
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigations on non-classical silylium ions leading to a cyclobutenyl cation†
Arthur
Martens
 ,
Marvin
Kreuzer
,
Alexander
Ripp
,
Marius
Schneider
,
Daniel
Himmel
,
Harald
Scherer
and
Ingo
Krossing
*
,
Marvin
Kreuzer
,
Alexander
Ripp
,
Marius
Schneider
,
Daniel
Himmel
,
Harald
Scherer
and
Ingo
Krossing
*
Institut für Anorganische und Analytische Chemie, Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
First published on 10th January 2019
Abstract
Instead of yielding the desired non-classical silylium ions, the reactions of different alkenes/alkynes with several [Me3Si]+ sources mostly led to oligomerization, or – in the presence of Me3SiH – hydrosilylation of the alkenes/alkynes. Yet, from the reaction of 2-butyne with ion-like Me3Si–F–Al(ORF)3 (RF = C(CF3)3) the salt of the silylated tetramethyl cyclobutenyl cation [Me4C4–SiMe3]+[al–f–al]−1 ([al–f–al]− = [(RFO)3Al–F–Al(ORF)3]−) was obtained in good yield (NMR, scXRD, Raman, and IR). All the experimental and calculated evidence suggest a mechanism in which 1 was formed via a non-classical silylium ion as an intermediate. The removal of the [Me3Si]+ moiety from the cation in 1 was investigated as a means to provide free tetramethyl cyclobutadiene (CBD). However, the addition of [NMe4]F, in order to release Me3SiF and form CBD, led to the unexpected deprotonation of the cation. The addition of 4-dimethylaminopyridine to remove the [Me3Si]+ cation as a Lewis acid/base adduct, led to an adduct with the four-membered ring in the direct neighborhood of the Me3Si group. By the addition of Et2O to a solution of 1, the [F–Al(ORF)3]− anion (and Et2O–Al(ORF)3) was generated from the [al–f–al]− counterion. Subsequently, the [F–Al(ORF)3]− anion abstracted the [Me3Si]+ moiety from [Me4C4–SiMe3]+, probably releasing CBD. However, due to the immediate reaction of CBD with [Me4C4–SiMe3]+ and subsequent oligomerization, it was not possible to use CBD in follow-up chemistry.
Introduction
Carbocations play an important role as intermediates, e.g. in SN1 reactions or in Wagner–Meerwein rearrangements.1 The classical carbenium ions, e.g. [C(CH3)3]+, feature a tricoordinate electron deficient carbon atom with 2e–2c bonds.2 While their existence has long been accepted, there has been a long dispute about the existence of the (pentacoordinate) non-classical carbonium ions.3 Only in 2013 the final crystal structure evidence for the existence of non-classical carbocations was provided by our group.4 Motivated by this result, we were interested to see if analogous non-classical silylium ions also exist in the condensed phase, i.e. silylium ions stabilized by a 3c–2e bond (Scheme 1a and b). Although adducts of [Me3Si]+ with ethylene and acetylene were detected in the gas phase by mass spectrometry, their structures in condensed phases hitherto remain unknown.5–7 Yet, quantum-chemical calculations suggest a non-classical structure for adducts between [Me3Si]+ and C2H4/C2H2. However, it appears that the latter are only intermediates and react further by a methyl shift and formation of a vinyl silylium ion (Scheme 1c).6,7 By contrast, with substituted alkenes, classical carbenium ions as in Scheme 1b were calculated to be favored.6 Adducts of silylium ions with benzene or toluene could also be considered to possess non-classical structures, but crystallographic studies and also quantum-chemical calculations suggest a classical structure.8,9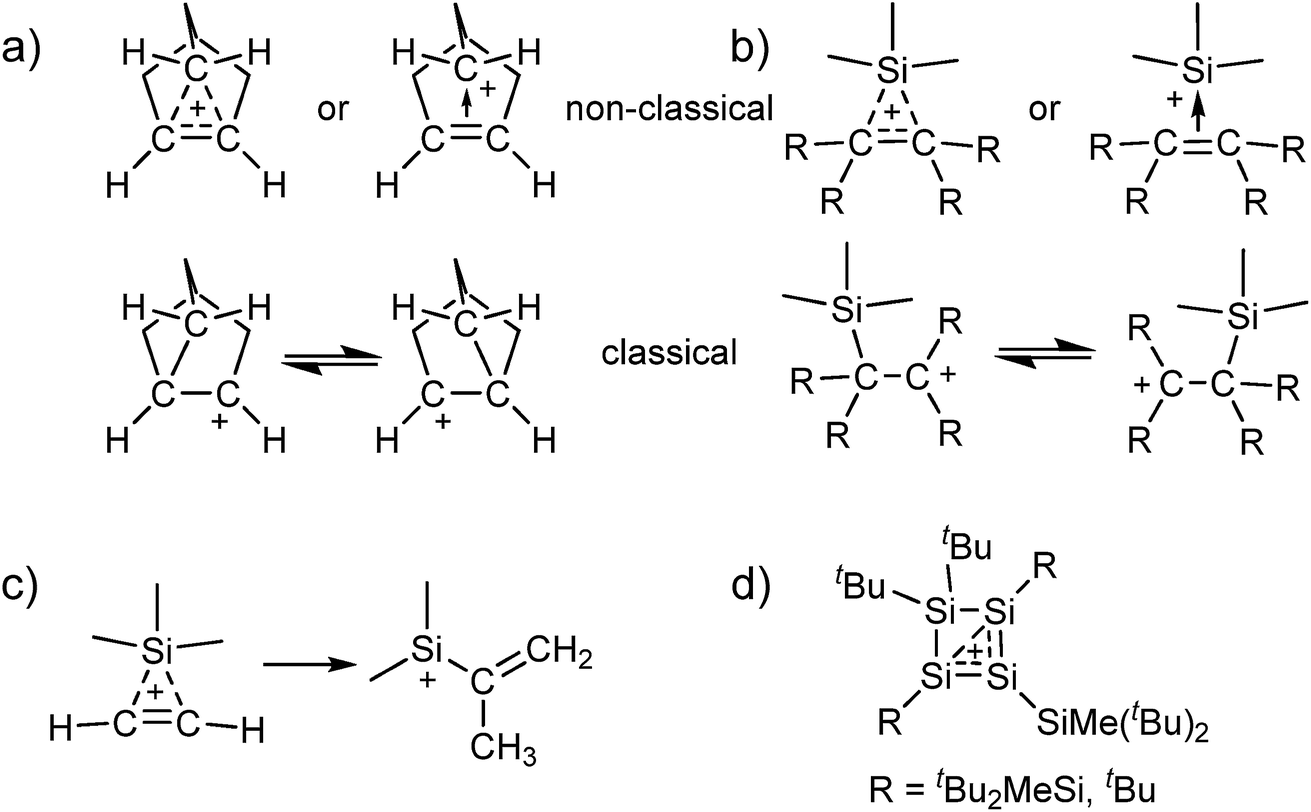 | ||
| Scheme 1 Classical and non-classical structures of (a) the 2-norbornyl cation and (b) analogous silylium ions (although we refer to “classical silylium ions” here, these are rather to be seen as carbenium ions stabilized by the β-Si effect); (c) the calculated methyl shift for the [Me3Si(C2H2)]+ cation;7 (d) the reaction products of silylium ions with di- and trisilenes.10,11 | ||
The all-silicon analogue to the non-classical carbonium ions would be a silylium ion, which is coordinated by a disilene R2Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiR2 or by a (formal) disilyne RSi
SiR2 or by a (formal) disilyne RSi![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiR. To the best of our knowledge, for the [Si3R5]+ cations no experimental data exist and computational analyses are limited to thermodynamics and do not discuss structural properties.12 Reactions of silylium ions with di- and trisilenes yielded cyclotetrasilenylium ions [(RSi)3SiR2]+ as part of more complicated rearrangement reactions (Scheme 1d; R = tBu2MeSi, tBu).10,11 In any event, the stabilization of disilenes and disilynes against oligomerization requires large substituents R, which hinder their – classical or non-classical – coordination to a silylium ion.10,11,13 Although calculations at the MP2/def2-TZVPP level suggest a non-classical adduct between [Me3Si]+ and the sterically unhindered Me2SiSiMe2, this disilene would not be isolable due to the discussed oligomerization. Therefore, we set out to synthesize non-classical silylium ions by reaction of sources of the small [Me3Si]+ silylium ion with alkenes or alkynes. With small substituents, their π bond is more accessible than that in sterically hindered room temperature stable disilenes and disilynes. Additionally, alkenes and alkynes would allow for a homogeneous delocalization of the positive charge among the carbon atoms, which would require more reorganization for the disilenes/disilynes due to their trans-bent structure. However, the fluoride ion affinity (FIA), as a measure of Lewis acidity,14 of silylium ions is significantly higher than that of carbenium ions (FIA = 952 vs. 836 kJ mol−1 for [Me3E]+; E = Si, C; calculated like in ref. 15; BP86/def-SV(P)). As a result, the addition of an alkene or alkyne to a silylium ion may also result in the formation of a carbenium ion that is stabilized in the classical structure by the so-called β-Si effect. This implies a hyperconjugative stabilization due to electron density transfer from the occupied σ(Si–C) orbital into the empty p-orbital of the cationic C atom (Scheme 1b).
SiR. To the best of our knowledge, for the [Si3R5]+ cations no experimental data exist and computational analyses are limited to thermodynamics and do not discuss structural properties.12 Reactions of silylium ions with di- and trisilenes yielded cyclotetrasilenylium ions [(RSi)3SiR2]+ as part of more complicated rearrangement reactions (Scheme 1d; R = tBu2MeSi, tBu).10,11 In any event, the stabilization of disilenes and disilynes against oligomerization requires large substituents R, which hinder their – classical or non-classical – coordination to a silylium ion.10,11,13 Although calculations at the MP2/def2-TZVPP level suggest a non-classical adduct between [Me3Si]+ and the sterically unhindered Me2SiSiMe2, this disilene would not be isolable due to the discussed oligomerization. Therefore, we set out to synthesize non-classical silylium ions by reaction of sources of the small [Me3Si]+ silylium ion with alkenes or alkynes. With small substituents, their π bond is more accessible than that in sterically hindered room temperature stable disilenes and disilynes. Additionally, alkenes and alkynes would allow for a homogeneous delocalization of the positive charge among the carbon atoms, which would require more reorganization for the disilenes/disilynes due to their trans-bent structure. However, the fluoride ion affinity (FIA), as a measure of Lewis acidity,14 of silylium ions is significantly higher than that of carbenium ions (FIA = 952 vs. 836 kJ mol−1 for [Me3E]+; E = Si, C; calculated like in ref. 15; BP86/def-SV(P)). As a result, the addition of an alkene or alkyne to a silylium ion may also result in the formation of a carbenium ion that is stabilized in the classical structure by the so-called β-Si effect. This implies a hyperconjugative stabilization due to electron density transfer from the occupied σ(Si–C) orbital into the empty p-orbital of the cationic C atom (Scheme 1b).
Results and discussion
Before turning to the experiments, we investigated the principle feasibility of the planned reactions by assessing the cations sought for with DFT and ab initio calculations and including solvation energies for the polar and weakly basic solvent ortho-difluorobenzene (o-DFB, εr = 13.4).16Preliminary assessment of the stabilities of non-classical silylium ions
Calculations on the reaction of the free [Me3Si]+ cation with different alkenes and alkynes were performed at the MP2/def2-TZVPP level of theory according to eqn (1) and (2) (Table 1).| [Me3Si]+ + RCCR → [Me3Si(RCCR)]+ (R = H, Me, Ph) | (1) |
| [Me3Si]+ + R2CCR2 → [Me3Si(R2CCR2)]+ (R = H, Me, Ph) | (2) |
| Ligand L | ΔrH0gas/ΔrG0gas | C–C–Si/° | Gas phase structure |
|---|---|---|---|
| (ΔG0o-DFB) | d SiC/pm | ||
| a Experimental data from ref. 8. b Cipso and Cortho were used for the measurements. | |||
| H2CCH2 |
−109/−60 | 73.4 |

|
| (−38) | 236.7 | ||
| Me2CCMe2 |
−174/−113 | 72.9 |

|
| (−56) | 235.6 | ||
| Ph2CCPh2 |
−143/−67 | 93.8/54.6 |

|
| (+20) | 221.6/271.4 | ||
| HCCH |
−98/−53 | 78.3/71.6 |

|
| (−28) | 231.0/238.4 | ||
| MeCCMe |
−157/−103 | 74.3 |

|
| (−58) | 228.3 | ||
| PhCCPh |
−188/−132 | 74.3 |

|
| (−52) | 229.5 | ||
| C6H6a | −134/−86 | 98.3/51.3b |
|
| (−39) | 217.3/275.8b | ||
For comparison, the structure of the (classical) benzene complex [Me3Si(C6H6)]+ is also included in Table 1. In order to evaluate whether the calculated molecules are non-classical silylium ions or classical carbenium ions, we analyzed both relevant C–C–Si bond angles. For a classical carbenium ion, one of these angles is expected to be larger than at least 90°, while for non-classical silylium ions both angles should be (almost) equal and smaller than 90°. The complex [Me3Si(Ph2CCPh2)]+ was calculated to be a classical carbenium ion stabilized by the β-Si effect with a C–C–Si angle of 93.8°, possibly due to the resonance of the phenyl moieties and also for steric reasons. The C–C–Si angles in the H2CCH2 (73.4°), Me2CCMe2 (72.9°), MeCMe (74.3°) and PhCCPh (74.3°) complexes combined with the symmetric C–Si distances suggest a non-classical structure for these cations. Regarding its structure, the [Me3Si(HCCH)]+ cation is a special case with asymmetric C–C–Si angles of 78.3° and 71.6°, most likely induced by steric repulsion of the H-atom with one methyl group. Yet, we will refer to its structure as being non-classical. It should be noted that the non-classical structure of this cation with symmetric C–C–Si angles is disfavored by only ΔG0gas = 0.01 kJ mol−1 and therefore these two structures would be expected to be indistinguishable. The structures calculated at the simpler BP86-D3(BJ)/def-TZVP level of theory are similar, except for [Me3Si(PhCCPh)]+. Here, the PhCCPh adduct is calculated to be a classical carbenium ion. It was not possible to calculate similar classical structures for the other adducts, as these are not even local minima or transition states on the respective energy hypersurface. This was exemplarily verified by calculating the energy of [Me3Si(MeCCMe)]+ dependent on the C–C–Si angle in the range of 60 to 140° (see the ESI† for details). Therefore, classical starting structures also collapse to the non-classical structures. Thus no clear energy difference between a formally classical and a non-classical structure can be given.
The gas phase reaction enthalpies ΔrH0gas and Gibbs energies ΔrG0gas with alkynes become more favorable when replacing the H substituents of the parent alkyne HCCH with Me, and even more so with Ph (MP2/def2-TZVPP). This can be explained by an increased stabilization of the positive charge due to hyperconjugation and resonance, respectively (ΔrH0gas = −98 (H) vs. −157 (Me) and −188 kJ mol−1 (Ph)). For alkenes, the same trend was expected. However, ΔrH0gas of the reaction with Ph2CCPh2 (−143 kJ mol−1) was calculated to be less favored than that with Me2CCMe2 (−174 kJ mol−1). We attribute this to steric reasons, as the phenyl groups cannot be coplanar as in the alkyne case. It should be noted that the non-classical structure of [Me3Si(Ph2CCPh2)]+ with equivalent Si–C distances was calculated to be a transition state for the [Me3Si]+ migration between both carbon atoms. This transition state is higher in energy than the calculated minimum structure by only ΔG0o-DFB = +7 kJ mol−1 (ΔG0gas = +11 kJ mol−1). Therefore, it might not be possible to differentiate between the classical and non-classical structure by NMR spectroscopy (coalescence) or single crystal X-ray diffraction (dynamic disorder).
Including COSMO17 Gibbs solvation energies, all calculated reaction energies become less favored and the reaction of [Me3Si]+ with the larger PhCCPh was also calculated to be less exergonic than that with the smaller MeCCMe. This is attributed to the fact that smaller ions are generally better solvated than larger ones. Since upon reaction with [Me3Si]+ the product cation always increases in size, reactions (1) and (2) become less favored the larger the ligand. Due to this effect, the formation of [Me3Si(Ph2CCPh2)]+ was calculated to be endergonic in solution by ΔG0o-DFB = +20 kJ mol−1.
Reactions of Me3Si–F–Al(ORF)3 with alkenes and alkynes
Since the calculations suggested that the formation of non-classical silylium ions should be possible, in part also in solution, we turned towards their synthesis. Ethylene and acetylene are gases and only weakly bound to [Me3Si]+ (cf.Table 1). Therefore, we performed the reactions of Me3Si–F–Al(ORF)3 with the substituted compounds R2CCR2 and RCCR (R = Me, Ph). However, the reactions with Me2CCMe2, Ph2CCPh2 and PhCCPh in o-DFB or CH2Cl2 and at r.t. or −40 °C did not yield any silylium ions, but only led to exergonic oligomerization of the alkenes and alkynes. However, this signals activation of the unsaturated hydrocarbons by Me3Si–F–Al(ORF)3. By contrast, reactions with MeCCMe in o-DFB or CH2Cl2 at various temperatures (−40 °C to r.t.; eqn (3)), reproducibly gave a 28% yield of [Me4C4–SiMe3]+[al–f–al]−1 ([al–f–al]− = [(RFO)3Al–F–Al(ORF)3]−; NMR, scXRD, Raman, IR) after crystallization. This compound will be discussed later in more detail.
Reactions of [Ph3C]+[al–f–al]− with Me3SiH and alkenes/alkynes
By exchanging Me3Si–F–Al(ORF)3 for more reactive silylium ions, we hoped to form the desired products faster and thereby prevent oligomerization. In order to generate the [Me3Si]+ cation as an intermediate, we mixed the starting materials (liquid alkenes or alkynes) with [Ph3C]+[al–f–al]− in the absence of solvent. An excess of Me3SiH was condensed onto this mixture to generate silylium ions or their Me3SiH adducts18in situ and allowed to warm to r.t. Although NMR spectra of these reaction mixtures in CD2Cl2 showed complete conversion of the starting materials, the non-classical silylium ions could not be detected. Instead, hydrosilylation of PhCCPh and Me2CCMe2 (see the ESI† for details) and formation of neutral 2 for MeCCMe were observed (eqn (4)). Upon reaction of MeCCMe with [Ph3C]+[al–f–al]− and Me3SiH, presumably 1 was formed, which then reacted with excess Me3SiH to yield 2 and [Me3Si(solv)]+ (solv = Me3SiH, CH2Cl2). Only after complete conversion of the alkenes and alkynes, the anion was attacked and decomposed.19 It should be noted that the evolving [Me3Si–H–SiMe3]+[al–f–al]− is not isolable as a pure material. Yet, the [al–f–al]− anion was shown to be fairly stable against silylium ions in CH2Cl2.19
Reactions of bromo-ethyl/vinyl silanes with Ag+[al–f–al]−
In order to prevent oligomerization of the alkenes and alkynes, we reacted bromo-ethyl and bromo-vinyl silanes with Ag+[al–f–al]− in CH2Cl2 at −40 °C, respectively. The abstraction of Br− from these silanes by Ag+ should result in the formation of ethylene/acetylene adducts of [Me3Si]+ (cf. eqn (5a) for the bromo-ethyl example). Here the reaction with a second equivalent of an alkene/alkyne is not possible and the non-classical silylium ions should become isolable. Upon reaction, no solid AgBr visibly formed. Nevertheless, NMR spectra of these reactions did not show signals of the starting silanes, but of Me3SiBr in both cases. This signal is slightly shifted to a lower field by 2 ppm and suggests a coordination of Me3SiBr to Ag+.19 It seems likely that the non-classical silylium ions [Me3Si(C2H4/C2H2)]+ were generated in situ (eqn (5a)). However, apparently these silylium ions then reacted further with the (solvated) AgBr under the formation of [(Me3SiBr)Ag(solv)x]+ and release of C2H4/C2H2 (see eqn (5b) for the ethene example; the evolving C2H4 could be identified by gas phase IR spectroscopy).20 Similar silver–halosilane adducts were observed and structurally characterized upon reaction of tBu3SiBr with Ag+[al–f–al]− – although halide abstraction and AgBr formation were desired.19 Additionally, partial decomposition of the anion and formation of Me3SiF and Me3SiCl were observed, which is further evidence for the generation of silylium ions.
Formation of [Me3Si(MeC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CMe)]+vs. [Me4C4–SiMe3]+
CMe)]+vs. [Me4C4–SiMe3]+
The reactions of 2-butyne with Me3Si–F–Al(ORF)3 and silylium ions only yielded 1 and 2, respectively. Both compounds were formed from two 2-butyne units. This led to the question of whether it is at least theoretically possible to isolate the desired silylium ion [Me3Si(MeCCMe)]+ by these routes, or if such non-classical ions reside in shallow potential wells that allow for a simple and low-barrier subsequent reaction with MeCCMe to give 1. Therefore, we calculated the reaction mechanism for the formation of [Me4C4–SiMe3]+ (P-4) at the BP86-D3(BJ)/def-TZVP level of theory (Fig. 1). In order to reduce computational cost, we started from 2-butyne and [Me3Si]+, instead of Me3Si–F–Al(ORF)3.
 | ||
| Fig. 1 Calculated reaction path, geometries and relative energies in kJ mol−1 for the formation of [Me4C4–SiMe3]+ (P-4, black) and the 5-membered ring (P-5, red); BP86-D3(BJ)/def-TZVP. | ||
Since 2-butyne itself does not dimerize under standard conditions,21 the silylation of 2-butyne can be assumed to be the first step in the formation of [Me4C4–SiMe3]+. The Gibbs activation energy ΔG≠ (TS1) for this exothermic process was calculated to be only 10 kJ mol−1. The silylated 2-butyne (I1) then reacts with a second equivalent of 2-butyne to exothermically form the intermediate I2. We were not able to determine the Gibbs activation energy (TS2) for this reaction with the method used. This indicates that this barrier is either very small or not existent at all. Based on thermodynamics, this intermediate should rearrange to yield a 5-membered ring (P-5). However, the barrier for this rearrangement (TS3-5) was calculated to be 45 kJ mol−1 – which is almost triple the barrier (TS3-4) for the formation of the observed [Me4C4–SiMe3]+ (P-4, 16 kJ mol−1). Thus, the calculations are in agreement with the experimental observation and suggest that P-4 (=1) is the kinetic product.
When Me3Si–F–Al(ORF)3 is used for the silylation of 2-butyne, the Gibbs activation energy (transition state similar to TS1) for this reaction is expected to be higher, due to its lower reactivity compared to [Me3Si]+. However, this will not change the entire picture. In conclusion it seems impossible to isolate the silylated 2-butyne (I1) by use of Me3Si–F–Al(ORF)3 or even (non-existent) free [Me3Si]+, as the reaction with the second molecule of 2-butyne appears to be much faster than the silylation.
Properties of [Me4C4–SiMe3]+[al–f–al]−
From the reaction solution in o-DFB at r.t. single crystals of 1 were obtained by the addition of less polar CH2Cl2. The molecular cation structure of salt 1 is shown in Fig. 2. With Si–C1–C2/C2′ angles being 113°–118°, the stabilization of this cation by the β-silicon effect is not evident. Otherwise, these angles would be expected to lie around 90°.22 | ||
| Fig. 2 Molecular structure of the cation in [Me4C4–SiMe3][al–f–al] (1) with thermal ellipsoids set at 50% probability level. The [al–f–al]− anion was omitted for clarity. Scheme: Si (yellow), C (grey), H (white). Selected distances (pm), bond angles (deg), and torsion angles (deg): Si–C1 192.2(2), C1–C2 151.8(2), C1–C2′ 152.6(2), C2–C3 138.9(2), C2′–C3 138.5(2), C2–C2′ 179.4(2), Si–C1–C2 117.9(1), Si–C1–C2′ 113.0(1), C2–C1–C2′ 72.2(1), C2–C3–C2′ 80.6(1), and C1–C2–C3–C2′ 25.5(1). | ||
This cation can be seen as a silylated cyclobutadiene and is closely related to the already known homoaromatic [R4C4–H]+, [R4C4–Cl]+, [tBu4C4–OH]+ and the neutral [R4C4–AlX3] (R = H, Me, tBu, Ph; X = Cl, Br).23–29 Of these, the protonated cations [R4C4–H]+ with R = H, Me, Ph were only characterized by NMR at −40 to −70 °C,27 while with R = tBu they were shown to be isolable with a large variety of anions, like Br− or [SbF6]−.26 Also the chlorinated and hydroxylated cations [Ph4C4–Cl]+ and [tBu4C4–OH]+ and the neutral [Me4C4–AlCl3] were shown to be stable at r.t. and could be isolated.23,24,26,29
A comparison of selected structural parameters, calculated NPA and PABOON partial charges and π(*)(C2–C3–C2′)-orbital energies of the compounds is found in Table 2. In order to allow for a better comparison, these calculations were performed on the methyl-substituted derivatives for all compounds. The [(RSi)3SiR2]+ cations (R = tBu2MeSi, tBu) can also be seen as silicon analogues of these compounds but will not be discussed.10,11 When comparing the convolution angles (C1–C2–C2′–C3) of 1, [tBu4C4–H]+, [Ph4C4–Cl]+, [tBu4C4–OH]+ and [Me4C4–AlX3], their absolute values are relatively similar and range between 37.3° and 31.5°, except for [Ph4C4–Cl][Nb2OCl9] with a dihedral angle of only 4.3°. However, the dihedral angles in 1 and [Me4C4–AlX3] are positive, while in [tBu4C4–H]+, [tBu4C4–OH]+ and [Ph4C4–Cl]+ they are negative. This is a consequence of the (C2–)Me groups being bent, which leads to repulsion of the sterically most demanding group. The relatively small dihedral angle in [Ph4C4–Cl]+ results from resonance of the phenyl moieties with the (C2–C2′) orbital, which is an intermediate between a σ and a π orbital. This leads to an increased C2–C2′ distance compared to that in the other compounds (203 vs. ∼180 pm).27,30
| 1 | [tBu4aC4–H]+ (ref. 26) | [Ph4C4–Cl]+ (ref. 23) | [tBu4C4–OH]+ (ref. 26) | [Me4C4–AlCl3] (ref. 31) | [Me4C4–AlBr3] | Me4C4 | |
|---|---|---|---|---|---|---|---|
| a HOMO/LUMO+1. b HOMO−2/LUMO+1. c HOMO−1/LUMO+1. d HOMO−6/LUMO+1. e HOMO/LUMO. | |||||||
| Bond distances (pm) and dihedral angles (deg) | |||||||
| d(C1–C2) | 151.8/152.6 (152.7) | 152.4 (153.4) | 152.9/154.4 (153.2) | 152.3/153.8 (154.2) | 151.0 (151.5) | (151.6) | (159.1) |
| d(C2–C2′) | 179.4 (183.2) | 180.6 (187.5) | 203.3 (195.5) | 183.3 (190.7) | 177.4 (183.1) | (184.4) | (208.5) |
| d(C2–C3) | 138.5/138.9 (140.2) | 140.7 (140.2) | 138.7/140.4 (140.6) | 139.4/139.6 (140.2) | 138.7 (140.2) | (140.2) | (134.7) |
| C1–C2–C2′–C3 | 31.7 (29.3) | −37.3 (−27.7) | −4.3 (−22.1) | −36.3 (−26.2) | 31.5 (28.5) | (27.3) | (0.0) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| NPA/PABOON partial charges for the methyl derivatives [Me 4 C 4 –EY x ] (+) | |||||||
| δ(C1) | (−0.43/−0.14) | (−0.27/−0.05) | (−0.09/0.09) | (0.26/0.15) | (−0.52/−0.14) | (−0.54/−0.13) | (0.00/0.01) |
| δ(C2) | (0.27/0.21) | (0.28/0.19) | (0.30/0.15) | (0.17/0.15) | (0.27/0.27) | (0.28/0.27) | (0.00/0.01) |
| δ(C3) | (0.00/−0.02) | (0.02/0.01) | (−0.01/0.01) | (0.02/0.03) | (−0.04/−0.05) | (−0.05/−0.05) | (0.00/0.01) |
| δ(C1 + 2 × C2 + C3) | (0.11/0.26) | (0.31/0.34) | (0.50/0.40) | (0.62/0.48) | (−0.02/0.35) | (−0.03/0.36) | (−0.01/0.04) |
| δ(Me4C4) | (0.51/0.82) | (0.75/0.90) | (1.00/1.03) | (1.16/1.01) | (0.25/0.67) | (0.25/0.67) | (0.00/0.00) |
| δ(E) | (0.49/0.18) | (0.25/0.10) | (0.00/−0.03) | (−0.16/−0.01) | (−0.25/−0.67) | (−0.25/−0.67) | (—) |
|
|||||||
| Energies of the (anti-)bonding π(C2–C3–C2′)-orbitals of the methyl derivatives [Me 4 C 4 –EY x ] (+) | |||||||
| E π*(C2–C3–C2′) | (−5.60)a | (−6.04)a | (−6.14)b | (−6.02)c | (−1.92)d | (−1.92)d | (−1.70)e |
| E π(C2–C3–C2′) | (−10.36)a | (−11.75)a | (−12.12)b | (−11.90)c | (−7.33)d | (−7.04)d | (−3.74)e |
When looking at the bonding π(C2–C3–C2′) orbitals of the methylated derivatives of the discussed compounds (Table 2), a correlation between their energies and the partial charge of the Me4C4 moiety is evident, with a higher partial charge leading to lower orbital energies. From a frontier orbital point of view, this implies that the cations [Me4C4–SiMe3]+, [R4C4–H]+, [R4C4–Cl]+, and [tBu4C4–OH]+ are more electron-deficient than the neutral [Me4C4–AlX3]. This is also evident when simply looking at the total charge of these molecules. Interestingly, the energies of the π*(C2–C3–C2′) orbitals seem to be less affected by the partial charge of the Me4C4 moiety, but mostly by the total charge of the molecules. Therefore, the energy of this orbital is nearly the same for [Me4C4–AlCl3] and Me4C4, while for the cationic species it is lower by ∼4 eV.
Evaluating the homoaromatic character of [R4C4–E](+)
Since the structural parameters of the discussed compounds are very similar, especially d(C2–C2′) and d(C2–C3), an unambiguous comparison of the C2–C2′ interactions is not possible (except for [Ph4C4–Cl]+, which does not show this interaction). This interaction is a measure of homoaromaticity and can be determined from the difference in NMR chemical shifts between C2/C2′ and C3 (Table 3).26,27 In allylic systems, the terminal C atoms (here C2/C2′) bear a positive charge, resulting in the deshielding of these. The interaction between C2 and C2′ leads to their shielding, combined with the transfer of the positive charge to C3 (deshielding). For homoaromatic systems a negative value for Δ(δ13C(C2) − δ13C(C3)) is expected, while for allylic systems this difference should be positive.26| Cation | δ(C1) | δ(C2/C2′) | δ(C3) | δ(C2) − δ(C3) |
|---|---|---|---|---|
| a Cyclopentenylium cation as reference for an allylic cation. | ||||
| [Me4C4–SiMe3]+ | 66.8 | 166.0 | 170.4 | −4.4 |
| [tBu4C4–OH]+ (ref. 26) | 101.0 | 161.5 | 184.9 | −23.4 |
| [Me4C4–Cl]+ (ref. 27) | 76.0 | 191.5 | 174.4 | +17.1 |
| [Me4C4–AlCl3] (ref. 29) | — | 162.0 | 164.3 | −2.3 |
| [Me4C4–H]+ (ref. 27) | 57.8 | 171.3 | 171.3 | 0.0 |
| [tBu4C4–H]+ (ref. 26) | 78.5 | 156.6 | 196.6 | −40.0 |
| [Ph4C4–H]+ (ref. 27) | 52.5 | 190.0 | 152.3 | +38.6 |
| [H4C4–H]+ (ref. 27) | 54.0 | 133.5 | 187.6 | −54.1 |
| [H6C5–H]+a (ref. 27) | 48.7 | 234.7 | 145.7 | +89.0 |
With [H4C4–H]+ and the cyclopentenylium cation [H6C5–H]+ being the prototypes for homoaromatic and allylic cations, respectively, the homoaromatic character of the discussed compounds and influence of the substituents can be evaluated. Exchanging the H substituents for Me moieties in [R4C4–H]+ leads to hyperconjugation of the C–H bond to C2. As a result, the C2–C2′ interactions are decreased and C2 is deshielded. Therefore the difference between δ13C(C2) and δ13C(C3) is more positive (0.0 vs. −54.1 ppm for [H4C4–H]+). The methylated compounds [Me4C4–SiMe3]+ and [Me4C4–AlCl3] show similar differences in the shielding of C2 and C3 and therefore are considered to have a similar homoaromatic character as [Me4C4–H]+. Only [Me4C4–Cl]+ shows a comparatively high deshielding of C2 due to low homoaromaticity, which is a consequence of the hyperconjugation of the σ(C–Cl) orbital to the (C2–C2′) orbital.
When looking at the C2–C2′ interactions of [tBu4C4–H]+ and [tBu4C4–OH]+, they would be expected to be even weaker than in [Me4C4–H]+ and [Me4C4–Cl]+, respectively, due to the stronger electron-donating properties of the tBu moieties. However, the NMR chemical shifts suggest a significantly increased homoaromatic character. We assign this to the bulkiness of the tBu moieties leading to a repulsion of the C3–tBu and C2–tBu groups. As a result, the C1–C2–C2′–C3 dihedral angle is higher than that for the methylated derivatives (Table 2). Phenyl substituents seem to completely prevent the C2–C2′ interactions due to resonance and charge delocalization onto the phenyl residue(s). This follows from the NMR chemical shifts of [Ph4C4–H]+ and from the structural parameters of [Ph4C4–Cl]+ (dihedral angle and d(C2–C2′)).
Taking a closer look at the dihedral angles in the discussed compounds, a correlation between these and Δ(δ13C(C2) − δ13C(C3)) is evident. The higher the dihedral angle, the stronger the C2–C2′ interaction in the compound. For [H4C4–H]+ this angle was calculated to be 33.5° (BP86-D3(BJ)/def-TZVP), which is in agreement with this thesis. Interestingly, the distances d(C2–C2′) and d(C2–C3) are only slightly affected by these C2–C2′ interactions (Table 2).
Investigations towards release of Me4C4 from 1
While information on the reactivities of most of the discussed compounds is scarce, [R4C4–AlX3] was shown to be a source of cyclobutadienes R4C4 (CBDs) by use of coordinating solvents, like DMSO, due to abstraction of AlCl3.28,32–34 Besides the discussed AlX3 adducts cyclobutene dicarboxylic acid anhydrides and [Fe(CO3)(C4H4)] can also be used for the generation of CBDs.35 CBDs are anti-aromatic and, therefore, unsubstituted CBD undergoes dimerization readily at T > 35 K.36 Thus, small CBDs have to be generated in situ. Only when they bear large substituents, dimerization can be prevented.37By abstraction of the [Me3Si]+ moiety, 1 might be used as a tetramethylcyclobutadiene (Me4C4) donor as well. The advantage of 1 over [Me4C4–AlX3] as a CBD donor would be its increased stability, which allows for storage in a glove box for more than a year. NMR spectra of [Me4C4–AlX3] show partial coalescence of the signals already at 20 °C.29 In contrast, all signals of 1 are resolved, including the 5JH–H coupling of 0.46 Hz. The increased stability very likely results from the higher Lewis acidity of [Me3Si]+ over AlX3 (FIA = 539 vs. 425/438 kJ mol−1, X = Cl/Br). These FIAs were calculated in an environment with the polarity of CH2Cl2 (εr = 8.9) using COSMO,17 in order to account for the different charges of the Lewis acids. The effect is also evident by addition of AlX3 to [Me4C4–AlX3] in order to increase the Lewis acidity, i.e. the formation of [Me4C4–Al2X6]. It shows an increased thermal stability and reduced coalescence compared to [Me4C4–AlX3].29
Reaction with [NMe4]F
Due to the high stability of the Si–F bond, we reacted 1 with [NMe4]F in o-DFB/CH2Cl2 at r.t., in order to generate Me3SiF and Me4C4. However, only negligible amounts of Me3SiF formed. The main products were [NMe4]+[F1+x–Al(ORF)3−x]− (x = 0, 1), HORF and 3 due to deprotonation of 1 (eqn (6a)). The formation of the anions [F1+x–Al(ORF)3−x]− occurs due to the high Lewis acidity of [al–f–al]−.19 The deprotonation of 1 suggested that a non-charged nucleophile, like 4-dimethylaminopyridine (DMAP), could be helpful to prevent deprotonation of the cation.Reaction with DMAP
The reaction of 1 with an excess of DMAP in a mixture of o-DFB and CH2Cl2 at r.t. yielded mainly DMAP–Al(ORF)3, [F–Al(ORF)3]− (reaction with the anion) and the adduct of [Me4C4–SiMe3]+ with DMAP (4, eqn (6b)). We were able to obtain single crystals of both compounds at −40 °C from the reaction solution (see the ESI† for details). The crystal structure and NMR spectra of 4 showed that the cation is obtained as an enantiomeric mixture, with the DMAP and the [Me3Si]+ moieties being in syn-conformation, i.e. in (S,R) and (R,S) configurations, which is attributed to the orientation of the LUMO of 1 (see Fig. S-60†). A similar adduct between Me4C4–AlCl3 and PPhCl2 was already proposed as an intermediate in the synthesis of phosphole oxides.33 Due to the syn-conformation of the DMAP and Me3Si moieties it seemed possible to remove both of them in the form of [DMAP–SiMe3]+ under the release of Me4C4 by gentle warming/heating. However, heating of 4 to 100 °C did not result in the elimination of [DMAP–SiMe3]+, but only led to the formation of 3, [DMAP–H]+[f–al]− and other unidentified products.
Reaction with diethylether
Unexpectedly, one of the minor side products in the reaction of 1 with DMAP was Me3SiF, providing evidence for the reaction of [Me4C4–SiMe3]+ with a fluoride ion source. Since 1 is stable in o-DFB over several days, the Lewis basic [F–Al(ORF)3]− anion ([f–al]−)19 has to be considered as the fluoride ion source. This is in agreement with the findings that the NMR spectra of 1 never showed the presence of [f–al]−, independent of the reaction stoichiometry.38 Therefore, we dissolved 1 in Et2O at r.t., which is known to induce the dissociation of [al–f–al]− into Et2O–Al(ORF)3 and [f–al]−.19 NMR spectra of this reaction mixture revealed the formation of Me3SiF and complete decomposition of 1 (eqn (6c)), but no evidence for the expected dimerization product of Me4C4 could be found.34 Instead, the NMR spectra suggested the formation of oligomerization products. When this reaction was performed in the presence of 2-butyne, partial formation of C6Me6 was observed, similar to the reaction of [Me4C4–AlCl3] with 2-butyne and DMSO.34 Therefore, we assume this reaction is (weak) evidence for the intermediate release of CBD from 1. However, analogous reactions of 1 with other electron-deficient or electron-rich dienophiles (MeO2C–CC–CO2Me, Me3Si–CC–SiMe3) never gave the desired Diels–Alder products,29 but only the known oligomerization products and complete retention of the alkynes.
It should be noted that the reaction solution for the synthesis of 1 also contained C6Me6 and these oligomerization products. The reason for this is that in the first stage of the reaction of Me3Si–F–Al(ORF)3 with 2-butyne the [f–al]− anion is formed. Therefore, 1, [f–al]− and 2-butyne are present in solution at the same time. Thus, 1 readily reacts with [f–al]− upon its initial synthesis, resulting in its decomposition and accounting for the relatively low yield of 28%. Eventually, the formation of [al–f–al]− from [f–al]− and Me3Si–F–Al(ORF)3 suppresses this decomposition reaction.
Rationalization
Why is there such a difference in the reactivity of alkynes with Me4C4 released from 1 and from [Me4C4–AlX3]? Me4C4 is an electron-rich diene (high HOMO energy) and therefore should react with electron-deficient dienophiles (low LUMO energy). While the π-LUMO energy of −1.92 eV for [Me4C4–AlX3] is rather high and similar to that for free Me4C4, it is much lower for 1 with an energy of −5.60 eV (Table 1). For this reason [Me4C4–AlX3] is a poor dienophile and the released Me4C4 alone undergoes Diels–Alder reactions with the added alkynes. In contrast, the π-LUMO energy of even very electron-deficient alkynes, like F3C–CC–CF3 (−2.70 eV), is significantly higher than that of 1. As a result, the evolving Me4C4 reacts with [Me4C4–SiMe3]+ instead of other dienophiles, leading to oligomerization. The same problem is likely to arise when using the other cationic cyclobutenyl cations as their π-LUMO energies are expected to be in the same region as those of 1.
Conclusion
By the reaction of Me3Si–F–Al(ORF)3 with different alkenes and alkynes the synthesis of non-classical silylium ions was investigated. However, most of these reactions only led to oligomerizations in which such silylium ions may be intermediates; they were assessed by calculations (Table 1). Replacing Me3Si–F–Al(ORF)3 with silylium ions, generated in situ from [Ph3C]+[al–f–al]− and Me3SiH, also did not yield the desired non-classical silylium ions. Instead, hydrosilylation of the alkenes and alkynes was observed. Preliminary tests suggest that these hydrosilylation reactions can be performed with only catalytic amounts of [Ph3C]+[al–f–al]−.39 However, this should be investigated in a separate study by specialists. Halide abstraction reactions from bromo-vinyl/alkyl silanes by Ag+[al–f–al]− seemed to be successful, but resulted in the formation of bromo silanes and C2H2/C2H4 due to the reaction of the silylium ions with solvated AgBr. From the reaction of Me3Si–F–Al(ORF)3 with 2-butyne, a salt of the stable silylated tetramethyl cyclobutenyl cation [Me4C4–SiMe3]+ was obtained. Attempts to release the CBD Me4C4 from this compound did not result in the planned Diels–Alder reactions, but only in oligomerization products. We attribute this to the comparatively low π-LUMO energy of [Me4C4–SiMe3]+, leading to the reaction of this cation with the evolving Me4C4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg, the ERC project UniChem and the DFG. We would like to acknowledge the MagRes facility of the University of Freiburg and Fadime Bitgül for the measurement of the NMR spectra and Daniel Kratzert for the valuable discussions of the crystallographic data.References
- (a) G. A. Olah and G. K. Surya Prakash, ed. Carbocation Chemistry, John Wiley & Sons, Hoboken, New Jersey, 2004 Search PubMed; (b) H. Meerwein and K. van Emster, Ber. Dtsch. Chem. Ges. A/B, 1922, 55, 2500 CrossRef.
- G. A. Olah, J. Am. Chem. Soc., 1972, 94, 808 CrossRef CAS.
- (a) P. v. R. Schleyer, D. Lenoir, P. Mison, G. Liang, G. K. S. Prakash and G. A. Olah, J. Am. Chem. Soc., 1980, 102, 683 CrossRef CAS; (b) H. C. Brown, Acc. Chem. Res., 1986, 19, 34 CrossRef CAS; (c) S. Winstein and D. S. Trifan, J. Am. Chem. Soc., 1949, 71, 2953 CrossRef CAS; (d) H. C. Brown, The Nonclassical Ion Problem, Springer US, Boston, MA, 1977 CrossRef.
- F. Scholz, D. Himmel, F. W. Heinemann, P. v. R. Schleyer, K. Meyer and I. Krossing, Science, 2013, 341, 62 CrossRef CAS PubMed.
- (a) B. Chiavarino, M. E. Crestoni, J. Lemaire, P. Maitre and S. Fornarini, J. Chem. Phys., 2013, 139, 71102 CrossRef PubMed; (b) O. Mó, M. Yáñez, J.-F. Gal, P. C. Maria and J.-C. Guillemin, J. Phys. Org. Chem., 2002, 15, 509 CrossRef; (c) B. Chiavarino, M. E. Crestoni and S. Fornarini, Chem. Commun., 2002, 1418 RSC.
- X. Li and J. A. Stone, J. Am. Chem. Soc., 1989, 111, 5586 CrossRef CAS.
- H.-U. Siehl, S. Brixner, C. Coletti, N. Re, B. Chiavarino, M. E. Crestoni, A. de Petris and S. Fornarini, Int. J. Mass Spectrom., 2013, 334, 58 CrossRef CAS.
- M. F. Ibad, P. Langer, A. Schulz and A. Villinger, J. Am. Chem. Soc., 2011, 133, 21016 CrossRef CAS PubMed.
- (a) T. Müller, C. Bauch, M. Ostermeier, M. Bolte and N. Auner, J. Am. Chem. Soc., 2003, 125, 2158 CrossRef PubMed; (b) S. Duttwyler, Q.-Q. Do, A. Linden, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2008, 47, 1719 CrossRef CAS PubMed; (c) J. B. Lambert, S. Zhang and S. M. Ciro, Organometallics, 1994, 13, 2430 CrossRef CAS; (d) J. B. Lambert, S. Zhang, C. L. Stern and J. C. Huffman, Science, 1993, 260, 1917 CrossRef CAS PubMed.
- S. Inoue, M. Ichinohe, T. Yamaguchi and A. Sekiguchi, Organometallics, 2008, 27, 6056 CrossRef CAS.
- A. Sekiguchi, T. Matsuno and M. Ichinohe, J. Am. Chem. Soc., 2000, 122, 11250 CrossRef CAS.
- A. Burcat and E. Goos, Int. J. Chem. Kinet., 2018, 7, 793 Search PubMed.
- (a) A. Sekiguchi, Science, 2004, 305, 1755 CrossRef CAS PubMed; (b) R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343 CrossRef CAS PubMed; (c) I. Bejan and D. Scheschkewitz, Angew. Chem., Int. Ed., 2007, 46, 5783 CrossRef CAS PubMed.
- T. E. Mallouk, G. L. Rosenthal, G. Mueller, R. Brusasco and N. Bartlett, Inorg. Chem., 1984, 23, 3167 CrossRef CAS.
- K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis, Boca Raton, 90th edn, 2010 Search PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC.
- S. P. Hoffmann, T. Kato, F. S. Tham and C. A. Reed, Chem. Commun., 2006, 767 RSC.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058 RSC.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 325 CrossRef CAS.
- (a) V. G. Baonza, O. R. Montoro, M. Taravillo, M. Cáceres and J. Núñez, J. Chem. Phys., 2004, 121, 11156 CrossRef CAS PubMed; (b) C. Mediavilla, J. Tortajada and V. G. Baonza, Chem. Phys. Lett., 2008, 454, 387 CrossRef CAS.
- S. G. Wierschke, J. Chandrasekhar and W. L. Jorgensen, J. Am. Chem. Soc., 1985, 107, 1496 CrossRef CAS.
- E. Hey, F. Weller and K. Dehnicke, Z. Anorg. Allg. Chem., 1983, 502, 45 CrossRef CAS.
- R. F. Bryan, J. Am. Chem. Soc., 1964, 86, 733 CrossRef CAS.
- H. H. Freedman and A. E. Young, J. Am. Chem. Soc., 1964, 86, 734 CrossRef CAS.
- G. Maier, R. Emrich, K.-D. Malsch, K.-A. Schneider, M. Nixdorf and H. Irngartinger, Chem. Ber., 1985, 118, 2798 CrossRef CAS.
- G. A. Olah, J. S. Staral, R. J. Spear and G. Liang, J. Am. Chem. Soc., 1975, 97, 5489 CrossRef CAS.
- J. B. Koster, G. J. Timmermans and H. van Bekkum, Synthesis, 1971, 1971, 139 CrossRef.
- P. B. J. Driessen and H. Hogeveen, J. Am. Chem. Soc., 1978, 100, 1193 CrossRef CAS.
- M. Simonetta and S. Winstein, J. Am. Chem. Soc., 1954, 76, 18 CrossRef CAS.
- C. Krüger, P. J. Roberts, Y.-H. Tsay and J. B. Koster, J. Organomet. Chem., 1974, 78, 69 CrossRef.
- (a) Š. Janková, J. Schulz, S. Hybelbauerová, I. Císařová, P. Štěpnička and M. Kotora, Eur. J. Org. Chem., 2013, 2013, 44 CrossRef; (b) P. B. J. Driessen and H. Hogeveen, J. Organomet. Chem., 1978, 156, 265 CrossRef CAS; (c) M. J. Marsella, S. Estassi, L.-S. Wang and K. Yoon, Synlett, 2004, 192 CrossRef CAS.
- K. S. Fongers, H. Hogeveen and R. F. Kingma, Tetrahedron Lett., 1983, 24, 1423 CrossRef CAS.
- H. Hogeveen, H. Jorritsma, P. A. Wade, F. van Rantwijk, J. B. Koster, J. J. Prooi, A. Sinnema and H. van Bekkum, Tetrahedron Lett., 1974, 15, 3915 CrossRef.
- T. Bally and S. Masamune, Tetrahedron, 1980, 36, 343 CrossRef CAS.
- A. Fattahi, L. Lis, Z. Tian and S. R. Kass, Angew. Chem., Int. Ed., 2006, 45, 4984 CrossRef CAS PubMed.
- (a) H. Irngartinger and H. Rodewald, Angew. Chem., Int. Ed., 1974, 13 Search PubMed; (b) H. Kimling and A. Krebs, Angew. Chem., Int. Ed., 1972, 11, 932 CrossRef CAS.
- (a) J. Possart, A. Martens, M. Schleep, A. Ripp, H. Scherer, D. Kratzert and I. Krossing, Chem.–Eur. J., 2017, 23, 12305 CrossRef CAS; (b) M. Rohde, L. O. Müller, D. Himmel, H. Scherer and I. Krossing, Chem.–Eur. J., 2014, 20, 1218 CrossRef CAS PubMed.
- (a) R. J. Wehmschulte and L. Wojtas, Inorg. Chem., 2011, 50, 11300 CrossRef CAS PubMed; (b) M. Kira, T. Hino and H. Sakurai, Chem. Lett., 1992, 21, 555 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, procedures, weights, and 1D- and 2D-NMR spectra of the reactions are displayed. Details of the quantum chemical calculations are given together with crystallographic details. CCDC 1868136–1868138. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04591g |
| This journal is © The Royal Society of Chemistry 2019 |