition to this signal, also a rhombic spin system consistent with low-spin Fe(III) 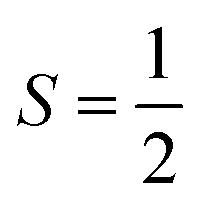 species (Table 1 and Fig. S8†). The latter is similar to the spectrum for FeL1 in the presence of 20 equiv. of 1-MeIm (Fig. S8†) and to reported EPR data for other 6-coordinate low spin Fe(III) corroles (with CN− and pyridine as axial ligands).54 These data are consistent with contributions from both mono- and bis-axial ligated FeL1 in the EPR experiment, likely due to the increased ligand affinity upon freezing the solutions for EPR analysis. Increased bis-axial ligation to FeL1 is observed for both 1-MeIm and Aβ1–16 in solution at lower temperatures (10 °C) by UV-Vis (Fig. S10†), and freezing a 5-coordinate (OEC)Fe(III)(py) corrole (OEC = trianion of 2,3,7,8,12,13,17,18-octaethylcorrole) in pyridine results in a similar spectral pattern with both intermediate and low spin signals.42b Due to the distinct temperature-dependence of signal intensity for the EPR spectra of the Fe species it is not possible to accurately determine their ratios from these experiments.55
species (Table 1 and Fig. S8†). The latter is similar to the spectrum for FeL1 in the presence of 20 equiv. of 1-MeIm (Fig. S8†) and to reported EPR data for other 6-coordinate low spin Fe(III) corroles (with CN− and pyridine as axial ligands).54 These data are consistent with contributions from both mono- and bis-axial ligated FeL1 in the EPR experiment, likely due to the increased ligand affinity upon freezing the solutions for EPR analysis. Increased bis-axial ligation to FeL1 is observed for both 1-MeIm and Aβ1–16 in solution at lower temperatures (10 °C) by UV-Vis (Fig. S10†), and freezing a 5-coordinate (OEC)Fe(III)(py) corrole (OEC = trianion of 2,3,7,8,12,13,17,18-octaethylcorrole) in pyridine results in a similar spectral pattern with both intermediate and low spin signals.42b Due to the distinct temperature-dependence of signal intensity for the EPR spectra of the Fe species it is not possible to accurately determine their ratios from these experiments.55
Incubation of both Cu(II) and FeL1 with Aβ1–16 affords the ternary species FeL1–Aβ1–16–Cu(II) with an EPR spectrum that is essentially the sum of the components Aβ1–16–Cu(II) and FeL1–Aβ1–16 (Fig. 6). The simulation parameters are detailed in Table 1, with the two Cu(II) species Component I and Component II in a 0.7:0.3 ratio. While this differs slightly from the ratio for Aβ1–16–Cu(II) alone, the limited resolution of the spectra suggests minimal change to the Cu-site upon binding of FeL1 to the peptide (vide infra). Similarly, the simulation parameters for the Fe species present in FeL1–Aβ1–16–Cu(II) (Table 1) are identical to FeL1–Aβ1–16 suggesting that while both Cu(II) and FeL1 bind to Aβ1–16, the binding sites are independent of one another as far as can be determined through the EPR experiments. This is reminiscent of reported EPR analysis of Cu(II) and heme with Aβ1–16, which also suggested that while both did bind to the peptide there was no observable interaction between the two metal centres.17a
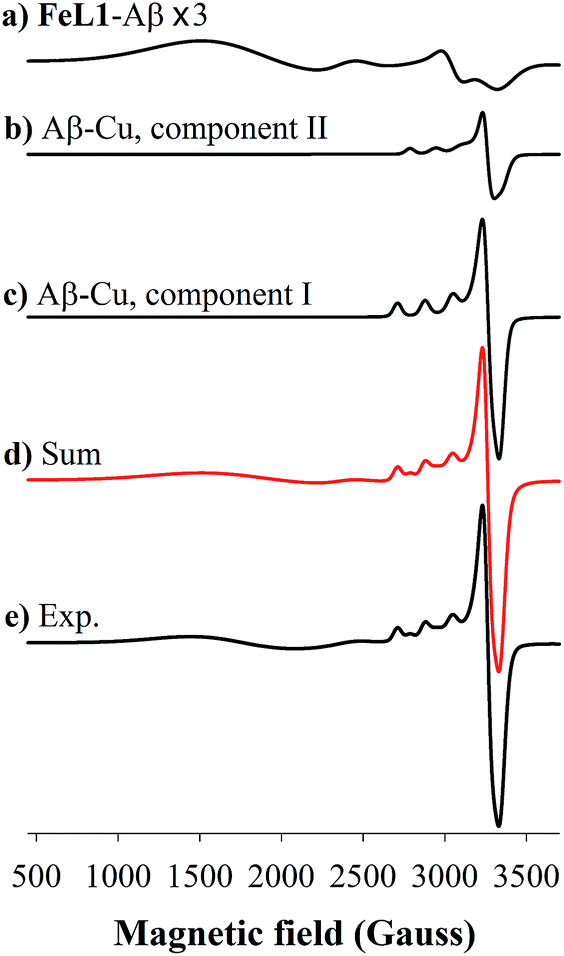 | ||
| Fig. 6 Frozen-solution EPR spectrum (bottom), corresponding simulation (red) and spectral deconvolution of FeL1 in the presence of 1 equiv. of both Aβ1–16 and Cu(II) at 20 K. (a) FeL1–Aβ component simulation (multiplied by a factor of 3), (b) Aβ–Cu(II) Component II simulation (c) Aβ–Cu(II) Component I simulation (d) sum of all simulated species and (e) experimental spectrum. Conditions: [Aβ1–16] = 550 μM, [FeL1] = [Cu(II)] = 500 μM, in PBS buffer (0.05 M, pH 7.4). EPR parameters: frequency = 9.38 GHz, microwave power = 2.0 mW, time constant = 40.96 ms, modulation amplitude = 5G, average of five 1 min scans. | ||
Influence of FeL1 on Aβ aggregation
Gel electrophoresis and western blotting, in combination with Transmission Electron Microscopy (TEM), were used to investigate if binding of FeL1 to the Aβ peptide would alter the size distribution of Aβ species and the morphology of the resulting aggregates. The longer Aβ1–42 peptide was employed for this study, as it is most aggregation prone and neurotoxic.13b,56 Incubation with low concentrations of FeL1 (0.1 to 1 equiv., for 24 h) significantly affected the aggregation pattern (Fig. 7A). While the Aβ1–42 peptide forms mostly high molecular weight aggregates (lane 1) in the absence of FeL1, consistent with previous reports,19a,24b,57FeL1 exhibits a concentration-dependent effect on the aggregation pattern (lanes 2–5). Only low molecular weight species were observed after 24 h with one equiv. of FeL1 (lane 5). The influence of FeL1 on Aβ1–42 aggregation was further confirmed by TEM (Fig. 7B–D). The TEM image for peptide alone shows long fibrils and large aggregate size, matching previous reports.24b,57 However, as the concentration of FeL1 is increased, a reduction in aggregate size is observed, with only small aggregates present with 1 equiv. of FeL1 (Fig. 7D). We also investigated the effect of the free ligand L1 on Aβ1–42 peptide aggregation. Under the same experimental conditions, L1 also displays a concentration-dependent effect on aggregation (Fig. S11†), however aggregate species are observed over a broad molecular weight range. We hypothesize that L1 alters the aggregation pattern via hydrophobic interactions with the Aβ peptide,19a,37,58 while the covalent interaction of FeL1 with Aβ His residues results in the preferential formation of low molecular weight species (Fig. 7).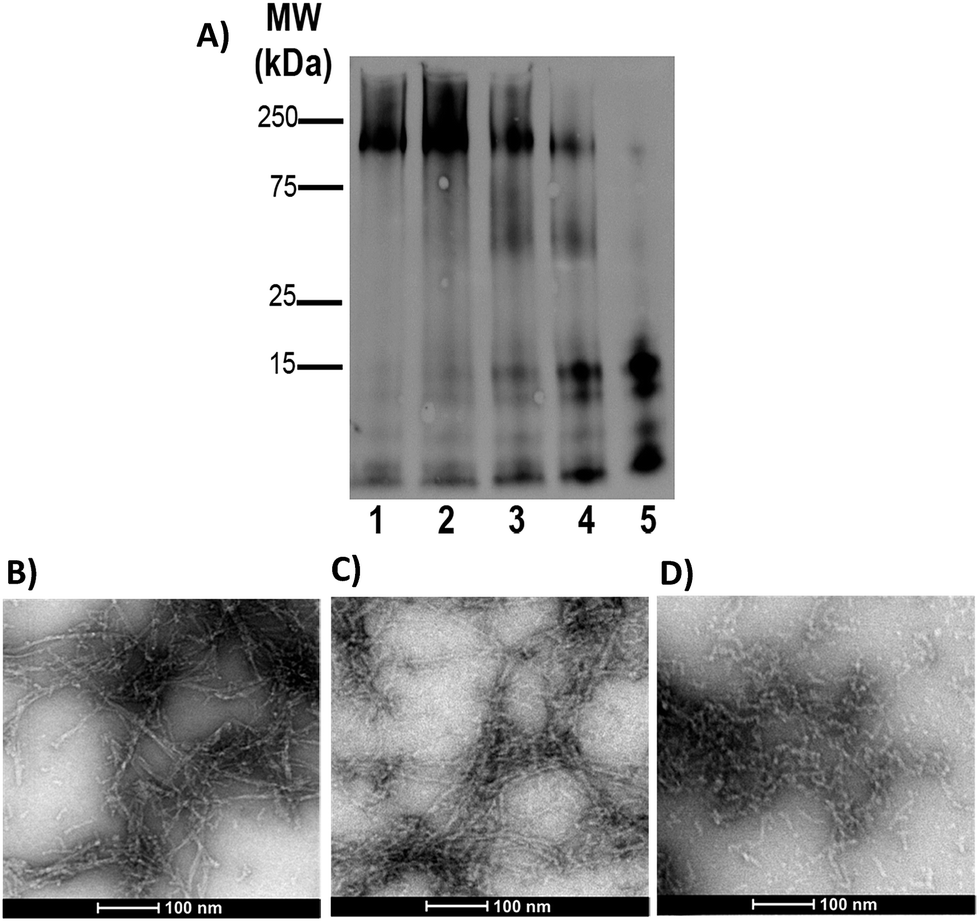 | ||
| Fig. 7 Influence of FeL1 on the aggregation profile of Aβ1–42. (A) Gel electrophoresis/Western blot of 25 μM Aβ1–42 and different concentrations of FeL1 in PBS buffer (0.01 M, pH 7.4) after 24 h incubation with agitation at 37 °C, using anti-Aβ antibody 6E10. Lane 1: Aβ1–42; lane 2: Aβ1–42 + FeL1 (0.1 equiv.); lane 3: Aβ1–42 + FeL1 (0.25 equiv.); lane 4: Aβ1–42 + FeL1 (0.5 equiv.); lane 5: Aβ1–42 + FeL1 (1 equiv.). And TEM images of (B) Aβ1–42; (C) Aβ1–42 + 0.1 equiv. FeL1; and (D) Aβ1–42 + 1 eq FeL1. | ||
Catalytic antioxidant activity
FeL1 has been previously reported to exhibit exceptional antioxidant activity for the disproportionation of H2O2,30d dismutation of O2˙−,30a and catalytic activity for the decomposition of peroxynitrite (PN, ONOO−).32 In addition, the antioxidant activity of FeL1 is maintained, and even enhanced, when bound to albumin, lipoproteins, or imidazole since this minimizes formation of the less catalytically-active μ-oxo iron(IV) dimer.30c,30d This work highlighted that the FeL1–Aβ species could act as a potent antioxidant, and possibly minimize ROS generation from Aβ–Cu(II) when both FeL1 and Cu(II) are bound to the peptide simultaneously.:1 FeL1–Aβ1–16 adduct displayed good catalase activity, with the latter being superior. This shows that His binding of the Aβ peptide to FeL1 results in an enhancement of catalase activity at all concentrations studied (1–5 μM).
 | ||
| Scheme 3 Reactive oxygen species generated by Aβ–Cu(II) in the presence of ascorbate and the possible assays to detect them, modified from C. Cheignon et al. 2018.38b | ||
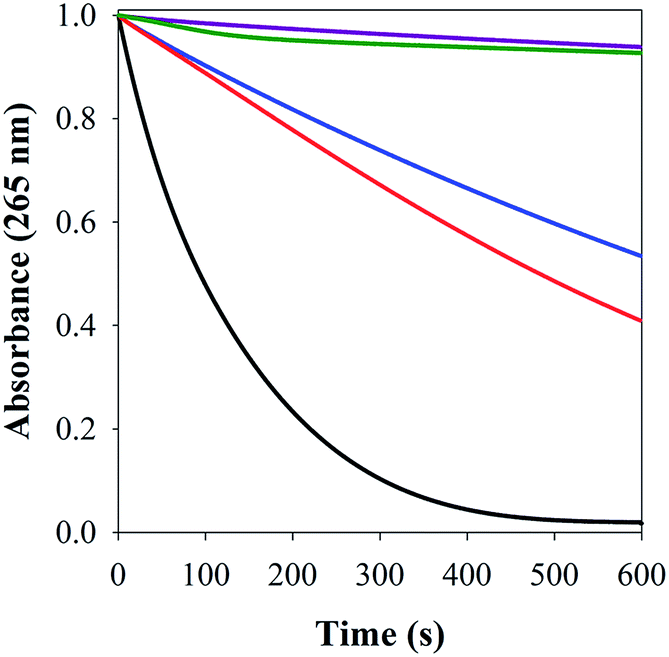 | ||
| Fig. 8 Ascorbate consumption by Cu(II) (black), Aβ–Cu(II) (blue); FeL1–Aβ–Cu(II) (red); FeL1–Aβ (purple); FeL1 (green), measured using UV-Vis spectroscopy (λ = 265 nm) as a function of time in PBS buffer (0.01 M, pH 7.4). [Asc] = 100 μM, [Aβ1–16] = [FeL1] = 10 μM and [CuCl2] = 9 μM. | ||
Cu–Aβ species can transform O2 to H2O2 through a series of steps, which can be detected via its reaction with Amplex Red, which forms the intensively colored resorufin (Scheme 3).60 Following this process by monitoring the formation of resorufin (Fig. 9A) revealed that: (a) Cu(II) alone induces the fastest rate of formation of H2O2; (b) the binding of Cu(II) to Aβ slows down the process, as reported previously;61 and (c) the ternary FeL1–Aβ1–16–Cu(II) species shows a reduced rate of H2O2 formation and lower amount overall. The latter phenomenon is consistent with FeL1 quenching the H2O2 that is produced by the bound Cu(II), due to the good catalase-like activity of FeL1 in both its free form and when bound to the Aβ peptide (Fig. S12B†). In addition to catalase activity, FeL1 displays exceptional antioxidant activity for the dismutation of O2˙−,30a and thus the complex may also quench superoxide formed as shown in Scheme 3. Detection of this reactivity using a cytochrome c assay9b,17b was challenging due to interference of FeL1 absorption bands.
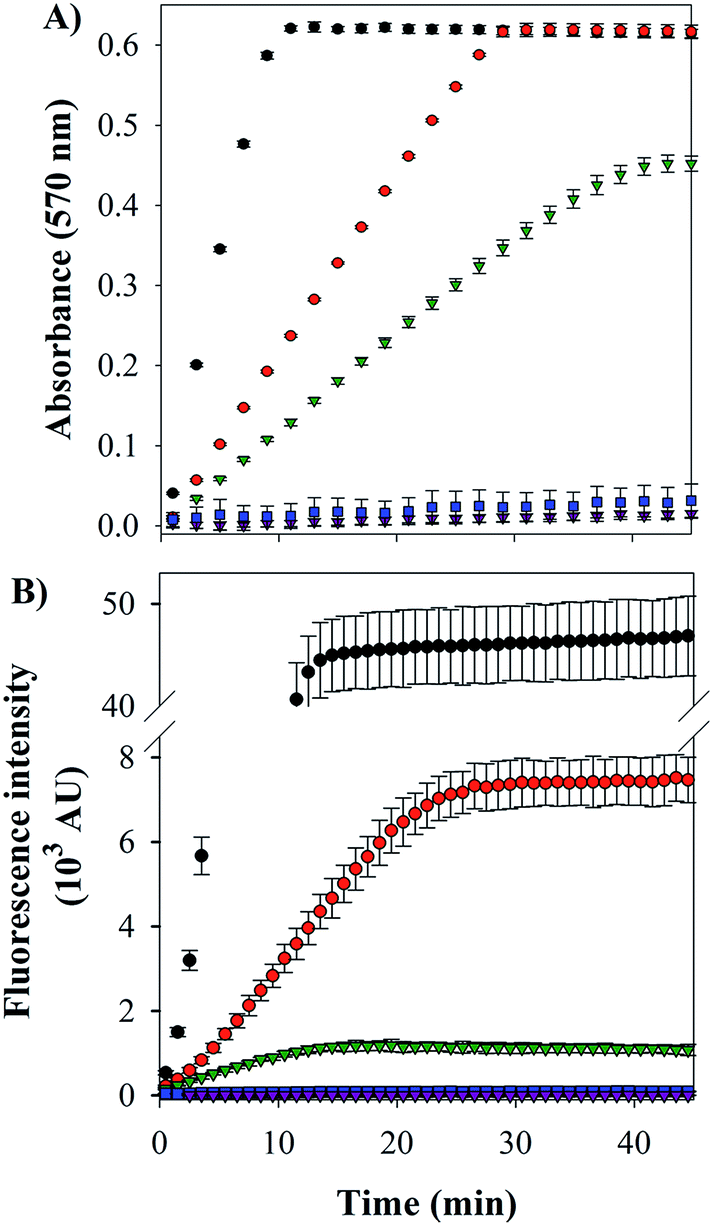 | ||
Fig. 9 ROS production by Aβ–Cu(II) in the presence of ascorbate. (A) Amplex red assay for H2O2 formation by UV-Vis absorbance at 570 nm. [Asc] = 400 μM, [Aβ1–16] = [FeL1] = [Cu(II)] = 20 μM, [Amplex red] = 100 μM; [HRP] = 0.4 U mL−1 (where 1 unit is defined as the amount of enzyme that will form 1.0 mg purpurogallin from pyrogallol in 20 seconds at pH 6.0 at 20 °C). (B) CCA assay for ·OH formation measured by fluorescence, λex 395 nm and λem 450 nm. [Asc] = 300 μM, [Aβ1–16] = [FeL1] = [Cu(II)] = 40 μM, [CCA] = 100 μM.  Cu(II); Cu(II);  Aβ–Cu(II); Aβ–Cu(II);  FeL1–Aβ–Cu(II); FeL1–Aβ–Cu(II);  FeL1–Aβ; FeL1–Aβ;  FeL1. FeL1. | ||
The last and most damaging step in Scheme 3 occurs via the formation of the hydroxyl radical (·OH) from the reaction of Cu(I) with H2O2 (Scheme 3), which may be detected by the reaction of 3-coumarin carboxylic acid (3-CCA) with ·OH to form the fluorescent 7-hydroxy-3-coumarin-carboxylic acid (7-OH-3-CCA).62 Consistent with previous reports,12e ·OH production is quite fast and significant for Aβ–Cu(II) albeit much less than for non-His-coordinated Cu(II) (Fig. 9B). The addition of FeL1, to form the ternary FeL1–Aβ1–16–Cu(II) species, resulted in a further 6-fold reduction in the amount of 7-OH-3-CCA. In principle, this may reflect either the lower availability of H2O2 due to the catalase-like activity or the direct quenching of ·OH by FeL1, or a combination of both. Another possible interpretation is that Cu in the ternary complex is less reactive, which is unlikely considering the minimal interaction of FeL1 with Aβ1–16–Cu(II) binding motif. In any case, the almost complete elimination of hydroxyl radical formation demonstrates that the potent antioxidant activity of FeL1 is maintained when bound to the Aβ peptide.
Summary
This study underlines the ability of FeL1 to target both Aβ peptide aggregation and ROS formation, two factors influencing AD progression. FeL1 and the free corrole ligand L1 influence Aβ aggregation differently; FeL1 stabilizes low molecular weight species while L1 stabilizes aggregates over a broad MW range. FeL1 forms a 1:1 adduct with Aβ via axial binding of one His residue with a moderate binding affinity (Kd of ∼ 10−7 M), which is weaker in comparison to Cu binding to Aβ (Kd of ∼ 10−10 M) but still stronger than Zn(II) binding (Kd of ∼10−5 M).10b,14a,38a,51 It is interesting to note that FeL1 has a much higher affinity for Aβ peptide His residues than 1-MeIm or free His, suggesting significant non-covalent interactions between FeL1 and the Aβ peptide. These results are in agreement with the specific binding of FeL1 to HDL2 proteins in comparison to other serum constituents, due to the amphipolar character of FeL1.30c Indeed, L1 was also shown to influence Aβ peptide aggregation likely due to hydrophobic interactions, albeit to a significantly lower extent. In a similar manner, non-covalent π–π stacking interactions, in addition to covalent binding, have been shown to dictate the association of Pt(II)(phenanthroline) complexes with the Aβ peptide.21a,29,63
We have also shown herein that FeL1 binds to the Aβ peptide concurrently with Cu(II). Our EPR data suggests no significant change in the Cu-binding site with FeL1 His coordination. This is further corroborated by the ascorbate oxidation assay, which displays only minor changes to the rate of ascorbate oxidation for Aβ–Cu(II) and the ternary species FeL1–Aβ–Cu(II). However, the bound FeL1 acts as an efficient catalase, and decomposes a significant fraction of the H2O2 generated by Aβ–Cu(II) (Fig. 9A). In the presence of FeL1 we also observe a decrease in the formation of ·OH (Fig. 9B), consistent with the result from the amplex red assay. Our results show that amphipolar FeL1 binds specifically to the Aβ peptide via a His residue in a 1:1 stoichiometry, and this interaction modulates the peptide aggregation pathway. In addition, the peptide-bound FeL1 maintains its exceptional antioxidant activity, limiting ROS formation from Aβ–Cu(II). Overall, our results highlight the promising multifunctional character of FeL1 to limit Aβ peptide aggregation and the formation of damaging ROS, two hallmarks of AD.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council (NSERC) Discovery Grant (T. S., M. A. S., and C. J. W.), a Michael Smith Career Investigator Award (T. S.), and a New Investigator Grant from the Alzheimer's Association (NIRG-15-362537), Brain Canada (to T. S.) and Science without borders (CAPES – Proc. no. 0711/13-6, Brazil to L. M. F. G.). Dr Andrew Lewis is thanked for assistance with NMR measurements. K. E. P. acknowledges a NSERC Vanier CGS for support. Z. G. and A. M. acknowledge The Israel Science Foundation for support.Notes and references
- A. W. Martin Prince, M. Guerchet, G.-C. Ali, Yu-T. Wu and M. Prina, World Alzheimer Report 2015, The Global Impact on Dementia. An Analysis of Prevalence, Incidence, Cost and Trends, 2015 Search PubMed.
- Dementia statistics, http://www.alz.co.uk/research/statistics, accessed May 24th.
- (a) V. H. Finder, J. Alzheimer's Dis., 2010, 22, S5–S19 CAS; (b) E. D. Roberson and L. Mucke, Science, 2006, 314, 781–784 CrossRef PubMed; (c) P. A. Adlard, S. A. James, A. I. Bush and C. L. Masters, Drugs Today, 2009, 45, 293–304 CrossRef CAS PubMed; (d) M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS; (e) D. J. Selkoe, Nat. Med., 2011, 17, 1693 CrossRef CAS.
- J. L. Hickey and P. S. Donnelly, Coord. Chem. Rev., 2012, 256, 2367–2380 CrossRef CAS.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed.
- M. P. Mazanetz and P. M. Fischer, Nat. Rev. Drug Discovery, 2007, 6, 464–479 CrossRef CAS PubMed.
- (a) G. K. W. Kong, J. J. Adams, H. H. Harris, J. F. Boas, C. C. Curtain, D. Galatis, C. L. Masters, K. J. Barnham, W. J. McKinstry, R. Cappai and M. W. Parker, J. Mol. Biol., 2007, 367, 148–161 CrossRef CAS PubMed; (b) D. J. Selkoe and D. Schenk, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 545–584 CrossRef CAS PubMed.
- C. A. McLean, R. A. Cherny, F. W. Fraser, S. J. Fuller, M. J. Smith, K. Beyreuther, A. I. Bush and C. L. Masters, Ann. Neurol., 1999, 46, 860–866 CrossRef CAS.
- (a) C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald and K. Beyreuther, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 4245–4249 CrossRef CAS; (b) M. Mital, E. Wezynfeld Nina, T. Frączyk, Z. Wiloch Magdalena, E. Wawrzyniak Urszula, A. Bonna, C. Tumpach, J. Barnham Kevin, L. Haigh Cathryn, W. Bal and C. Drew Simon, Angew. Chem., Int. Ed., 2015, 54, 10460–10464 CrossRef CAS PubMed; (c) V. Borghesani, B. Alies and C. Hureau, Eur. J. Inorg. Chem., 2018, 2018, 7–15 CrossRef CAS PubMed.
- (a) D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed; (b) M. C. Heffern, P. T. Velasco, L. M. Matosziuk, J. L. Coomes, C. Karras, M. A. Ratner, W. L. Klein, A. L. Eckermann and T. J. Meade, ChemBioChem, 2014, 15, 1584–1589 CrossRef CAS PubMed; (c) S. Lesne, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, Nature, 2006, 440, 352–357 CrossRef CAS; (d) A. D. Watt, V. L. Villemagne and K. J. Barnham, J. Alzheimer's Dis., 2013, 33(suppl. 1), S283–S293 Search PubMed.
- D. H. Cho, T. Nakamura, J. G. Fang, P. Cieplak, A. Godzik, Z. Gu and S. A. Lipton, Science, 2009, 324, 102–105 CrossRef CAS PubMed.
- (a) E. Nam, J. Han, J.-M. Suh, Y. Yi and M. H. Lim, Curr. Opin. Chem. Biol., 2018, 43, 8–14 CrossRef CAS; (b) S. C. Drew and K. J. Barnham, Acc. Chem. Res., 2011, 44, 1146–1155 CrossRef CAS PubMed; (c) E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed; (d) J. A. Duce and A. I. Bush, Prog. Neurobiol., 2010, 92, 1–18 CrossRef CAS PubMed; (e) C. Cheignon, M. Jones, E. Atrian-Blasco, I. Kieffer, P. Faller, F. Collin and C. Hureau, Chem. Sci., 2017, 8, 5107–5118 RSC; (f) Y. Yuan, F. Niu, Y. Liu and N. Lu, Neurol. Sci., 2014, 35, 923–928 CrossRef.
- (a) M. G. Savelieff, S. Lee, Y. Liu and M. H. Lim, ACS Chem. Biol., 8, 856–865 CrossRef CAS PubMed; (b) K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS PubMed; (c) T. D. Rae, P. J. Schmidt, R. A. Pufahl, V. C. Culotta and T. V. O'Halloran, Science, 1999, 284, 805–808 CrossRef CAS.
- (a) F. Hane and Z. Leonenko, Biomolecules, 2014, 4, 101–116 CrossRef; (b) C. C. Curtain, F. Ali, I. Volitakis, R. A. Cherny, R. S. Norton, K. Beyreuther, C. J. Barrow, C. L. Masters, A. I. Bush and K. J. Barnham, J. Biol. Chem., 2001, 276, 20466–20473 CrossRef CAS; (c) R. J. Ward, D. T. Dexter and R. R. Crichton, J. Trace Elem. Med. Biol., 2015, 31, 267–273 CrossRef CAS.
- (a) M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurosci., 1998, 158, 47–52 CAS; (b) L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, J. Struct. Biol., 2006, 155, 30–37 CrossRef CAS PubMed; (c) R. Squitti, Front. Biosci., 2012, 17, 451–472 CrossRef CAS; (d) A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 67–73 CrossRef CAS PubMed; (e) P. Faller and C. Hureau, Dalton Trans., 2009, 1080–1094, 10.1039/b813398k.
- (a) A. Lakatos, B. Gyurcsik, N. V. Nagy, Z. Csendes, E. Weber, L. Fulop and T. Kiss, Dalton Trans., 2012, 41, 1713–1726 RSC; (b) S. L. Leong, T. R. Young, K. J. Barnham, A. G. Wedd, M. G. Hinds, Z. Xiao and R. Cappai, Metallomics, 2014, 6, 105–116 RSC; (c) A. S. Pithadia, A. Kochi, M. T. Soper, M. W. Beck, Y. Z. Liu, S. Lee, A. S. DeToma, B. T. Ruotolo and M. H. Lim, Inorg. Chem., 2012, 51, 12959–12967 CrossRef CAS PubMed; (d) F. Bousejra-ElGarah, C. Bijani, Y. Coppel, P. Faller and C. Hureau, Inorg. Chem., 2011, 50, 9024–9030 CrossRef CAS PubMed.
- (a) D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545–15552 CrossRef CAS; (b) K. Reybier, S. Ayala, B. Alies, J. V. Rodrigues, S. B. Rodriguez, G. L. Penna, F. Collin, C. M. Gomes, C. Hureau and P. Faller, Angew. Chem., Int. Ed., 2016, 55, 1085–1089 CrossRef CAS PubMed; (c) X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. A. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glahe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1999, 274, 37111–37116 CrossRef CAS PubMed; (d) G. F. Z. da Silva and L. J. Ming, Angew. Chem., Int. Ed., 2005, 44, 5501–5504 CrossRef CAS; (e) X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS PubMed; (f) C. Cheignon, P. Faller, D. Testemale, C. Hureau and F. Collin, Metallomics, 2016, 8, 1081–1089 RSC.
- (a) M. G. Savelieff, A. S. DeToma, J. S. Derrick and M. H. Lim, Acc. Chem. Res., 2014, 47, 2475–2482 CrossRef CAS; (b) M. C. Carreiras, E. Mendes, M. J. Perry, A. P. Francisco and J. Marco-Contelles, Curr. Top. Med. Chem., 2013, 13, 1745–1770 CrossRef CAS; (c) Z. Liu, A. Zhang, H. Sun, Y. Han, L. Kong and X. Wang, RSC Adv., 2017, 7, 6046–6058 RSC.
- (a) L. M. F. Gomes, R. P. Vieira, M. R. Jones, M. C. P. Wang, C. Dyrager, E. M. Souza-Fagundes, J. G. Da Silva, T. Storr and H. Beraldo, J. Inorg. Biochem., 2014, 139, 106–116 CrossRef CAS; (b) L. Hao, Q. Yunwei and W. Xiaohui, Future Med. Chem., 2018, 10, 679–701 CrossRef; (c) M. R. Jones, E. L. Service, J. R. Thompson, M. C. P. Wang, I. J. Kimsey, A. S. DeToma, A. Ramamoorthy, M. H. Lim and T. Storr, Metallomics, 2012, 4, 910–920 RSC; (d) X. Wang, X. Wang and Z. Guo, Coord. Chem. Rev., 2018, 362, 72–84 CrossRef CAS; (e) S. Lee, X. Zheng, J. Krishnamoorthy, M. G. Savelieff, H. M. Park, J. R. Brender, J. H. Kim, J. S. Derrick, A. Kochi, H. J. Lee, C. Kim, A. Ramamoorthy, M. T. Bowers and M. H. Lim, J. Am. Chem. Soc., 2014, 136, 299–310 CrossRef CAS.
- (a) J. J. Miller, L. M. F. Gomes, T. Storr and A. Casini, The Interaction of Metal Compounds with Protein Targets: New Tools in Medicinal Chemistry and Chemical Biology, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2017, DOI:10.1002/9781119951438.eibc2499; (b) K. Chen and M. Cui, MedChemComm, 2017, 8, 1393–1407 RSC; (c) D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- (a) K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS PubMed; (b) I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404–6407 RSC; (c) V. A. Streltsov, V. Chandana Epa, S. A. James, Q. I. Churches, J. M. Caine, V. B. Kenche and K. J. Barnham, Chem. Commun., 2013, 49, 11364–11366 RSC; (d) F. Collin, I. Sasaki, H. Eury, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 2130–2132 RSC.
- (a) M. R. Jones, C. Mu, M. C. P. Wang, M. I. Webb, C. J. Walsby and T. Storr, Metallomics, 2015, 7, 129–135 RSC; (b) L. Messori, M. Camarri, T. Ferraro, C. Gabbiani and D. Franceschini, ACS Med. Chem. Lett., 2013, 4, 329–332 CrossRef CAS PubMed.
- (a) G. S. Yellol, J. G. Yellol, V. B. Kenche, X. M. Liu, K. J. Barnham, A. Donaire, C. Janiak and J. Ruiz, Inorg. Chem., 2015, 54, 470–475 CrossRef CAS; (b) L. Lu, H.-J. Zhong, M. Wang, S.-L. Ho, H.-W. Li, C.-H. Leung and D.-L. Ma, Sci. Rep., 2015, 5, 14619 CrossRef CAS PubMed.
- (a) J. Suh, S. H. Yoo, M. G. Kim, K. Jeong, J. Y. Ahn, M.-s. Kim, P. S. Chae, T. Y. Lee, J. Lee, J. Lee, Y. A. Jang and E. H. Ko, Angew. Chem., Int. Ed., 2007, 46, 7064–7067 CrossRef CAS PubMed; (b) J. S. Derrick, J. Lee, S. J. C. Lee, Y. Kim, E. Nam, H. Tak, J. Kang, M. Lee, S. H. Kim, K. Park, J. Cho and M. H. Lim, J. Am. Chem. Soc., 2017, 139, 2234–2244 CrossRef CAS PubMed.
- C. Y. Chan, A. Noor, C. A. McLean, P. S. Donnelly and P. J. Barnard, Chem. Commun., 2017, 53, 2311–2314 RSC.
- B. Y.-W. Man, H.-M. Chan, C.-H. Leung, D. S.-H. Chan, L.-P. Bai, Z.-H. Jiang, H.-W. Li and D.-L. Ma, Chem. Sci., 2011, 2, 917–921 RSC.
- (a) C. D. Amandine, A. Vinita, G. Régis, D. Nicolas, P. Clotilde and H. Christelle, Chem.–Eur. J., 2018, 24, 5095–5099 CrossRef; (b) R. Walke Gulshan, S. Ranade Dnyanesh, M. Bapat Archika, R. Srikanth and P. Kulkarni Prasad, ChemistrySelect, 2016, 1, 3497–3501 CrossRef.
- L. He, X. Wang, D. Zhu, C. Zhao and W. Du, Metallomics, 2015, 7, 1562–1572 RSC.
- V. B. Kenche, L. W. Hung, K. Perez, I. Volitakes, G. Ciccotosto, J. Kwok, N. Critch, N. Sherratt, M. Cortes, V. Lal, C. L. Masters, K. Murakami, R. Cappai, P. A. Adlard and K. J. Barnham, Angew. Chem., Int. Ed., 2013, 52, 3374–3378 CrossRef CAS.
- (a) M. Eckshtain, I. Zilbermann, A. Mahammed, I. Saltsman, Z. Okun, E. Maimon, H. Cohen, D. Meyerstein and Z. Gross, Dalton Trans., 2009, 7879–7882, 10.1039/b911278b; (b) Z. Gross, G. Golubkov and L. Simkhovich, Angew. Chem., Int. Ed., 2000, 4045–4047 CrossRef CAS; (c) A. Haber, M. Aviram and Z. Gross, Chem. Sci., 2011, 2, 295–302 RSC; (d) A. Mahammed and Z. Gross, Chem. Commun., 2010, 46, 7040–7042 RSC; (e) Z. Okun and Z. Gross, Inorg. Chem., 2012, 51, 8083–8090 CrossRef CAS.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS.
- A. Mahammed and Z. Gross, Angew. Chem., Int. Ed., 2006, 45, 6544–6547 CrossRef CAS PubMed.
- A. Haber, A. Mahammed, B. Fuhrman, N. Volkova, R. Coleman, T. Hayek, M. Aviram and Z. Gross, Angew. Chem., Int. Ed., 2008, 47, 7896–7900 CrossRef CAS PubMed.
- (a) C. Ghosh, M. Seal, S. Mukherjee and S. Ghosh Dey, Acc. Chem. Res., 2015, 48, 2556–2564 CrossRef CAS PubMed; (b) M. Seal, S. Mukherjee and S. G. Dey, Metallomics, 2016, 8, 1266–1272 RSC; (c) S. Mukherjee, M. Seal and S. G. Dey, J. Biol. Inorg Chem., 2014, 19, 1355–1365 CrossRef CAS PubMed.
- A. Haber, M. Aviram and Z. Gross, Inorg. Chem., 2012, 51, 28–30 CrossRef CAS PubMed.
- P. Lim, A. Mahammed, Z. Okun, I. Saltsman, Z. Gross, H. B. Gray and J. Termini, Chem. Res. Toxicol., 2012, 25, 400–409 Search PubMed.
- A. Mahammed, H. B. Gray, J. J. Weaver, K. Sorasaenee and Z. Gross, Bioconjugate Chem., 2004, 15, 738–746 CrossRef CAS PubMed.
- (a) L. Q. Hatcher, L. Hong, W. D. Bush, T. Carducci and J. D. Simon, J. Phys. Chem. B, 2008, 112, 8160–8164 CrossRef CAS PubMed; (b) C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed; (c) B. Alies, A. Conte-Daban, S. Sayen, F. Collin, I. Kieffer, E. Guillon, P. Faller and C. Hureau, Inorg. Chem., 2016, 55, 10499–10509 CrossRef CAS PubMed; (d) I. Zawisza, M. Rózga and W. Bal, Coord. Chem. Rev., 2012, 256, 2297–2307 CrossRef CAS.
- D. Valensin, P. Anzini, E. Gaggelli, N. Gaggelli, G. Tamasi, R. Cini, C. Gabbiani, E. Michelucci, L. Messori, H. Kozlowski and G. Valensin, Inorg. Chem., 2010, 49, 4720–4722 CrossRef CAS PubMed.
- (a) Y. Miller, B. Ma and R. Nussinov, Coord. Chem. Rev., 2012, 256, 2245–2252 CrossRef CAS; (b) V. Wineman-Fisher, D. N. Bloch and Y. Miller, Coord. Chem. Rev., 2016, 327–328, 20–26 CrossRef CAS; (c) S. Parthasarathy, F. Long, Y. Miller, Y. Xiao, D. McElheny, K. Thurber, B. Ma, R. Nussinov and Y. Ishii, J. Am. Chem. Soc., 2011, 133, 3390–3400 CrossRef CAS PubMed; (d) Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- (a) C. A. Joseph and P. C. Ford, J. Am. Chem. Soc., 2005, 127, 6737–6743 CrossRef CAS PubMed; (b) E. Van Caemelbecke, S. Will, M. Autret, V. A. Adamian, J. Lex, J.-P. Gisselbrecht, M. Gross, E. Vogel and K. M. Kadish, Inorg. Chem., 1996, 35, 184–192 CrossRef CAS PubMed; (c) E. Vogel, S. Will, A. S. Tilling, L. Neumann, J. Lex, E. Bill, A. X. Trautwein and K. Wieghardt, Angew. Chem., Int. Ed., 1994, 33, 731–735 CrossRef.
- F. A. Walker, M.-W. Lo and M. T. Ree, J. Am. Chem. Soc., 1976, 98, 5552–5560 CrossRef CAS PubMed.
- D. Sellmann, S. Emig and F. W. Heinemann, Angew. Chem., Int. Ed., 1997, 36, 1734–1736 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, Ann. Chim., 1999, 89, 45–49 CAS.
- T. Kowalik-Jankowska, M. Ruta, K. Wiśniewska and L. Łankiewicz, J. Inorg. Biochem., 2003, 95, 270–282 CrossRef CAS PubMed.
- (a) A. E. Martell and R. D. Hancock, Metal Complexes in Aqueous Solutions, Plenum Press, New York, 1996 CrossRef; (b) J.-S. Choi, J. J. Braymer, R. P. R. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21990–21995 CrossRef CAS PubMed; (c) W. R. Harris, C. J. Carrano and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 2722–2727 CrossRef CAS.
- Z. Xiao and A. G. Wedd, Nat. Prod. Rep., 2010, 27, 768–789 RSC.
- T. S. Choi, H. J. Lee, J. Y. Han, M. H. Lim and H. I. Kim, J. Am. Chem. Soc., 2017, 139, 15437–15445 CrossRef CAS PubMed.
- H. Eury, C. Bijani, P. Faller and C. Hureau, Angew. Chem., Int. Ed. Engl., 2011, 50, 901–905 CrossRef CAS PubMed.
- E. Atrián-Blasco, M. del Barrio, P. Faller and C. Hureau, Anal. Chem., 2018, 90, 5909–5915 CrossRef PubMed.
- (a) B. Alies, I. Sasaki, O. Proux, S. Sayen, E. Guillon, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 1214–1216 RSC; (b) S. C. Drew, C. J. Noble, C. L. Masters, G. R. Hanson and K. J. Barnham, J. Am. Chem. Soc., 2009, 131, 1195–1207 CrossRef CAS PubMed; (c) C. Hureau and P. Dorlet, Coord. Chem. Rev., 2012, 256, 2175–2187 CrossRef CAS.
- G. Palmer, Iron Porphyrins. Part II, Addison-Wesley Publ. Co., Reading, MA, 1983 Search PubMed.
- (a) S. Cai, S. Licoccia and F. A. Walker, Inorg. Chem., 2001, 40, 5795–5798 CrossRef CAS PubMed; (b) L. Simkhovich, I. Goldberg and Z. Gross, Inorg. Chem., 2002, 41, 5433–5439 CrossRef CAS PubMed.
- L. V. Liu, S. Hong, J. Cho, W. Nam and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 3286–3299 CrossRef CAS PubMed.
- (a) D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS PubMed; (b) C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed; (c) Y. S. Gong, L. Chang, K. L. Viola, P. N. Lacor, M. P. Lambert, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10417–10422 CrossRef CAS PubMed; (d) R. Jakob-Roetne and H. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 3030–3059 CrossRef CAS PubMed.
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. Wee Yong, S. Tsutsui, P. K. Stys and T. Storr, Chem. Sci., 2017, 8, 5636–5643 RSC.
- (a) H. Kroth, A. Ansaloni, Y. Varisco, A. Jan, N. Sreenivasachary, N. Rezaei-Ghaleh, V. Giriens, S. Lohmann, M. P. Lopez-Deber, O. Adolfsson, M. Pihlgren, P. Paganetti, W. Froestl, L. Nagel-Steger, D. Willbold, T. Schrader, M. Zweckstetter, A. Pfeifer, H. A. Lashuel and A. Muhs, J. Biol. Chem., 2012, 287, 34786–34800 CrossRef CAS PubMed; (b) M. G. Savelieff, Y. Liu, R. R. P. Senthamarai, K. J. Korshavn, H. J. Lee, A. Ramamoorthy and M. H. Lim, Chem. Commun., 2014, 50, 5301–5303 RSC.
- A. Mahammed and Z. Gross, Catal. Sci. Technol., 2011, 1, 535–540 RSC.
- M. Zhou, Z. Diwu, N. Panchuk-Voloshina and R. P. Haugland, Anal. Biochem., 1997, 253, 162–168 CrossRef CAS PubMed.
- S. Chassaing, F. Collin, P. Dorlet, J. Gout, C. Hureau and P. Faller, Curr. Top. Med. Chem., 2012, 12, 2573–2595 CrossRef CAS PubMed.
- M. Jensen, A. Canning, S. Chiha, P. Bouquerel, J. T. Pedersen, J. Østergaard, O. Cuvillier, I. Sasaki, C. Hureau and P. Faller, Chem.–Eur. J., 2012, 18, 4836–4839 CrossRef CAS PubMed.
- (a) S. G. Yao, R. A. Cherny, A. I. Bush, C. L. Masters and K. J. Barnham, J. Pept. Sci., 2004, 10, 210–217 CrossRef CAS PubMed; (b) G. Ma, F. Huang, X. Pu, L. Jia, T. Jiang, L. Li and Y. Liu, Chem.–Eur. J., 2011, 17, 11657–11666 CrossRef CAS PubMed; (c) M. Turner, J. A. Platts and R. J. Deeth, J. Chem. Theory Comput., 2016, 12, 1385–1392 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04660c |
| This journal is © The Royal Society of Chemistry 2019 |