
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low overpotential water oxidation at neutral pH catalyzed by a copper(II) porphyrin†
Yanju
Liu
a,
Yongzhen
Han
a,
Zongyao
Zhang
a,
Wei
Zhang
 b,
Wenzhen
Lai
a,
Yong
Wang
cd and
Rui
Cao
*ab
b,
Wenzhen
Lai
a,
Yong
Wang
cd and
Rui
Cao
*ab
aDepartment of Chemistry, Renmin University of China, Beijing 100872, China. E-mail: ruicao@ruc.edu.cn
bKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China
cState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
dInstitute of Drug Discovery Technology, Ningbo University, Ningbo 315211, China
First published on 7th January 2019
Abstract
Low overpotential water oxidation under mild conditions is required for new energy conversion technologies with potential application prospects. Extensive studies on molecular catalysis have been performed to gain fundamental knowledge for the rational designing of cheap, efficient and robust catalysts. We herein report a water-soluble CuII complex of tetrakis(4-N-methylpyridyl)porphyrin (1), which catalyzes the oxygen evolution reaction (OER) in neutral aqueous solutions with small overpotentials: the onset potential of the catalytic water oxidation wave measured at current density j = 0.10 mA cm−2 is 1.13 V versus a normal hydrogen electrode (NHE), which corresponds to an onset overpotential of 310 mV. Constant potential electrolysis of 1 at neutral pH and at 1.30 V versus NHE displayed a substantial and stable current for O2 evolution with a faradaic efficiency of >93%. More importantly, in addition to the 4e water oxidation to O2 at neutral pH, 1 can catalyze the 2e water oxidation to H2O2 in acidic solutions. The produced H2O2 is detected by rotating ring–disk electrode measurements and by the sodium iodide method after bulk electrolysis at pH 3.0. This work presents an efficient and robust Cu-based catalyst for water oxidation in both neutral and acidic solutions. The observation of H2O2 during water oxidation catalysis is rare and will provide new insights into the water oxidation mechanism.
Introduction
The growing threat of anthropogenic climate change and the intense global energy demand on fossil fuels are among the biggest challenges faced by human beings in the current century, which makes seeking efficient carbon-neutral sources of renewable energy the most significant and urgent task of the scientific society.1–3 The global total primary energy demand will exceed 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mtoe (million tons of oil equivalent) in 2025 and reach 18000 Mtoe in 2040. Among all the renewable energy sources, sunlight is the ideal energy resource to meet such a demand. One of the biggest challenges of sunlight utilization is the large-scale economy efficient conversion and storage.4–8 Nature, however, presents a fascinating scenario to solve this problem by using photosystem II (PSII) to store solar energy as chemical energy in a scale of sustaining the whole life circle on the earth.9–11 PSII is a membrane protein complex located in the thylakoid membranes and performs a series of light-induced electron transfer reactions leading to the splitting of water into electrons/protons and molecular dioxygen.6,12,13 Upon an initial light-induced charge separation, chlorophyll (P680) with a magnesium porphyrin core is converted to a cation radical species P680˙+, which subsequently abstracts an electron from the Mn4CaOx cluster of the oxygen-evolving complex (OEC). After four such successive steps, water is oxidized at the OEC to release dioxygen, and the generated electrons/protons are used for the fixation of carbon dioxide. Therefore, water oxidation holds the central place of the natural energy-conversion scheme.
000 Mtoe (million tons of oil equivalent) in 2025 and reach 18000 Mtoe in 2040. Among all the renewable energy sources, sunlight is the ideal energy resource to meet such a demand. One of the biggest challenges of sunlight utilization is the large-scale economy efficient conversion and storage.4–8 Nature, however, presents a fascinating scenario to solve this problem by using photosystem II (PSII) to store solar energy as chemical energy in a scale of sustaining the whole life circle on the earth.9–11 PSII is a membrane protein complex located in the thylakoid membranes and performs a series of light-induced electron transfer reactions leading to the splitting of water into electrons/protons and molecular dioxygen.6,12,13 Upon an initial light-induced charge separation, chlorophyll (P680) with a magnesium porphyrin core is converted to a cation radical species P680˙+, which subsequently abstracts an electron from the Mn4CaOx cluster of the oxygen-evolving complex (OEC). After four such successive steps, water is oxidized at the OEC to release dioxygen, and the generated electrons/protons are used for the fixation of carbon dioxide. Therefore, water oxidation holds the central place of the natural energy-conversion scheme.
Extensive studies on artificial molecular catalysts for homogeneous water oxidation have been performed to unveil its mechanistic mysteries, particularly the pivotal O–O bond formation process.1,3,12,14 Such mechanistic information provides fundamental knowledge for the rational design of efficient and robust water oxidation catalysts. The natural Mn4CaOx of the OEC encourages scientists to design biomimetic multinuclear metal clusters with redox flexibility suitable for the multi-electron water oxidation reaction. In this context, several notable metal clusters have been studied as OER catalysts, including tetramanganese,15,16 dimanganese,17,18 tetracobalt,19 tetraruthenium,20,21 diruthenium,22 and pentanuclear iron23 complexes. In these examples, intramolecular O–O bond formation through the coupling of two metal–oxo units is usually proposed. On the other hand, since the first report of mononuclear Ru complexes as active catalysts for water oxidation,24 many mononuclear metal complexes have been identified to be active OER catalysts.25–52 Unlike multinuclear systems, for mononuclear catalysts nucleophilic water attack to terminal metal–oxo/oxyl units is the likely O–O bond formation route. Despite these achievements, however, cheap catalysts functioning at neutral pH with low overpotentials for water oxidation are still under development.
As an alternative approach, metal porphyrins are promising to serve as water oxidation catalysts. The cation radical species P680˙+ is the most oxidizing redox cofactor known in biology with an estimated redox potential of 1.30 V versus NHE.6,12 It provides the initial oxidizing power for water oxidation (2H2O → O2 + 4H+ + 4e−, ΔE = 1.23 V versus NHE). The oxidizing equivalent is stored in the porphyrin macrocycle. Thus, it makes sense to design metal porphyrins to directly catalyze water oxidation, given that the metal core with the porphyrin macrocycle can provide one binding site for an aqua ligand. Bioinspired by this, we and others have reported a series of metal-porphyrin OER catalysts.3,49,53,54
Herein, we present a significant improvement on water oxidation catalysis with water-soluble cationic CuII porphyrin 1 (Fig. 1). Copper plays vital roles in biological and biomimetic O2 chemistry to mediate O–O bond formation and cleavage.55–59 As a consequence, Cu complexes have attracted increasing attention as catalysts for water oxidation.60–75 CuII porphyrin 1 can catalyze water oxidation in neutral aqueous solution with impressively small overpotentials. The onset potential of the catalytic water oxidation wave measured at the current density j = 0.10 mA cm−2 is 1.13 V versus NHE, which corresponds to an onset overpotential η = 310 mV. This value is remarkable in comparison with that of any other reported molecular OER catalysts working at neutral pH. More importantly, 1 can also catalyze the 2e water oxidation to H2O2 in acidic solutions. The selective H2O2 generation is significant because H2O2 has been extensively used as an oxidant in industry and is regarded as an alternative fuel with the benefits of easy storage and transportation.76–78 The property of 1 to catalyze both O2 evolution at neutral pH and H2O2 formation at acidic pH during water oxidation is very rare and will provide new insights into the catalyst design and also the mechanism for water oxidation.
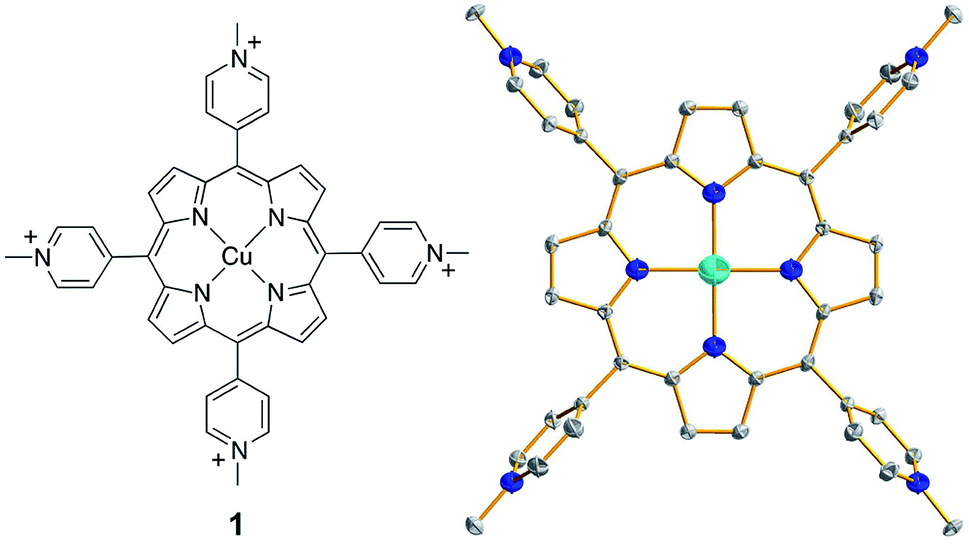 | ||
| Fig. 1 (Left) Molecular structure of 1. (Right) Thermal ellipsoid plot of the X-ray structure of 1 (50% probability). Hydrogen atoms and counter anions are omitted for clarity. | ||
Results
Synthesis and characterization
Complex 1 was synthesized by the reaction of CuII salts with a porphyrin ligand (see Experimental section for details). Crystals of a triflate salt were isolated by slow evaporation from a water/acetonitrile solution at room temperature, and were used for characterization and electrochemical measurements. The high-resolution mass spectrum (HRMS) of 1 showed ions at mass-to-charge ratios of 184.8086 and 518.5703, which matched the calculated values of 184.8090 for the tetravalent ion [Por-Cu]4+ and of 518.5700 for the divalent ion [Por-Cu-(OTf)2]2+, respectively (Fig. S1†). Importantly, the observed isotopic distributions matched those expected for these ions with separations of 0.25 and 0.50 units between each peak for tetravalent and divalent ions, respectively.The structure of 1 was determined by single crystal X-ray diffraction. Crystallographic studies revealed that 1 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 3 (crystal data and structure refinement details are summarized in Table S1†). In the X-ray structure of 1, one molecule is located at the special position with the central Cu ion situated on the crystallographically required inversion center. The other two molecules are located at the general position and are symmetry-related through the inversion center. As a consequence, the asymmetric unit contains one and a half molecules. The Cu ion is coordinated by the porphyrin ligand through four N atoms to give a distorted square-planar geometry (Fig. 1, right). No axial ligands were found for the Cu ion in the solid state, although several co-crystallized solvent water molecules were located in the crystal lattice. Moreover, no changes were observed in the UV-vis spectrum of 1 in propylene carbonate (PC, a weak coordinating solvent) with the addition of water (Fig. S2†), further confirming that water will not bind to the Cu ion of 1. The average Cu–N bond distance of 1.998(4) Å indicates a d9 CuII electronic structure. In addition, for each molecule of 1, four CF3SO3− counter anions were successfully located in the X-ray structure, further confirming the CuII oxidation state.
with Z = 3 (crystal data and structure refinement details are summarized in Table S1†). In the X-ray structure of 1, one molecule is located at the special position with the central Cu ion situated on the crystallographically required inversion center. The other two molecules are located at the general position and are symmetry-related through the inversion center. As a consequence, the asymmetric unit contains one and a half molecules. The Cu ion is coordinated by the porphyrin ligand through four N atoms to give a distorted square-planar geometry (Fig. 1, right). No axial ligands were found for the Cu ion in the solid state, although several co-crystallized solvent water molecules were located in the crystal lattice. Moreover, no changes were observed in the UV-vis spectrum of 1 in propylene carbonate (PC, a weak coordinating solvent) with the addition of water (Fig. S2†), further confirming that water will not bind to the Cu ion of 1. The average Cu–N bond distance of 1.998(4) Å indicates a d9 CuII electronic structure. In addition, for each molecule of 1, four CF3SO3− counter anions were successfully located in the X-ray structure, further confirming the CuII oxidation state.
Electrocatalytic water oxidation
The cyclic voltammogram (CV) of 1.0 mM 1 in 0.10 M pH 7.0 phosphate buffer obtained using a fluorine-doped tin oxide (FTO) working electrode displayed a pronounced catalytic wave. The onset of this catalytic wave, measured at j = 0.10 mA cm−2, is at 1.13 V versus NHE (all potentials reported in this work are referenced to NHE unless otherwise noted), which corresponds to an onset η = 310 mV (Fig. 2a). CVs of 1 in neutral H2O and D2O phosphate buffers showed a kinetic isotope effect (KIE = 1.81, Fig. S3†). The normalized catalytic currents (icat/ν1/2) decreased with increasing scan rates (Fig. 2b), indicating that the current is associated with a catalytic process. Comparison of 1 with reported Cu-based molecular catalysts underpins its remarkable efficiency. As shown in Table 1, Cu complexes usually require strong alkaline conditions and large onset η values >400 mV to function as active OER catalysts. Only a few Cu catalysts are able to work at neutral pH. However, they always require large onset η > 600 mV to exhibit evident catalytic activities.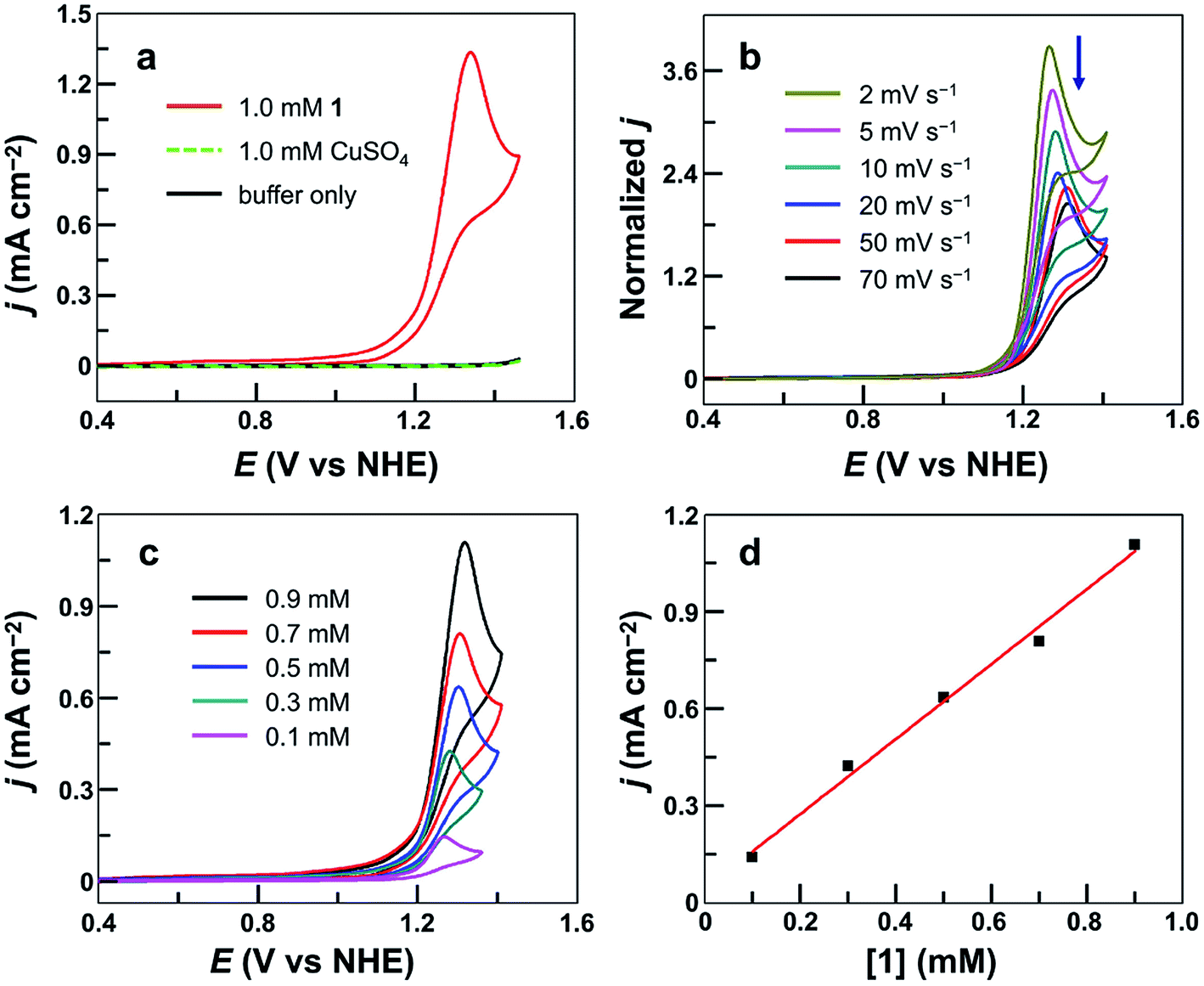 | ||
| Fig. 2 (a) CVs of 1, CuSO4, and buffer-only solution at a 100 mV s−1 scan rate. (b) Normalized CVs of 0.50 mM 1 at different scan rates in mA cm−2 V−1/2 s1/2. (c) CVs of 1 at different concentrations. (d) Plot of catalytic peak current versus [1]. Conditions: FTO working electrode (S = 0.25 cm2), 0.10 M pH 7.0 phosphate buffer, and 20 °C. | ||
| Catalyst | Catalyst concentration (mM) | pH | Onset ηa (mV) | TOF (s−1) | Reference |
|---|---|---|---|---|---|
| a The onset overpotential values for water oxidation mediated by molecular catalysts are generally reported and are used for comparison in the literature. Abbreviations used in this table: bpy, 2,2′-bipyridine; TGG, triglycylglycine; Py3P, N,N-bis(2-(2-pyridyl)ethyl)pyridine-2,6-dicarboxamidate; dhbp, 6,6′-dihydroxy-2,2′-bipyridine; L2 and L4, N1,N1′-(1,2-phenylene)bis(N2-methyloxalamide) derivatives; TMC, 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; en, 1,2-ethylenediamine; pyalk, 2-pyridyl-2-propanoate; pdca, N,N′-2,6-dimethylphenyl-2,6-pyridinedicarboxamidate; BPMAN, 2,7-[bis(2-pyridylmethyl)-aminomethyl]-1,8-naphthyridine; TMPA, bis((6-methyl-2-pyridyl)methyl)(2-pyridylmethyl)amine. | |||||
| Cu porphyrin 1 | 1.0 | 7.0 | 310 | 30 | This work |
| [(bpy)Cu(OH)2] | 1.0 | 12.5 | 540 | 100 | 60 |
| [(TGG4−)CuII(OH2)]2− | 0.89 | 11 | 520 | 33 | 62 |
| [(Py3P)Cu(OH)]− | 0.7 | 8.0 | 400 | 20 | 67 |
| [(dhbp)Cu(OH)2] | 1.0 | 12.4 | 330 | 0.4 | 61 |
| [(L2)Cu]2− | 1.0 | 11.5 | 400 | 3.58 | 64 |
| [(L4)Cu]2− | 1.0 | 11.5 | 170 (not stable) | 0.16 | |
| [Cu(TMC)(H2O)]2+ | 1.0 | 7.0 | 600 | 30 | 73 |
| [Cu(en)2]2+ | 1.0 | 8.0 | 530 | 0.4 | 72 |
| Cu(pyalk)2 | 1.0 | 13.3 | 460 | 0.7 | 66 |
| [pdca–CuII–CO3H]− | 1.0 | 10.0 | 650 | 20.1 | 69 |
| [Cu2(BPMAN)(μ-OH)]3+ | 1.0 | 7.0 | 800 | 0.6 | 63 |
| [(TMPA–CuII)2–(μ-OH)2]2+ | 1.0 | 12.5 | 720 | 33 | 68 |
The molecular nature of 1 for water oxidation was verified. No catalytic currents were observed up to 1.45 V in a buffer-only solution and in a 1.0 mM CuSO4 solution under identical conditions (Fig. 2a). The catalytic currents increased with increasing concentration of 1 (Fig. 2c), exhibiting a linear dependence (Fig. 2d). Further analysis of the catalytic water oxidation wave by using foot of the wave analysis (FOWA,64,79–83 Fig. S4†) showed that the observed reaction rate constant remained almost unchanged at different concentrations of 1. These results together indicate the first-order dependence of the catalytic reaction kinetics on 1. In addition, the coordination-caused inhibition by either phosphate anions (Fig. S5†) or acetonitrile molecules (Fig. S6†) is strong evidence for Cu-centered homogeneous catalysis.
The stability of 1 under catalytic conditions was evaluated. Successive CV scans of 1 showed no current increase (Fig. 3a). The peak current slightly decreased in the first several scans and then remained almost constant. The initial small drop of the peak current is due to the generation and accumulation of O2 gas bubbles on the surface of the FTO working electrode. Constant potential electrolysis (CPE) of 1 for 10 h at 1.30 V displayed a substantial and stable current (Fig. 3b). The FTO working electrodes after 20 CV scans and after CPE displayed no catalytic current in freshly prepared buffer-only solutions (Fig. 3c). Surface analysis of the electrode after CPE by scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX) showed no sign of the deposition of any heterogeneous Cu-based materials (Fig. S7†). In addition, the solution after CPE was analyzed by UV-vis spectroscopy, showing a negligible change as compared to that before CPE (Fig. 3d). These results strongly supported the molecular nature of 1 and its stability under catalytic OER conditions.
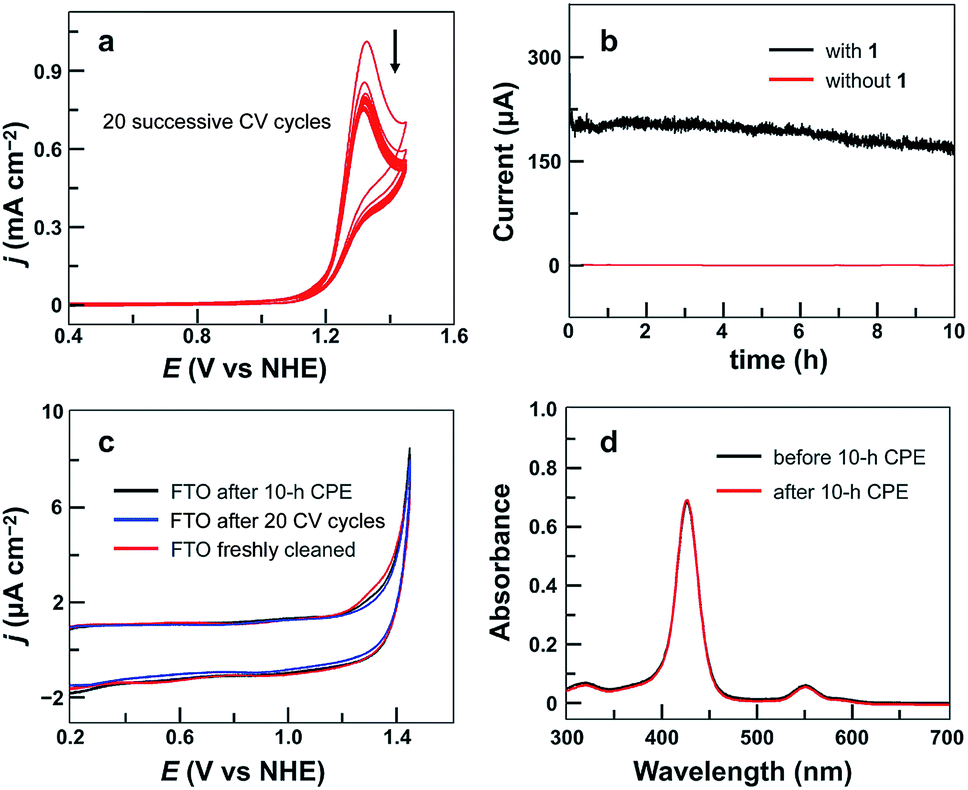 | ||
| Fig. 3 (a) 20 successive CV cycles of 0.75 mM 1 with the FTO working electrode (S = 0.25 cm2) at a 50 mV s−1 scan rate. (b) Catalytic currents in CPE with or without 1 (0.75 mM) using the FTO electrode (S = 1.0 cm2) at an applied potential of 1.30 V. (c) CVs of the FTO electrode after 10 h CPE with 1, the FTO electrode after 20 successive CV cycles with 1, and a freshly cleaned FTO electrode in the buffer-only solution. Conditions: 0.10 M pH 7.0 phosphate buffer, 100 mV s−1 scan rate, and 20 °C. (d) UV-vis spectra of 1 before and after 10 h CPE. | ||
The evolved O2 during CPE was then determined, revealing a faradaic efficiency of >93% for O2 evolution (Fig. S8†). The turnover number was calculated to be 5.0 based on all catalyst molecules in the solution. A diffusion-controlled CuII/CuI redox couple of 1 was found at E1/2 = 0.06 V (Fig. S9†). With this information, the plot of icat/idversus ν−1/2 shows a linear relationship (Fig. S10†), which is consistent with a pure kinetic behavior in this scan rate range. A turnover frequency (TOF) value of 30 s−1 can be calculated using established methods.84 This value is remarkable because it is obtained at neutral pH and is comparable to the values presented by those Cu-based molecular catalysts working in alkaline solutions. Moreover, this TOF value with 1 is obtained at much smaller overpotentials as compared to that of reported Cu-based catalysts (Table 1).
pH-Dependent electrochemical studies
After establishing the molecular nature of 1 for catalysis, we next carried out pH-dependent studies. Fig. 4a displayed the linear sweep voltammetry (LSV) curves of 0.50 mM 1 in 0.10 M pH 2.0–7.0 phosphate buffers obtained using glassy carbon (GC) electrodes. Significantly, 1 was active even in strong acidic solutions (i.e., pH < 3). This result is additional support for the molecular nature of 1 as the real water oxidation catalyst because any Cu oxides that may possibly be generated upon the decomposition of 1 are unstable in acidic solutions. The plot of the potential as measured at j = 0.45 mA cm−2versus pH gives a linear relationship (Fig. 4b). The slope is −0.029 V per pH unit, indicating that the catalytic cycle involves an unusual 2e−/1H+ redox step. Notably, in strong acidic solutions at pH 2.0–3.0, a shoulder peak at 1.29 V prior to the catalytic water oxidation wave can be identified, which is assigned to the 1e oxidation of 1.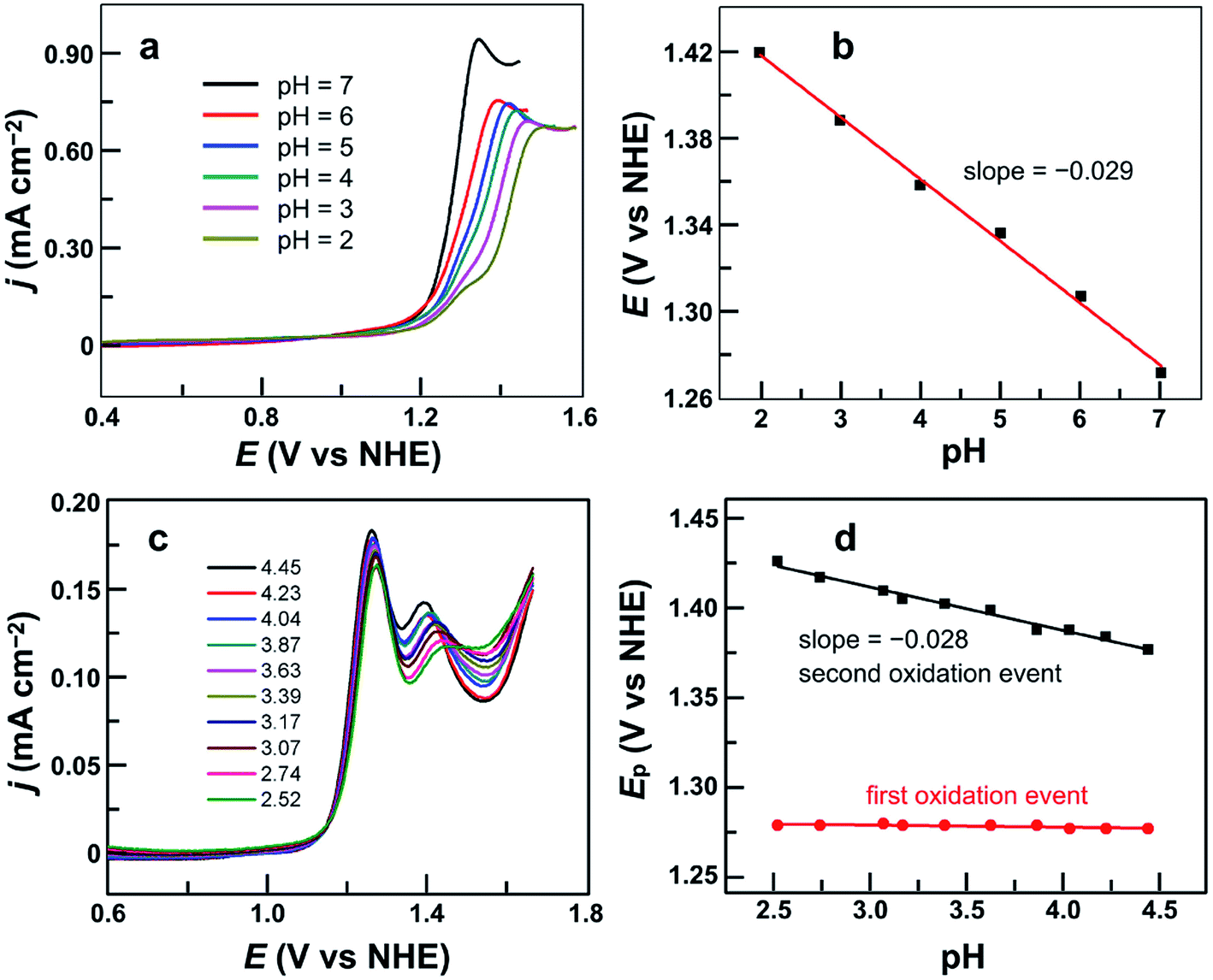 | ||
| Fig. 4 (a) LSV curves of 0.50 mM 1 in 0.10 M pH 2.0–7.0 phosphate buffers. (b) Plot of potential measured at j = 0.45 mA cm−2versus pH. (c) DPV curves of 0.50 mM 1 in 0.10 M pH 2.52–4.45 phosphate buffers. (d) Plots of the first and second oxidation peak potentials versus pH from 2.52 to 4.45. Conditions: GC working electrode, scan rate 100 mV s−1, and 20 °C. | ||
On the basis of these results, we studied the pH-dependent electrochemistry by using the differential pulse voltammetry (DPV) method. The DPV results of 1 displayed two oxidation events in 0.1 M pH 2.52–4.45 phosphate buffers (Fig. 4c). At pH 2.52, these two oxidation events are at Ep,a = 1.27 and 1.44 V, respectively. Significantly, the first oxidation is pH-independent, while the second one is pH-dependent. The plots of the first and second oxidation peak potentials versus pH values are depicted in Fig. 4d, showing that the Ep,a values of the first oxidation remain nearly identical but those of the second oxidation show a monotonic decrease with a slope of −0.028 V per pH unit. This number is the same as that obtained in the abovementioned LSV studies, suggesting that the second oxidation corresponds to the 2e−/1H+ process. With the further increase of the solution pH, the second oxidation wave steadily moves towards the first oxidation wave (Fig. S11†), and they eventually become superimposed under near neutral conditions. Notably, in the literature, overpotentials at the initial foot of the DPV oxidation wave and at the half-peak position of the catalytic CV oxidation wave are also reported. In the case of 1 at pH 7.0, the potential at the initial foot of the DPV oxidation wave is 1.12 V, while the half-peak potential of the catalytic CV oxidation wave is 1.26 V. These give η = 300 mV (at the initial foot of DPV) and 440 mV (at the half-peak position of the CV wave) for catalytic water oxidation, respectively. The overpotential values determined from the initial foot of both the CV and DPV oxidation waves of 1 are identical in our experiments.
It is worth noting that at pH 2.52, the current of the second oxidation is much smaller than that of the first oxidation in the DPV plot of 1 (Fig. 4c). With increasing pH, the second oxidation moves towards the first oxidation, and its current also increases significantly. This trend implies a pH-dependent chemical reaction/transformation of the 1e-oxidized species of 1. In order to get more insights into the first oxidation process, we measured the CV of 1 at pH 2.52 by reversing the scan prior to the catalytic water oxidation wave (i.e., <1.35 V). As shown in Fig. 5, almost no reduction peak can be observed at a 100 mV s−1 scan rate. However, a small but significant reduction peak appears at a 200 mV s−1 scan rate and the current increases with the scan rate. Moreover, the ratio of the reduction peak current versus the oxidation peak current increases with the scan rate (Fig. 5). This result further supports the chemical reaction/transformation of the 1e-oxidized species of 1, leading to its corresponding reduction wave only being detectable on reversing the scan with large scan rates.
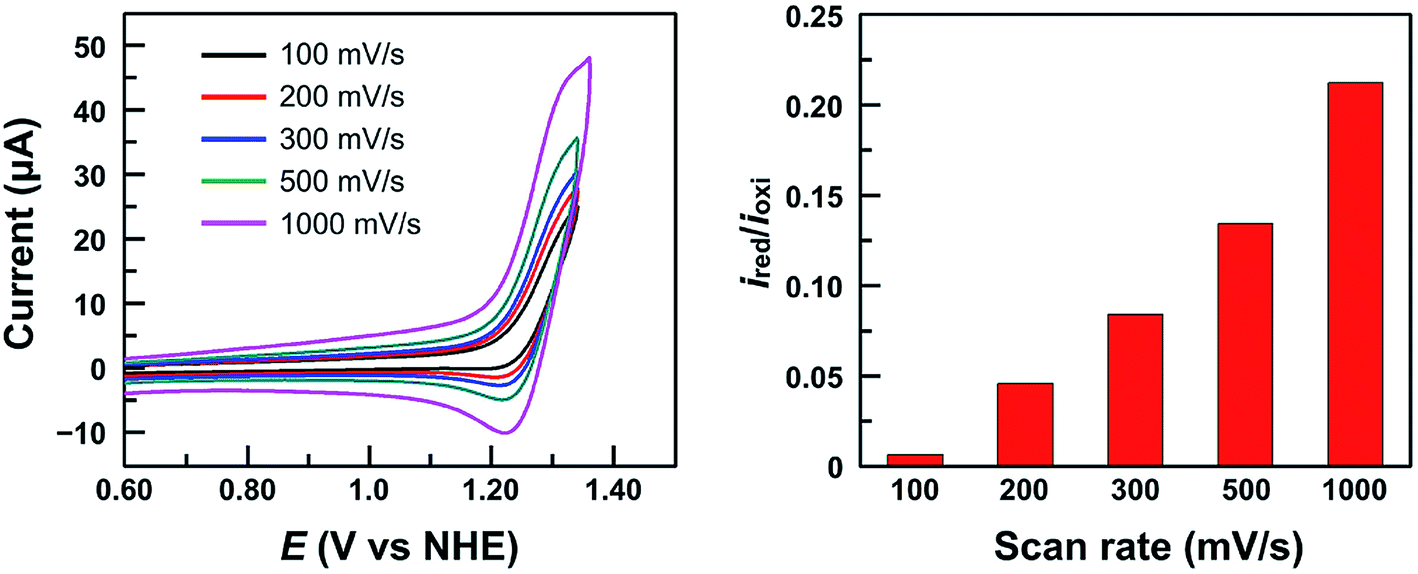 | ||
| Fig. 5 CVs of 1.0 mM 1 in 0.10 M pH 2.52 phosphate buffer by reversing the scan at 1.35 V with different scan rates (Left) and the ratio of the reduction peak current versus the oxidation peak current ired/ioxi (Right). Conditions: GC working electrode and 20 °C. | ||
Next, we performed electrolysis of 1 at 1.40 V in a pH 3.0 phosphate buffer solution. Thin-layer spectroelectrochemical analysis showed an absorbance decrease at 425 and 550 nm but the absorbance increases in the 600–800 nm range (Fig. S12†). These results together suggest that the first oxidation of 1 is ligand-centered to form a CuII porphyrin cation radical species. In addition, we can oxidize 1 in a PC solution using ceric ammonium nitrate (CAN) as the chemical oxidant. As shown in Fig. S13,† similar changes were observed in this chemical oxidation process: the absorbance decreases at 544 nm but increases in the 590–800 nm range, which is characteristic for the ligand-centered oxidation to give porphyrin cation radical species.
Interestingly, the electrochemical behaviors of this first oxidation event of 1 are different in aqueous solutions versus those in organic solutions. In aqueous solutions, the first oxidation wave is irreversible at a 100 mV s−1 scan rate. However, in organic solutions, complex 1 displays a quasi-reversible oxidation process even at slow scan rates (i.e., 30 mV s−1, Fig. S14†). We propose that upon the first oxidation, the 1e-oxidized species of 1 can bind an aqua ligand at the axial position of the Cu ion, which can then initiate the water oxidation process. This chemical reaction/transformation is unlikely to happen for the 1e-oxidized species of 1 in the organic PC solution.
H2O2-related electrochemical studies
Significantly, during the electrolysis of 1 in pH 3.0 buffer solution with applied potentials <1.40 V, H2O2 was generated and was detected by the addition of sodium iodide (Fig. S15†). In order to determine the faradaic efficiency of H2O2 generation, we used a rotating ring (Pt)–disk (GC) electrode (RRDE) with CPE of the GC disk electrode at 1.40 V and CPE of the Pt ring electrode at 0.70 V in a pH 3.0 phosphate buffer solution. Stable currents were observed on both electrodes (Fig. S16†), giving a faradaic efficiency of 45%. This value is much smaller than 100%, and is likely caused by some level of oxidation of H2O2 at the disk electrode upon its formation. This provides strong evidence for the electrocatalytic features of 1 to catalyze 2e water oxidation to generate H2O2 in acidic solutions.Moreover, the CVs of 1 with addition of authentic H2O2 show remarkably increased catalytic waves (Fig. 6a). As a control, the CV of H2O2 under the same conditions without 1 showed a negligible current (Fig. S17†). We next studied the DPV of 1 with H2O2 in 0.10 M pH 3.6 phosphate buffer (Fig. 6b). Significantly, with the addition of H2O2, the first oxidation wave remained almost unchanged, but the second oxidation wave became much more pronounced, confirming that the second oxidation was associated with the H2O2-related species.
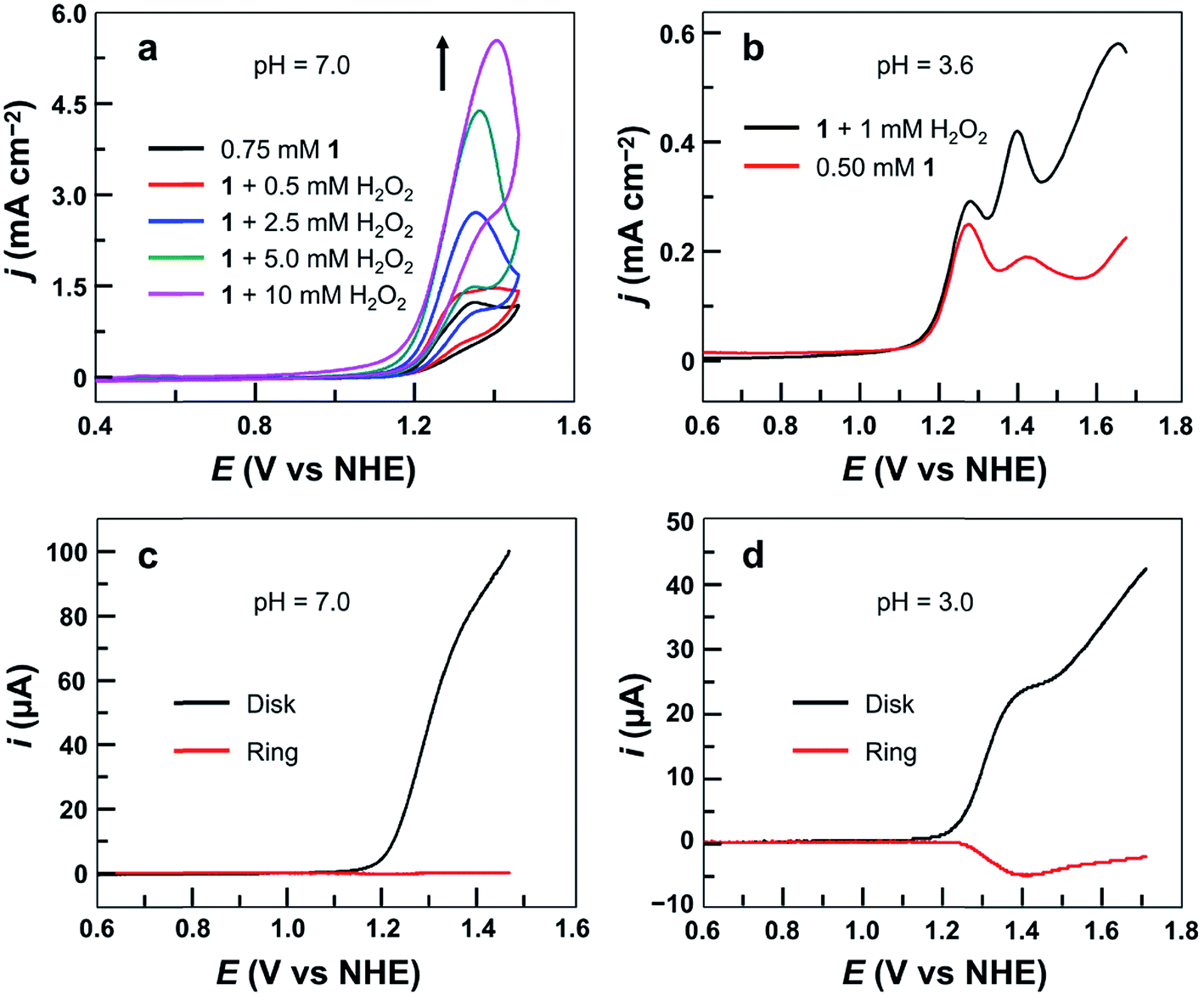 | ||
| Fig. 6 (a) CVs of 0.75 mM 1 with the addition of H2O2 using the GC electrode at a 100 mV s−1 scan rate. (b) DPV curves of 0.50 mM 1 with or without 1.0 mM H2O2. Conditions: 0.10 M phosphate buffer and 20 °C. (c) RRDE analysis of 0.5 mM 1 in 0.10 M pH 7.0 phosphate buffer. (d) RRDE analysis of 0.50 mM 1 in 0.10 M pH 3.0 phosphate buffer. The potential of the ring electrode is maintained at 0.70 V. Rotation rate is 1000 rpm, and scan rate is 10 mV s−1. | ||
More importantly, the association of the second oxidation in the DPV of 1 with H2O2-related species can be further verified by using RRDE analysis. Electrochemical studies of 1 were conducted in 0.10 M pH 7.0 or pH 3.0 phosphate buffer solutions using LSV of the GC disk electrode with a scan rate of 10 mV s−1 and CPE of the Pt ring electrode at 0.70 V. At pH 7.0, no anodic current of H2O2 oxidation could be detected at the Pt ring electrode (Fig. 6c). However, in the pH 3.0 solution, a large current due to H2O2 oxidation was observed at the Pt ring electrode (Fig. 6d). This difference is likely caused by the different potentials required for the second oxidation in pH 7.0 and pH 3.0 solutions. At neutral pH, the second oxidation wave is superimposed with the first oxidation wave of 1 (Fig. S11†). As a consequence, this H2O2-related species is instantly oxidized on the GC disk electrode upon its formation. At pH 3.0, however, more positive potentials (i.e., >1.40 V) are required for the oxidation of this H2O2-related species (Fig. 4c). Therefore, it can be detected at the Pt ring electrode. Importantly, as shown in Fig. 6d, along with the generation and accumulation of the H2O2-related species in LSV, the current at the Pt ring electrode constantly increases. However, at more positive potentials (i.e., >1.40 V), the current at the Pt ring electrode starts to decrease. This phenomenon is consistent with the oxidation/degradation of the H2O2-related species at the GC disk electrode at more positive potentials. The synchronization between the generation/degradation of the peroxide species at the disk electrode and the increase/decrease of the current at the ring electrode during RRDE measurements is supportive of (1) the formation of H2O2 during water oxidation with 1 in acidic media and (2) the association of the second oxidation in the DPV of 1 with H2O2-related species.
Discussion
Since the first report by Mayer and co-workers in 2012, Cu-based molecular OER catalysts have grown rapidly due to the advantages of high earth abundance, low cost and rich redox features of copper. On the other hand, copper enzymes play significant roles in numerous O2 activation processes in nature, and biomimetic Cu complexes have also been extensively investigated for such O2 chemistry. These features make Cu complexes the most attractive candidates as OER catalysts. For the reported mononuclear Cu complexes, however, their catalytic activity is mostly limited to functioning at high overpotentials and in alkaline solutions. A water nucleophilic attack (WNA) to high-valent CuIV–oxo or CuIII–oxyl units is commonly suggested for the O–O bond formation mechanism. Electrochemical evidence was provided by Meyer and co-workers to support the presence of the nascent CuII–OOH species. Very recently, Maseras, Llobet and co-workers proposed an alternative single electron transfer–water nucleophilic attack (SET–WNA) mechanism. In this mechanism, a formal CuIV–OH species was proposed as the active species to react with a hydroxyl ion for O–O bond formation. Instead of transferring two electrons in a direct WNA step, two single electron transfer steps from a hydroxyl ion to CuIV–OH take place with the involvement of an (HO⋯OH)− moiety. This mechanism avoids the involvement of high-energy CuIV–oxo, but alkaline conditions are still required. Kieber-Emmons and co-workers recently described a dinuclear Cu complex [CuII2–(μ-OH)2]2+, which catalyzed water oxidation with an onset η = 720 mV at pH = 12.5. The 2e-oxidized form [CuIII2–(μ-O)2]2+ was considered to couple the two bridging oxo atoms to form a “side-on” μ-(η2:η2)-peroxo dinuclear CuII intermediate. Subsequent redox isomerization converts this “side-on” structure to an “end-on” μ-(1,2)-peroxo structure. However, the authors mentioned that WNA to one oxo group of [CuIII2–(μ-O)2]2+ was also possible for O–O bond formation. Liao, Zhang and co-workers described another dinuclear Cu complex [CuII2–(μ-OH)]3+, which catalyzed water oxidation at neutral pH with an onset η = 800 mV. After 2e oxidation, the oxygen atom of the terminal CuIII–OH unit coupled with the bridging oxo atom of [CuIII2–(μ-O)]4+ to form the O–O bond. For these dinuclear Cu complexes, although high-energy CuIV (i.e., CuIV–oxo) active species were avoided in the proposed mechanisms, dinuclear forms are intrinsically more difficult to be oxidized than mononuclear forms because of their larger overall positive charges. As a consequence, these dinuclear Cu catalysts still worked at high overpotentials.In the present article, we report on a water-soluble cationic mononuclear CuII porphyrin 1 as a highly efficient and stable molecular electrocatalyst for water oxidation. Complex 1 can catalyze the 4e water oxidation to generate O2 in neutral aqueous solution with a small onset η = 310 mV. This value is remarkable in comparison with that with any other reported molecular OER catalysts working at neutral pH. More importantly, 1 can also catalyze the 2e water oxidation to produce H2O2 in acidic solutions. The impressively small overpotentials for water oxidation, and the features of both O2 evolution at neutral pH and H2O2 formation at acidic pH during water oxidation catalysis, are unique for complex 1 as a water oxidation catalyst.
Although the complete catalytic cycle of water oxidation with 1 is difficult to be fully understood based on the current experimental results, we can observe H2O2 generation during water oxidation with 1 in acidic solutions and can also provide strong evidence to support the fact that the second oxidation of 1 is associated with H2O2-related species. This result suggests that the 1e-oxidized species of 1 triggers O–O bond formation. This proposal may explain why complex 1 can catalyze water oxidation in neutral aqueous solutions with small overpotentials. For example, one can imagine that the initiation of O–O bond formation through 1e-oxidized species, instead of involving high-valent CuIV–oxo/CuIII–oxyl species, should certainly decrease the potentials required for water oxidation. More mechanistic studies are in progress with aims to better understand the catalytic cycle, in particular, O–O bond formation, of water oxidation with this CuII porphyrin catalyst.
Experimental section
General materials and methods
All reagents were purchased from commercial suppliers and used as received unless otherwise noted. Deionized water was used in all experiments. Tetrakis(4-N-methylpyridyl)porphyrin was prepared according to modified literature procedures.49 High-resolution mass spectrometric measurements were made on a Brüker Fourier transform ion cyclotron resonance mass spectrometer APEX IV. Electronic absorption spectra were recorded on a Cary 50 spectrophotometer. The morphologies of the FTO surface before and after electrolysis were examined using a JSM-6700F field emission scanning electron microscope (FE-SEM). The EDX spectra were collected from three randomly selected areas of each sample. In addition, the materials were analyzed at several local spots to ensure their chemical homogeneity. Images were obtained with an acceleration voltage of 5 kV.Synthesis of [Por-Cu](OTf)4 (1)
Tetrakis(4-N-methylpyridyl)porphyrin tetratosylate (0.68 g, 0.50 mmol) and CuCl2 (0.47 g, 3.5 mmol) were added into H2O (50 mL). The mixture solution was then refluxed for 5 h in the dark. After cooling to room temperature, the reaction solution was precipitated by adding it to a saturated aqueous NH4PF6 solution under stirring. The precipitate that formed was filtered off, washed with water, and dried in a vacuum to give red crystalline solids of [Por-Cu](PF6)4 (0.53 g, 0.40 mmol, yield 80%). These red solids were added into a mixture of CH3CN/H2O (1:1), and the PF6− anions were exchanged by CF3SO3− anions on an anion exchange resin by slow stirring at room temperature for three days. The solution was filtered, and the resin was washed with CH3CN. The solvents were then removed under reduced pressure to give red crystalline solids. Red needle-like crystals of 1 were obtained by slow evaporation from CH3CN and H2O at room temperature with a yield of 85% (0.45 g, 0.35 mmol). HRMS: calcd for the tetravalent ion [Por-Cu]4+, 184.8090; found, 184.8086; calcd for the divalent ion [Por-Cu-(OTf)2]2+ 518.5700; found, 518.5703 (Fig. S1†). Anal. calcd for [Cu(C44H36N8)](CF3SO3)4: C, 43.13; H, 2.72; N, 8.38. Found: C, 43.32; H, 2.81; N, 8.50.
Crystallographic studies
The complete dataset for 1 (CCDC 1835967†) was collected. A single crystal suitable for X-ray analysis was coated with Paratone-N oil, suspended in a small fiber loop, and placed on a Bruker D8 Venture X-ray diffractometer at 150(2) K. Diffraction intensities were measured using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data collection, indexing, data reduction, and final unit cell refinements were carried out using APEX2;85 absorption corrections were applied using the program SADABS.86 The structure was solved by direct methods using SHELXS87 and refined against F2 on all data by full-matrix least-squares using SHELXL,88 following established refinement strategies. In the single crystal X-ray structure of 1, all non-hydrogen atoms were refined anisotropically. All hydrogen atoms binding to carbon were included in the model at geometrically calculated positions and refined using a riding model. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms they are linked to (1.5 times for methyl groups). Details of the data quality and a summary of the residual values of the refinements are listed in ESI Table S1.†Electrochemical studies
All electrochemical experiments were carried out using a CH Instruments electrochemical analyzer (model CHI660E) at 20 °C unless otherwise noted. A three-compartment cell was used with a 0.25 cm2 FTO or a 0.07 cm2 GC electrode as the working electrode, saturated Ag/AgCl as the reference electrode, and a graphite rod as the auxiliary electrode. All potentials are reported versus the NHE through the addition of 0.199 V to the measured potential. The working electrode was ultrasonically washed with deionized water. Electrolysis was recorded in 0.10 M pH 7.0 phosphate buffer containing 0.75 mM 1 at 1.30 V in a three-compartment cell with a 1.0 cm2 FTO working electrode, a graphite rod auxiliary electrode, and an Ag/AgCl reference electrode. The produced O2 was analyzed using an Ocean Optics FOXY probe (Model NeoFox). The generated H2O2 was detected by electrochemical measurements using RRDE analysis. Electrochemical studies of 1 were conducted in 0.10 M pH 7.0 or pH 3.0 phosphate buffer using LSV of the disk electrode with a scan rate of 10 mV s−1 and electrolysis of the ring electrode at 0.70 V.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We are grateful for the support from the “Thousand Talents Program” in China, the Fok Ying-Tong Education Foundation for Outstanding Young Teachers in University (by the Ministry of Education of China), the National Natural Science Foundation of China under Grants 21101170, 21573139, 21773146 and 21873052, the Fundamental Research Funds for the Central Universities, the 3315-program of the Ningbo government, and the Research Funds of Renmin University of China.References
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- W. Zhang, W. Z. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- K. S. Joya, Y. F. Joya, K. Ocakoglu and R. van de Krol, Angew. Chem., Int. Ed., 2013, 52, 10426–10437 CrossRef CAS PubMed.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48–53 CrossRef CAS PubMed.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- X. Sala, S. Maji, R. Bofill, J. García-Antón, L. Escriche and A. Llobet, Acc. Chem. Res., 2014, 47, 504–516 CrossRef CAS PubMed.
- W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335–7338 CrossRef CAS PubMed.
- Y. Shimazaki, T. Nagano, H. Takesue, B. H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98–100 CrossRef PubMed.
- E. A. Karlsson, B. L. Lee, T. Åkermark, E. V. Johnston, M. D. Kärkäs, J. L. Sun, Ö. Hansson, J. E. Bäckvall and B. Åkermark, Angew. Chem., Int. Ed., 2011, 50, 11715–11718 CrossRef CAS PubMed.
- Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS PubMed.
- Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- Y. V. Geletii, C. Besson, Y. Hou, Q. S. Yin, D. G. Musaev, D. Quiñonero, R. Cao, K. I. Hardcastle, A. Proust, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 17360–17370 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- R. F. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Angew. Chem., Int. Ed., 2012, 51, 9740–9747 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS PubMed.
- L. L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. C. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed.
- D. E. Polyansky, J. T. Muckerman, J. Rochford, R. F. Zong, R. P. Thummel and E. Fujita, J. Am. Chem. Soc., 2011, 133, 14649–14665 CrossRef CAS PubMed.
- M. Murakami, D. C. Hong, T. Suenobu, S. Yamaguchi, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605–11613 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017–16029 CrossRef CAS.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473–10481 CrossRef CAS PubMed.
- D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M. C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024–19027 CrossRef CAS PubMed.
- W. Schöfberger, F. Faschinger, S. Chattopadhyay, S. Bhakta, B. Mondal, J. A. A. W. Elemans, S. Müllegger, S. Tebi, R. Koch, F. Klappenberger, M. Paszkiewicz, J. V. Barth, E. Rauls, H. Aldahhak, W. G. Schmidt and A. Dey, Angew. Chem., Int. Ed., 2016, 55, 2350–2355 CrossRef PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS.
- L. D. Wickramasinghe, R. W. Zhou, R. F. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260–13263 CrossRef CAS.
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS.
- M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531–5534 CrossRef CAS PubMed.
- B. M. Klepser and B. M. Bartlett, J. Am. Chem. Soc., 2014, 136, 1694–1697 CrossRef CAS PubMed.
- E. L. Demeter, S. L. Hilburg, N. R. Washburn, T. J. Collins and J. R. Kitchin, J. Am. Chem. Soc., 2014, 136, 5603–5606 CrossRef CAS.
- H. L. Sun, Y. Z. Han, H. T. Lei, M. X. Chen and R. Cao, Chem. Commun., 2017, 53, 6195–6198 RSC.
- L. Xu, H. T. Lei, Z. Y. Zhang, Z. Yao, J. F. Li, Z. Y. Yu and R. Cao, Phys. Chem. Chem. Phys., 2017, 19, 9755–9761 RSC.
- H. T. Lei, C. Y. Liu, Z. J. Wang, Z. Y. Zhang, M. N. Zhang, X. M. Chang, W. Zhang and R. Cao, ACS Catal., 2016, 6, 6429–6437 CrossRef CAS.
- X. L. Li, H. T. Lei, J. Y. Liu, X. L. Zhao, S. P. Ding, Z. Y. Zhang, X. X. Tao, W. Zhang, W. C. Wang, X. H. Zheng and R. Cao, Angew. Chem., Int. Ed., 2018, 57, 15070–15075 CrossRef CAS PubMed.
- D. K. Dogutan, R. McGuire Jr and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- H. Y. Du, S. C. Chen, X. J. Su, L. Jiao and M. T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- J. F. K. Kotyk, C. M. Hanna, R. L. Combs, J. W. Ziller and J. Y. Yang, Chem. Sci., 2018, 9, 2750–2755 RSC.
- Y. Z. Han, Y. Z. Wu, W. Z. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- M. Zhang, M.-T. Zhang, C. Hou, Z. F. Ke and T. B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042–13048 CrossRef CAS PubMed.
- J. Masud, P. C. Ioannou, N. Levesanos, P. Kyritsis and M. Nath, ChemSusChem, 2016, 9, 3128–3132 CrossRef CAS PubMed.
- M. P. Santoni, G. La Ganga, V. M. Nardo, M. Natali, F. Puntoriero, F. Scandola and S. Campagna, J. Am. Chem. Soc., 2014, 136, 8189–8192 CrossRef CAS PubMed.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- L. Quintanar, C. Stoj, A. B. Taylor, P. J. Hart, D. J. Kosman and E. I. Solomon, Acc. Chem. Res., 2007, 40, 445–452 CrossRef CAS PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1045 CrossRef CAS.
- C. Y. Liu, H. T. Lei, Z. Y. Zhang, F. F. Chen and R. Cao, Chem. Commun., 2017, 53, 3189–3192 RSC.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. B. Liu, J. L. Wang and W. B. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS.
- M.-T. Zhang, Z. F. Chen, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS.
- X. J. Su, M. Gao, L. Jiao, R. Z. Liao, P. E. M. Siegbahn, J. P. Cheng and M. T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- P. Garrido-Barros, I. Funes-Ardoiz, S. Drouet, J. Benet-Buchholz, F. Maseras and A. Llobet, J. Am. Chem. Soc., 2015, 137, 6758–6761 CrossRef CAS PubMed.
- I. Funes-Ardoiz, P. Garrido-Barros, A. Llobet and F. Maseras, ACS Catal., 2017, 7, 1712–1719 CrossRef CAS.
- K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384–3387 CrossRef CAS.
- M. K. Coggins, M.-T. Zhang, Z. F. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed.
- S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586–8600 CrossRef CAS PubMed.
- F. F. Chen, N. Wang, H. T. Lei, D. Y. Guo, H. F. Liu, Z. Y. Zhang, W. Zhang, W. Z. Lai and R. Cao, Inorg. Chem., 2017, 56, 13368–13375 CrossRef CAS.
- T. T. Li and Y. Q. Zheng, Dalton Trans., 2016, 45, 12685–12690 RSC.
- D. L. Gerlach, S. Bhagan, A. A. Cruce, D. B. Burks, I. Nieto, H. T. Truong, S. P. Kelley, C. J. Herbst-Gervasoni, K. L. Jernigan, M. K. Bowman, S. L. Pan, M. Zeller and E. T. Papish, Inorg. Chem., 2014, 53, 12689–12698 CrossRef CAS PubMed.
- C. Lu, J. L. Du, X. J. Su, M. T. Zhang, X. X. Xu, T. J. Meyer and Z. F. Chen, ACS Catal., 2016, 6, 77–83 CrossRef CAS.
- F. S. Yu, F. Li, J. X. Hu, L. C. Bai, Y. Zhu and L. C. Sun, Chem. Commun., 2016, 52, 10377–10380 RSC.
- L. A. Stott, K. E. Prosser, E. K. Berdichevsky, C. J. Walsby and J. J. Warren, Chem. Commun., 2017, 53, 651–654 RSC.
- P. Garrido-Barros, C. Gimbert-Suriñach, D. Moonshiram, A. Picón, P. Monge, V. S. Batista and A. Llobet, J. Am. Chem. Soc., 2017, 139, 12907–12910 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- S. Fukuzumi and Y. Yamada, ChemElectroChem, 2016, 3, 1978–1989 CrossRef CAS.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS.
- V. Artero and J. M. Savéant, Energy Environ. Sci., 2014, 7, 3808–3814 RSC.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS.
- R. Matheu, M. Z. Ertem, J. Benet-Buchholz, E. Coronado, V. S. Batista, X. Sala and A. Llobet, J. Am. Chem. Soc., 2015, 137, 10786–10795 CrossRef CAS PubMed.
- R. Matheu, M. Z. Ertem, C. Gimbert-Suriñach, J. Benet-Buchholz, X. Sala and A. Llobet, ACS Catal., 2017, 7, 6525–6532 CrossRef CAS.
- R. Matheu, M. Z. Ertem, M. Pipelier, J. Lebreton, D. Dubreuil, J. Benet-Buchholz, X. Sala, A. Tessier and A. Llobet, ACS Catal., 2018, 8, 2039–2048 CrossRef CAS.
- J. M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef.
- APEX2 v2009, Bruker AXS, Madison, WI, 2009 Search PubMed.
- G. M. Sheldrick, SADABS, 2008/1, University of Göttingen, Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S17, Table S1, crystallographic data in CIF format for 1, and the X-ray crystallographic coordinates for the structure reported in this article. CCDC 1835967. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04529a |
| This journal is © The Royal Society of Chemistry 2019 |