
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic polyimine macrobicyclic cryptands – self-sorting with component selection†
Michał
Kołodziejski
 abc,
Artur R.
Stefankiewicz
*bc and
Jean-Marie
Lehn
*a
abc,
Artur R.
Stefankiewicz
*bc and
Jean-Marie
Lehn
*a
aLaboratory of Supramolecular Chemistry, Institut de Science et d'Ingénierie Supramoléculaires (ISIS), UMR 7006, CNRS, Université de Strasbourg, 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: lehn@unistra.fr
bFaculty of Chemistry, Adam Mickiewicz University, Umultowska 89b, 61-614 Poznań, Poland. E-mail: ars@amu.edu.pl
cCenter for Advanced Technologies, Adam Mickiewicz University, Umultowska 89c, 61-614 Poznań, Poland
First published on 12th December 2018
Abstract
Self-assembling macrobicyclic cryptand-type organic cages display remarkable self-sorting behavior with efficient component selection. Making use of the dynamic covalent chemistry approach, eight different cages were synthesized by condensation of tris(2-aminopropyl)amine with structurally different dialdehydes. A series of self-sorting experiments were first carried out on simple dynamic covalent libraries. They reveal the influence of different structural features of the aldehyde components on the condensation with two triamine capping units. Subsequently, self-sorting experiments were performed on more complex systems involving several dialdehyde building blocks. Altogether, the results obtained describe the effect of the presence of a heteroatom, of electrostatic interactions, of delocalization and of the flexibility/stiffness of the propensity of a component to undergo formation of a macrobicyclic cage. In the presence of a catalytic amount of acid, the macrobicyclic structure undergoes dynamic component exchange.
Introduction
Since the first synthesis of the macrobicyclic cryptands and cryptates,1,2 studies on such compounds have been pursued in numerous laboratories and have represented a fascinating area of investigation in supramolecular chemistry.3,4 The wide interest in macrobicyclic and, by extension, in macropolycyclic entities resides in the rich set of properties (as molecular receptors, catalysts and carriers) enabled by their structural diversity and specific spatial forms, resulting from the use of a variety of building blocks, as well as in their potential uses in areas such as supramolecular engineering and nanotechnology. Much activity has been displayed in the wide area of cage-type compounds and molecular containers.5–9Cryptands have generally been synthesized by stepwise build-up of the macrobicyclic framework based on covalent bond formation.1–4 In addition, several such structures have later also been obtained by formation of multiple imine bonds.10–20 The advent and active development of dynamic covalent chemistry (DCC)21–30 which rests on the implementation of reversible covalent reactions (in particular imine formation) in molecular frameworks, prompts to revisit the area of macrobi(poly)cyclic cryptand-type cage compounds in the light of this approach, which opens up new vistas in synthesis, properties and applications of this class of substances.
Dynamic molecules and materials, molecular as well as supramolecular, have attracted increasing interest in the realm of constitutional dynamic chemistry (CDC)21,31,32 towards the emergence of adaptive chemistry.21,32 A particular intriguing feature of such constitutional dynamic systems is the ability to perform component selection in the buildup of their constitution through “self-sorting” processes, introduced initially for the case of “self-recognition” (“homo-self-sorting”) in the generation of double and triple stranded helicates.33 Self-sorting reduces the potential number of combinations of components (thus counteracting entropy) by arranging them into specific organized structures.34–36 Non-covalent self-sorting in artificial systems, assisted by hydrogen bonds37 or donor–acceptor interactions38,39 has been widely explored.40,41 On the other hand, the initial synthesis and exploration of macrobicyclic cryptand-type molecular cages based on the formation of multiple reversible imine bonds10,12,15–20 opened the possibility to extend self-sorting processes to the behavior of such dynamic cage architectures. Indeed, the successful high yield generation of these compounds by self-assembly in a single operation based on reversible covalent imine formation, may be considered to involve the operation of “behind-the-scene” dynamic self-sorting. More recently, the chemistry of dynamic covalent cage compounds42–69 has been subject to inventive developments by several groups based notably on polyimine,42–60 disulfide61–65 or orthoester formation.66–69
Dynamic covalent bond formation is thus clearly a means to generate dynamic cage-type compounds (dynamic cryptands) with various differentiated properties and to enable self-sorting in multicomponent libraries. The reversibility of dynamic covalent bonds, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of imines, enables removal of undesired intermediates (“errors”) and leads to the thermodynamically most stable structures. It also provides a pathway to cage-to-cage transformation in solution – a very useful tool that can be used to change the composition of a dynamic library on purpose.54
N of imines, enables removal of undesired intermediates (“errors”) and leads to the thermodynamically most stable structures. It also provides a pathway to cage-to-cage transformation in solution – a very useful tool that can be used to change the composition of a dynamic library on purpose.54
One may thus conclude that organic three-dimensional cage-like architectures can be obtained overall using dynamic covalent bond formation, avoiding complicated, kinetically controlled multistep pathways. Moreover, an important feature of such procedures is the ability to modulate the structural transformations within the component library under the influence of chemical (pH, guest molecule) or physical (temperature, light) stimuli. Such three-dimensional cages suitable for guest inclusion have attracted much attention recently related largely to their use as sensors, drug carriers and catalysts, as well as for storage or isolation of gases.70,71 However, from the perspective of synthetic self-assembling methodology as well as of functional features, self-sorting protocols add a new dimension to this area and deserve to be closely scrutinized, as they allow for the analysis of the behavior of multicomponent systems.
In the present work, we report the investigation of the effect of structural features of the molecular components on the self-sorting phenomenon in the formation of a variety of imine-based macrobicyclic cryptand-type cage constituents. Moreover, we describe acid-catalysed processes of component exchange between different cages. As a preliminary step, a variety of structurally distinct dialdehydes (A–H; Scheme 1) have been reacted with the triamine “tren” (tris(2-aminoethyl)-amine; T) to give hexaimine cages, extending the initial work on such processes.10–16,19 Following the dynamic covalent chemistry approach, dynamic libraries consisting of dialdehydes and triamine components were set up in order to analyse the self-sorting behaviour between the aldehydes employed. In order to be able to evaluate the effects of the structural features of the components, we have restricted the present work to systems involving just the different dialdehydes A–H and the triamine T in the absence of other agents, such as metal ions.
 | ||
| Scheme 1 Molecular structures of dialdehydes (A–H), the triamine (T) and the generated [3 + 2] homoleptic imine-based organic macrobicyclic cryptand cages. | ||
Results and discussion
Synthesis and characterisation of the homoleptic macrobicyclic cages
The reactions examined involved seven aromatic dialdehydes (A–G) of fairly similar type and size as well as one (H) (very different in both size and flexibility) (Scheme 1). Before performing competitive experiments, the dialdehydes (A–H) were separately reacted with triamine T in acetonitrile (MeCN) at room temperature. In all cases, substantial precipitates formed within 24 h and were shown subsequently to be the desired cages involving the bridging of two T units by three dialdehyde branches by hexa-imine formation, designated as X3T2, with X = A–H. ortho-Phthalaldehyde was also included in the initial list of dialdehydes to be tested but it did not react in the same way with T as the others and did not provide a cage. All the bulk products gave elemental analyses consistent with the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry and all showed 1H NMR spectra (in CDCl3) consistent with the D3 symmetry expected for the cage where all imine units have the trans configuration. The molecular structures of the products also agreed with their ESI-TOF mass spectrometry data. In two cases, E3T2 and F3T2, details of their solid-state structures were established by single-crystal X-ray crystallography (Fig. 1a and b, respectively). The crystallographic structures of A3T2, B3T2, C3T2 and D3T2 cages have been previously described in the literature16,46–48 as well as those of closely related compounds.10,15 Thus, crystals of E3T2·2CHCl3 were obtained as yellow rods by vapour diffusion of diethylether into a chloroform solution. The solid-state molecular structure confirms the structural assignment based on the solution data, showing an extended capsular form with a length of approximately 15 Å, as defined by the distance between the central N atoms of the tren residues, and a twisted chiral shape along this N-to-N axis. The chloroform of crystallisation is not included within the cage, indicating that the lateral dimensions of the cage are unsuited for inclusion, and the lone pairs on the oxygen atoms would appear to be directed out from the cage, indicating that external rather than internal interactions of the cage would be preferred.
:2 stoichiometry and all showed 1H NMR spectra (in CDCl3) consistent with the D3 symmetry expected for the cage where all imine units have the trans configuration. The molecular structures of the products also agreed with their ESI-TOF mass spectrometry data. In two cases, E3T2 and F3T2, details of their solid-state structures were established by single-crystal X-ray crystallography (Fig. 1a and b, respectively). The crystallographic structures of A3T2, B3T2, C3T2 and D3T2 cages have been previously described in the literature16,46–48 as well as those of closely related compounds.10,15 Thus, crystals of E3T2·2CHCl3 were obtained as yellow rods by vapour diffusion of diethylether into a chloroform solution. The solid-state molecular structure confirms the structural assignment based on the solution data, showing an extended capsular form with a length of approximately 15 Å, as defined by the distance between the central N atoms of the tren residues, and a twisted chiral shape along this N-to-N axis. The chloroform of crystallisation is not included within the cage, indicating that the lateral dimensions of the cage are unsuited for inclusion, and the lone pairs on the oxygen atoms would appear to be directed out from the cage, indicating that external rather than internal interactions of the cage would be preferred.
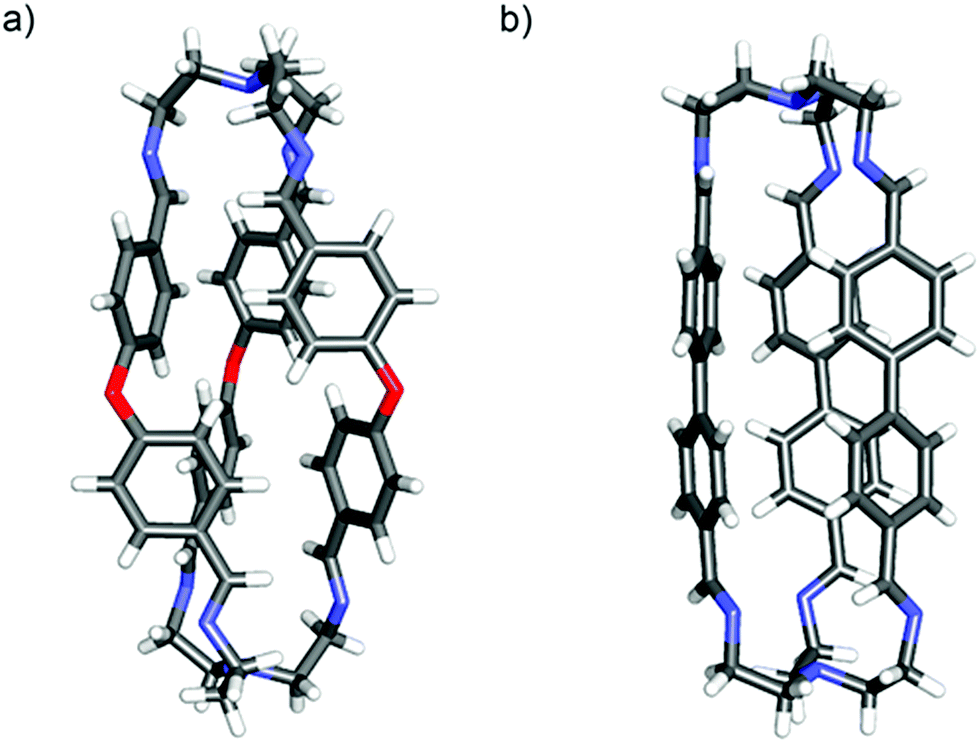 | ||
| Fig. 1 Solid state X-ray molecular structures of the cages E3T2·2CHCl3 (left) and F3T2·2CHCl3 (right). Colour code: red, O; blue, N; grey, C; white, H. | ||
Brownish, rod-like crystals of F3T2 were obtained by liquid diffusion of diisopropyl ether into a dichloromethane solution at room temperature in a closed vial. The cage unit is again a chiral capsule, though less twisted away from D3h symmetry than the E3T2 cage, and with a separation of the terminal N atoms close to 15 Å. There is no solvent included within the capsule.
Self-sorting experiments
| T | A | B | C | D | E | H |
| A | B3T2 | C3T2 | A3T2 | A3T2 | A3T2 | |
| B | B3T2 |
B3T2:C3T2 56:44 |
B3T2 | B3T2 | B3T2 | |
| C | C3T2 |
B3T2:C3T2 56:44 |
C3T2 | C3T2 | C3T2 | |
| D | A3T2 | B3T2 | C3T2 |
D3T2:E3T2 65:35 |
D3T2 | |
| E | A3T2 | B3T2 | C3T2 |
D3T2:E3T2 65:35 |
E3T2 | |
| H | A3T2 | B3T2 | C3T2 | D3T2 | E3T2 |
No formation of heteroleptic systems (mixed-aldehyde components cage species) was observed in any of the tested samples, whether at equilibrium or during its attainment. Thus, the species shown in Scheme 1 suffice to describe the formation behaviour in all systems.
:3:2 ratio was applied in all experiments) that was carried out, the influence of a heteroaromatic donor atom was investigated. The dialdehyde components A and B differ only by one atom. The carbon atom in position 2 of isophthalic dicarbaldehyde becomes a nitrogen atom in pyridine-2,6-dicarbaldehyde. Both dialdehydes and their imines contain a fully conjugated system of the aromatic ring and its substituents but only in B are attractive interactions possible between the two imine C–H bonds adjacent to the aromatic ring and the adjacent pyridine N atom of another cage arm.46 A possible additional factor that works in favour of cage B3T2 with respect to cage A3T2 are intramolecular interactions that may occur between imine C–H bonds and pyridine N lone pair (see ESI, Fig. S29†). Both cages, however, are stabilized through intramolecular hydrogen bonding interactions between the imine C–H bonds of one arm of the cage and the imine N atom of another.46 These latter interactions are also responsible for the almost identical position of the imine C–H signal in the 1H NMR spectra of both cages. Thus, the heteroaromatic cage B3T2 appears to be both kinetically and thermodynamically favoured in this self-sorting experiment and was formed in 92% yield. Cage A3T2 is not formed and component A remains unreacted in the reaction mixture (Fig. 2). At equilibrium, 8% of unreacted component B and triamine T are also present.
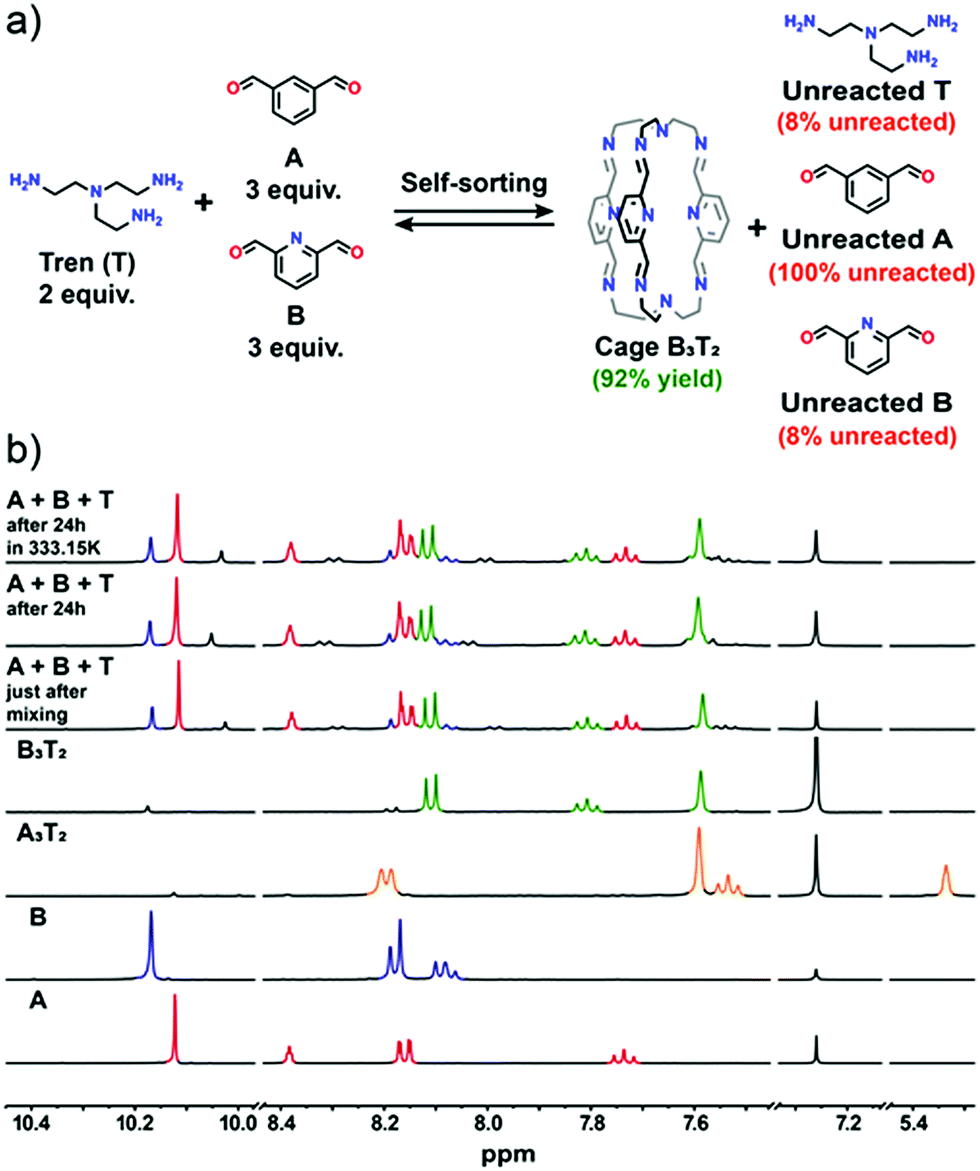 | ||
| Fig. 2 (a) General scheme of the self-sorting experiment between components A + B + T; (b) comparison of the 1H NMR spectra of all three components recorded over time. The 1H NMR spectra of reaction components and isolated cages are also shown for comparison. Low intensity signals observed in the spectra after component mixing may come from trace amounts of intermediate products such as open chain structures. All NMR spectra were recorded in CDCl3 at 400 MHz. | ||
 | ||
| Fig. 3 General schemes presenting product distribution of an equilibrium mixture for libraries consisting of (a) 3B + 3C + 2T and (b) 3D + 3E + 2T. | ||
In the case of the library comprising component D, E and T a mixture of two cages was again observed, though in this case the formation of D3T2 was favoured by about a factor of 2 at equilibrium, the product ratio D3T2:E3T2 being 65:35 (Fig. 3b). The aldehydes here differ in that the oxygen link in E breaks the conjugation of the two halves and introduces a degree of conformational flexibility that is absent in D (for NMR spectra see ESI, Fig. S21†).
 | ||
| Fig. 4 Changes in aldehyde proton signals of the 1H NMR spectra (400 MHz, CDCl3) during the self-sorting experiment involving titration of 3A + 3B + 3C + 3D + 3E + 3H with appropriate increasing amounts of T. The full spectra of dialdehyde components are shown in ESI.† | ||
This experiment led to the slightly modified order of effectiveness in cage formation of B = C > A = D > E ≫ H. Although it is not clear why the behaviour of A and D differs slightly in the full mixture from that in the binary one, the observations confirm that the most effective aldehydes of the chosen group are those (B, C) involving a heteroatom in a simple aromatic unit where there is full conjugation over both imine units. The presence of heteroatoms provides additional possibilities of hydrogen bond formation with the imine C–H, which may further stabilize the cage. Antiparallel orientation of the dipoles of the imine –N groups and the heteroatoms in the aromatic ring may also be a significant electrostatic factor. Next most effective are the aromatic dialdehydes (A, D) lacking a heteroatom in the ring but still enabling full conjugation of the imine units with the rings, while least effective are those (E, H) with some degree of flexibility and an inability to form a conjugated system. Obviously, other factors such as solvation differences may explain some degree of difference but the fact that in all cases it is the cage species that is formed requires that factors controlling the orientation of the imine substituents be considered most important, although here the conformational preferences of the triamine T also require consideration.
 | ||
| Fig. 5 Comparison of the 1H NMR spectra in the range of the dialdehyde signals for self-sorting experiment on libraries consisting in components (a) 3D + 3E + 3F + 2T and (b) 3B + 3C + 3G + 2T. The aldehyde proton signal of the dialdehyde components are also shown for comparison. All NMR spectra were recorded at 400 MHz in CDCl3 at room temperature. | ||
:C3T2 as 56:44). After addition of 4 molar amounts of triamine in total, these two cages were fully formed. With the final dose of T, the formation of G3T2 was also completed. At this point, there were three homoleptic cages in solution with once again no evidence of the formation of heteroleptic species, despite the similar equilibrium features shown by the dialdehydes B and C. Given the small advantage of dialdehyde B over C, the affinity of the dialdehydes for the triamine T is: B > C ≫ G. In all three cases, the macrobicyclic cage may be stabilized by H-bonding of the heteroatoms with the imino-CHN hydrogen. The interaction is expected to follow the sequence N > O > S, which corresponds to the sequence B > C ≫ G observed. In addition, ring size may have an effect when comparing B and C. Furthermore, the difference between C and G may result from the stronger H-bonding to O than to S as well as possibly to the smaller size of O (van der Waals radius – 48 ppm for O and 88 ppm for S) and to the difference in C–O(S) bond lengths (C–O: 0.136 nm; C–S: 0.182 nm). It would indicate that both the smaller radius of the oxygen atom and the shorter C–O bond length are structurally advantageous for cage formation. In addition, the significant difference in the electronegativity between the O and S heteroatoms may favour a more electronegative oxygen atom, enhancing the effect of antiparallel orientation of the dipoles of the imine –CHN– and of the aromatic heterocyclic groups.
In the first step, the less preferred cage A3T2 was broken into its components by the addition of MSA. The cage was then shown to reform rapidly after the addition of TEA. The dialdehyde B, known to form the cage preferentially to A, was then added and the 1H NMR spectrum of this mixture showed the absence of any perceptible exchange reaction, i.e. the cage A3T2 appeared to be stable in the presence of the alternative building block B.
Addition of MSA was once again used to dissociate the cage and this final mixture was thereafter made basic with TEA. The only cage species detectable in this solution was then B3T2 as expected. Additionally, these results also demonstrate the possibility to perform cage-to-cage mutation.54
These experiments established the lability of the cages in acidic media but involved their complete breakage. As an extension, component exchange from (A3T2 + B) to (B3T2 + A) was also investigated using a weak acid at catalytic concentrations to see if exchange of components could be observed in the absence of detectable cage dissociation. While reaction of A3T2 with B under such conditions (0.01 mol% catalyst) proved to be slow, cage breakup was not observed other than in that after 24 h reaction, the cage signals in the 1H NMR spectrum (Fig. 6) were those of B3T2 and were accompanied by those of free A. Thus, the strong equilibrium preference for formation of the cage involving B and not A was once more confirmed.
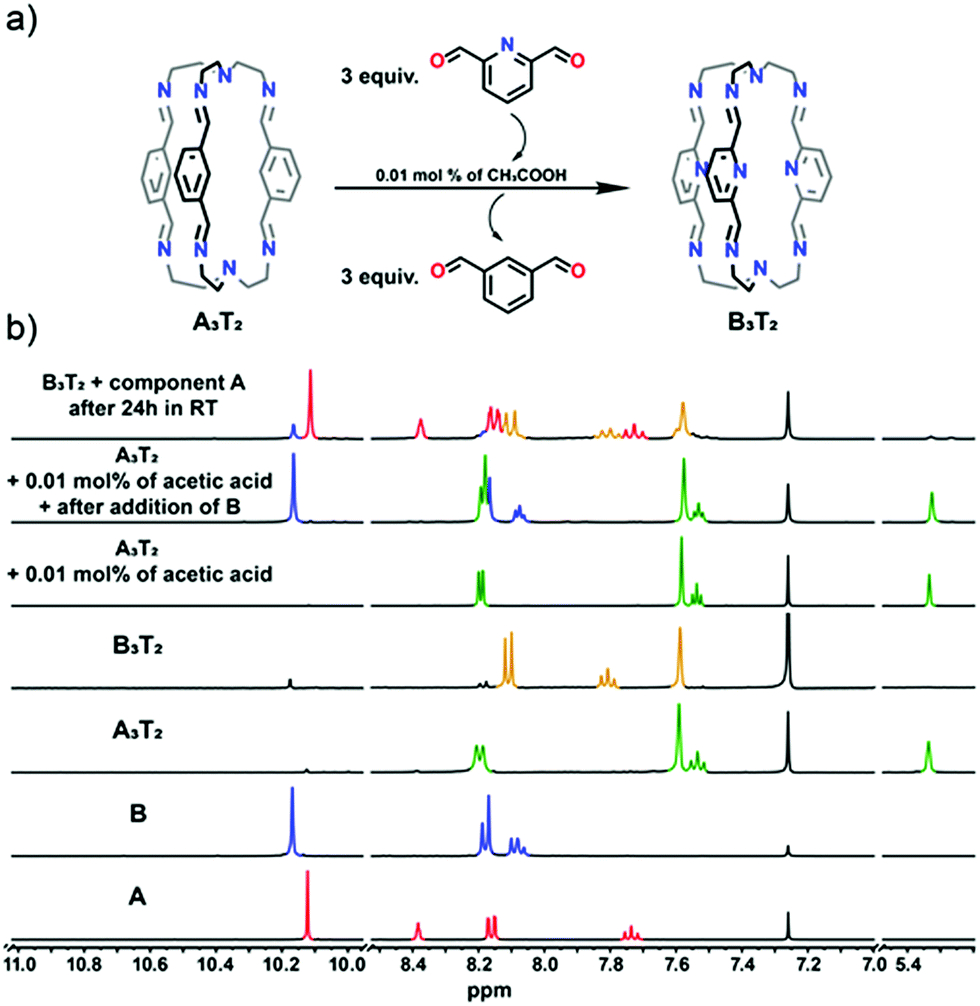 | ||
| Fig. 6 (a) General scheme of the exchange experiment (A3T2 + B → B3T2 + A) between components A + B + T; (b) 1H NMR spectra of acid catalysed exchange experiment between cage A3T2 and component B. For comparison 1H NMR spectra of components A and B as well as isolated cages A3T2 and B3T2 are also shown. All NMR spectra were recorded at room temperature in CDCl3. | ||
Conclusions
As demonstrated herein, remarkable self-sorting behavior of high selectivity takes place in the formation of imine-based macrobicyclic cryptand-type organic cages from mixtures of different dialdehyde components reacting with the same capping triamine (see also related processes in ref. 42–53). The structure of the components (their electronic properties, flexibility, presence of a heteroatom, etc.) plays a decisive role in the selective formation of given polyimine cages out of the different structural variations that may be generated from a mixture of very similar dialdehyde building blocks. Structural exploration supports the conclusion that the presence of a heteroatom in a simple aromatic unit favors the cage formation. The results point to the operation of different structural factors comprising attractive H-bonding interactions and electrostatic effects between the imine groups and heteroatomic centers as well as the generation of a conjugated system. Some degree of flexibility and hindrance to optimization of electronic interactions reduce the effectiveness of a component in the self-sorting process. Furthermore, once formed, a given cage may be kinetically trapped, thus forming an out-of-equilibrium mixture. However, in the presence of an agent capable of breaking the imine bonds in the hexaimine structure, exchange of components becomes possible, as expected from the dynamic covalent chemistry of imines, resulting in the generation of the equilibrium state. Developments towards the kinetic behavior of the self-sorting processes, the generation of kinetically trapped out-of-equilibrium states and the encapsulation of substrate molecules may be envisaged.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the ERC (Advanced Research Grant SUPRADAPT 290585), the ANR DYNAFUN grant No. ANR-15-CE29-0009-01 and the University of Strasbourg. Financial support was also provided by the National Science Centre in Poland (MK, grant PRELUDIUM UMO-2017/25/N/ST5/00942) and the National Centre for Research and Development (ARS, grant LIDER/024/391/L-5/13/NCBR/2014). We thank Li-Yuan Huang (Strasbourg, Guangzhou) for related work towards polyimine cage compounds. We also thank Prof. Jack Harrowfield for helpful discussions.Notes and references
- B. Dietrich, J. M. Lehn and J. P. Sauvage, Tetrahedron Lett., 1969, 10, 2885–2888 CrossRef.
- B. Dietrich, J. M. Lehn, J. P. Sauvage and J. Blanzat, Tetrahedron, 1973, 29, 1629–1645 CrossRef CAS.
- J. M. Lehn, Acc. Chem. Res., 1978, 11, 49–57 CrossRef CAS.
- L. Fabbrizzi, Cryptands and Cryptates, World Scientific Publishing Company, 2017 Search PubMed.
- P. Ballester, M. Fujita and J. Rebek, Chem. Soc. Rev., 2015, 44, 392–393 RSC.
- D. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 532–546 RSC.
- D. Ajami, L. Liu and J. Rebek Jr, Chem. Soc. Rev., 2015, 44, 490–499 RSC.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- G. Markiewicz, A. Jenczak, M. Kołodziejski, J. J. Holstein, J. K. M. Sanders and A. R. Stefankiewicz, Nat. Commun., 2017, 8, 15109 CrossRef PubMed.
- J. Jazwinski, J.-M. Lehn, D. Lilienbaum, R. Ziessel, J. Guilhem and C. Pascard, J. Chem. Soc., Chem. Commun., 1987, 1691–1694 RSC.
- R. Abidi, F. Arnaud-Neu, M. G. B. Drew, S. Lahely, D. Marrs, J. Nelson and M.-J. Schwing-Weill, J. Chem. Soc., Perkin Trans. 2, 1996, 2747–2755 RSC.
- D. MacDowell and J. Nelson, Tetrahedron Lett., 1988, 29, 385–386 CrossRef CAS.
- D. J. Marrs, V. McKee, J. Nelson, Q. Lu and C. J. Harding, Inorg. Chim. Acta, 1993, 211, 195–202 CrossRef CAS.
- Q. Lu, J.-M. Latour, C. J. Harding, N. Martin, D. J. Marrs, V. McKee and J. Nelson, J. Chem. Soc., Dalton Trans., 1994, 1471–1478 RSC.
- J.-M. Lehn, J.-P. Vigneron, I. Bkouche-Waksman, J. Guilhem and C. Pascard, Helv. Chim. Acta, 1992, 75, 1069–1077 CrossRef CAS.
- D. McDowell, J. Nelson and V. McKee, Polyhedron, 1989, 8, 1143–1145 CrossRef CAS.
- O. Kocian, R. J. Mortimer and P. D. Beer, Tetrahedron Lett., 1990, 31, 5069–5072 CrossRef CAS.
- R. Menif, A. E. Martell, P. J. Squattrito and A. Clearfield, Inorg. Chem., 1990, 29, 4723–4729 CrossRef CAS.
- J. de Mendoza, E. Mesa, J.-C. Rodríguez-Ubis, P. Vázquez, F. Vögtle, P.-M. Windscheif, K. Rissanen, J.-M. Lehn, D. Lilienbaum and R. Ziessel, Angew. Chem., Int. Ed. Engl., 1991, 30, 1331–1333 CrossRef.
- V. McKee, J. Nelson and R. M. Town, Chem. Soc. Rev., 2003, 32, 309–325 RSC.
- J.-M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 RSC.
- S. O. J. N. H. Reek, Dynamic Combinatorial Chemistry, Wiley-VCH, Weinheim, 2010 Search PubMed.
- B. L. Miller, Dynamic Combinatorial Chemistry, Wiley, Chichester, 2010 Search PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- Dynamic Covalent Chemistry: Principles, Reactions, and Applications, ed. W. Zhang and J. Yinghua, Wiley-VCH, Weinheim, 2018 Search PubMed.
- J.-M. Lehn, in Constitutional Dynamic Chemistry, ed. M. Barboiu, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–32 Search PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- R. Kramer, J. M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394–5398 CrossRef CAS.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- K. Osowska and O. Š. Miljanić, Synlett, 2011, 2011, 1643–1648 CrossRef.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- A. S. Singh and S.-S. Sun, Chem. Commun., 2012, 48, 7392–7394 RSC.
- K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef CAS PubMed.
- W. Drożdż, M. Kołodziejski, G. Markiewicz, A. Jenczak and A. Stefankiewicz, Int. J. Mol. Sci., 2015, 16, 16300 CrossRef PubMed.
- M. A. Squillaci, G. Markiewicz, A. Walczak, A. Ciesielski, A. R. Stefankiewicz and P. Samorì, Chem. Commun., 2017, 53, 9713–9716 RSC.
- K. Tahara, T. Fujita, M. Sonoda, M. Shiro and Y. Tobe, J. Am. Chem. Soc., 2008, 130, 14339–14345 CrossRef CAS PubMed.
- T. Jiao, G. Wu, L. Chen, C.-Y. Wang and H. Li, J. Org. Chem., 2018, 83, 12404–12410 CrossRef CAS PubMed.
- G. Zhu, Y. Liu, L. Flores, Z. R. Lee, C. W. Jones, D. A. Dixon, D. S. Sholl and R. P. Lively, Chem. Mater., 2018, 30, 262–272 CrossRef CAS.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276 CrossRef CAS PubMed.
- K. Ono and N. Iwasawa, Chem.–Eur. J., 2018, 24, 1–14 CrossRef.
- M. G. B. Drew, V. Felix, V. McKee, G. Morgan and J. Nelson, Supramol. Chem., 1995, 5, 281–287 CrossRef CAS.
- M. G. B. Drew, D. Marrs, J. Hunter and J. Nelson, J. Chem. Soc., Dalton Trans., 1992, 11–18 RSC.
- V. McKee, W. T. Robinson, D. McDowell and J. Nelson, Tetrahedron Lett., 1989, 30, 7453–7456 CrossRef CAS.
- N. Giri, C. E. Davidson, G. Melaugh, M. G. Del Pópolo, J. T. A. Jones, T. Hasell, A. I. Cooper, P. N. Horton, M. B. Hursthouse and S. L. James, Chem. Sci., 2012, 3, 2153–2157 RSC.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- P. Kieryk, J. Janczak, J. Panek, M. Miklitz and J. Lisowski, Org. Lett., 2016, 18, 12–15 CrossRef CAS PubMed.
- X.-Y. Hu, W.-S. Zhang, F. Rominger, I. Wacker, R. R. Schroder and M. Mastalerz, Chem. Commun., 2017, 53, 8616–8619 RSC.
- J. C. Lauer, W.-S. Zhang, F. Rominger, R. R. Schröder and M. Mastalerz, Chem.–Eur. J., 2018, 24, 1816–1820 CrossRef CAS PubMed.
- K. Acharyya, S. Mukherjee and P. S. Mukherjee, J. Am. Chem. Soc., 2013, 135, 554–557 CrossRef CAS PubMed.
- K. Acharyya and P. S. Mukherjee, Chem.–Eur. J., 2014, 20, 1646–1657 CrossRef CAS PubMed.
- P. Skowronek, B. Warzajtis, U. Rychlewska and J. Gawronski, Chem. Commun., 2013, 49, 2524–2526 RSC.
- P. Skowronek and J. Gawronski, Org. Lett., 2008, 10, 4755–4758 CrossRef CAS PubMed.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed.
- C. J. Pugh, V. Santolini, R. L. Greenaway, M. A. Little, M. E. Briggs, K. E. Jelfs and A. I. Cooper, Cryst. Growth Des., 2018, 18, 2759–2764 CrossRef CAS.
- T. Hasell, X. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed.
- D. Xu and R. Warmuth, J. Am. Chem. Soc., 2008, 130, 7520–7521 CrossRef CAS PubMed.
- N. Christinat, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2008, 47, 1848–1852 CrossRef CAS PubMed.
- A. R. Stefankiewicz, M. R. Sambrook and J. K. M. Sanders, Chem. Sci., 2012, 3, 2326–2329 RSC.
- W. Drożdż, C. Bouillon, C. Kotras, S. Richeter, M. Barboiu, S. Clément, A. R. Stefankiewicz and S. Ulrich, Chem.–Eur. J., 2017, 23, 18010–18018 CrossRef PubMed.
- A. R. Stefankiewicz and J. K. M. Sanders, Chem. Commun., 2013, 49, 5820–5822 RSC.
- M. von Delius, Synlett, 2016, 27, 177–180 CrossRef CAS.
- R.-C. Brachvogel, F. Hampel and M. von Delius, Nat. Commun., 2015, 6, 7129 CrossRef PubMed.
- R.-C. Brachvogel and M. von Delius, Chem. Sci., 2015, 6, 1399–1403 RSC.
- R.-C. Brachvogel, H. Maid and M. von Delius, Int. J. Mol. Sci., 2015, 16, 20641 CrossRef CAS PubMed.
- M. W. Schneider, H.-J. Siegfried Hauswald, R. Stoll and M. Mastalerz, Chem. Commun., 2012, 48, 9861–9863 RSC.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of obtained compounds, NMR spectra, CCDC deposition numbers. CCDC 1853998 and 1853999. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04598d |
| This journal is © The Royal Society of Chemistry 2019 |