
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular asymmetric reductive amination: synthesis of enantioenriched dibenz[c,e]azepines†
Tao
Yang
a,
Xiaochong
Guo
a,
Qin
Yin
 *ab and
Xumu
Zhang
*a
*ab and
Xumu
Zhang
*a
aShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, People's Republic of China. E-mail: zhangxm@sustc.edu.cn
bSUSTech Academy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen, Guangdong 518055, People's Republic of China. E-mail: yinq@sustc.edu.cn
First published on 27th December 2018
Abstract
An Ir-catalyzed intramolecular asymmetric reductive amination (ARA) of bridged biaryl derivatives has been described. Using this unprecedented approach, synthetically useful dibenz[c,e]azepines containing both central and axial chiralities are obtained with excellent enantiocontrol (up to 97% ee). This methodology represents a rare example of enantioselective chemocatalytic synthesis of chiral dibenz[c,e]azepines featuring a broad substrate scope, and their synthetic utilities are exhibited by derivatizing the products into a chiral amino acid derivative and chiral phosphoramidite ligands, which display excellent enantiocontrol in Rh-catalyzed asymmetric hydrogenation of α-dehydroamino acid derivatives. Remarkably, our method is also applicable to enantioselectively synthesize an allocolchicine analogue.
Introduction
Dibenz[c,e]azepines are a class of unique 7-membered cyclic amines featuring a biaryl bridge and have attracted increasing attention due to their importance in biological and synthetic chemistry.1 For instance, compound A is the active ingredient in an anti-obesity drug,2 and B is considered to be an allocolchicine analogue.3 In addition, dibenz[c,e]azepine-based compounds C, D, and E and compound F have found promising applications in either organocatalysis or asymmetric transition metal catalysis (eqn (a), Fig. 1). Of particular interest is the phenomenon that a central to axial chirality relay occurs when a stereogenic substituent is placed at the α-position of the nitrogen atom of 2a. Moreover, enantiomerically pure dibenz[c,e]azepines can function as a molecular switch with the axial chirality inverting upon N-Boc derivatisation caused by the center-axis relay effect (eqn (b), Fig. 1).4a | ||
| Fig. 1 (a) Selected examples containing a dibenz[c,e]azepine structural unit; (b) central to axial chirality relay phenomenon; (c) our proposed one pot N-Boc deprotection/intramolecular ARA sequence for the synthesis of enantioenriched dibenz[c,e]azepines. | ||
Despite their biological properties and promising applications in organic synthesis, asymmetric synthesis of this kind of framework lags behind and only a few examples were reported. In 2000, Kündig and coworkers developed a pioneering synthetic strategy towards chiral base C, featuring a Pd-catalyzed atropo-diastereoselective coupling to construct the Ar–Ar bond in a later stage with chiral-pool starting materials.5a Later, by making use of an auxiliary strategy,4 Wallace et al. prepared a conformationally labile secondary amine 2a involving a center-axis stereochemical relay.4a Very recently, Turner and coworkers described an elegant biocatalytic route to access enantiomerically pure dibenz[c,e]azepines by utilizing imine reductase (IRED) biocatalysts.6 To our knowledge, highly efficient chemical catalytic systems capable of directly providing structurally diverse chiral dibenz[c,e]azepines have never been reported.7 The lack of efficient methods toward enantioenriched dibenz[c,e]azepines has greatly impeded deeper studies on the structure–function relationship and synthetic applications of these unique chiral cyclic amines.
Transition metal-catalyzed asymmetric reductive amination represents a straightforward and step-economical method for the synthesis of chiral amines from readily available ketones.8 However, this field remains underdeveloped compared to the well-studied imine asymmetric hydrogenation.9
Recently, intramolecular ARA has exhibited great potential in the construction of chiral 5- and 6-membered N-heterocycles.10 In contrast, related applications for the synthesis of 7-membered N-heterocycles remain underdeveloped. To continue our interest in ARA with molecular H2,8k we wondered if this strategy was also applicable to synthesize chiral 7-membered dibenz[c,e]azepines efficiently. Herein, we report the first enantioselective synthesis of dibenz[c,e]azepines through a one pot N-Boc deprotection/intramolecular ARA sequence (eqn (c), Fig. 1). The synthetic utilities of the enantioenriched products are also presented.
Results and discussion
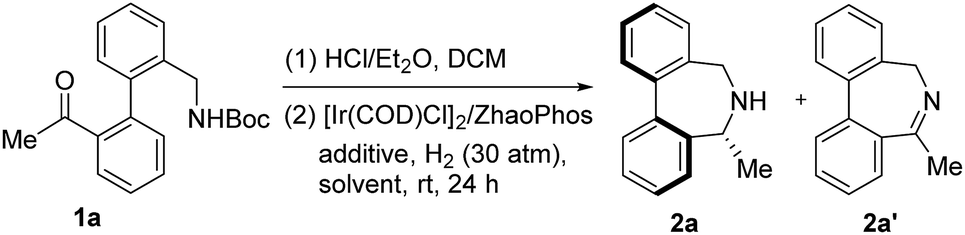
Inspired by Turner's elegant work6 and our recent discovery on ARA,11 aryl-bridged aminoketone 1a was prepared and selected as the model substrate for our research. We envisaged that 1a could undergo N-Boc deprotection promoted by HCl/Et2O and subsequent ARA enabled by the Ir/ZhaoPhos complex to provide enantioenriched product 2a. After the removal of all volatile components from the first step, the resulting mixture was treated with 30 atm of H2 for the ARA step in the presence of a catalyst combination of [Ir(COD)Cl]2 and ZhaoPhos using DCM as a solvent and Ti(OiPr)4 as an additive. Delightfully, the reaction gave the desired product 2a in moderate conversion (65%) albeit with promising enantioselectivity (36% ee), together with the formation of imine intermediate 2a′ (Table 1, entry 1). Unexpectedly, without Ti(OiPr)4, the reduction process did not work, suggesting the key role of Ti(OiPr)4 in accelerating the reduction process.12 Screening of other solvents, such as EtOAc, THF, iPrOH, MeOH, toluene and MeCN, disclosed that iPrOH was the best choice, providing the desired 2a in 77% ee.13 Despite moderate enantiocontrol in the Ir/ZhaoPhos catalytic system, these positive results confirmed our hypothesis and encouraged us to further modify the catalyst combination with the expectation of improving the ee to an excellent level.|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Solvent | Conv.b (%) | 2a/2a′b | 2a: eec (%) |
| a Reaction conditions: 1a (0.1 mmol), [Ir(COD)Cl]2 (0.5 mol%), ZhaoPhos (1.1 mol%), HCl/Et2O (4.0 equiv.), Ti(OiPr)4 (1 equiv.), and solvent (0.6 mL). b Determined by 1H NMR. c Determined by HPLC for the corresponding benzamides. | |||||
| 1 | Ti(OiPr)4 | DCM | >99 | 65/35 | 36 (R, aS) |
| 2 | — | DCM | >99 | 0/100 | — |
| 3 | Ti(OiPr)4 | EtOAc | >99 | 60/40 | 55 (R, aS) |
| 4 | Ti(OiPr)4 | iPrOH | >99 | 86/14 | 77 (R, aS) |
| 5 | Ti(OiPr)4 | THF | >99 | 79/21 | 66 (R, aS) |
| 6 | Ti(OiPr)4 | MeOH | >99 | 75/25 | 72 (R, aS) |
| 7 | Ti(OiPr)4 | Toluene | >99 | 72/28 | 12 (R, aS) |
| 8 | Ti(OiPr)4 | MeCN | >99 | 42/58 | 50 (R, aS) |
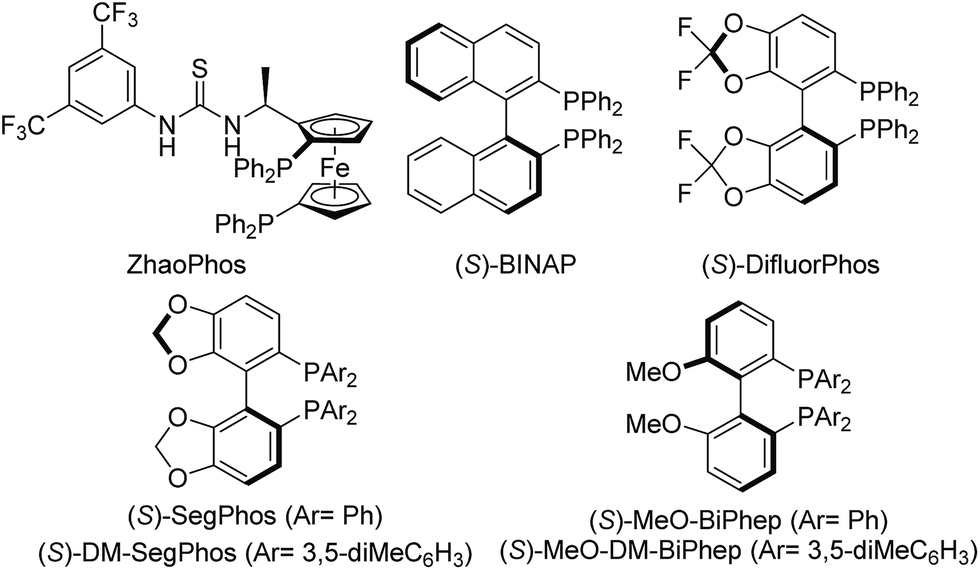
We subsequently evaluated a diverse array of commercially available chiral diphosphine ligands (Table 2). The most popular diphosphine ligand BINAP was first tried, giving the product 2a in high conversion (90%) but low enantioselectivity (40% ee) (Table 2, entry 2). Satisfactorily, double oxygenated diphosphine ligands including SegPhos, MeO–BiPhep and DifluorPhos displayed higher enantiocontrol (Table 2, entries 3–8). Particularly, DifluorPhos delivered the highest enantioselectivity (94% ee; Table 2, entry 8). Lowering the reaction temperature to 20 °C led to a slight improvement of the enantiocontrol (96% ee, 89% yield; Table 2, entry 9). The pressure of H2 was also evaluated, and the conversion slightly dropped to 90% with 20 atm of H2, without affecting the enantioselectivity (Table 2, entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Conv.b (%) | 2a/2a′b | 2a: eec (%) |
| a Reaction conditions: 1a (0.1 mmol), [Ir(COD)Cl]2 (0.5 mol%), ligand (1.1 mol%), HCl/Et2O (4.0 equiv.), Ti(OiPr)4 (1 equiv.) and solvent (0.6 mL). b Determined by 1H NMR; isolated yield of 2a in parentheses. c Determined by HPLC for the corresponding benzamides. d The reaction was conducted at 20 °C. e 20 atm of H2 was used. | ||||
| 1 | ZhaoPhos | >99 | 86/14 | 77 (R, aS) |
| 2 | (S)-BINAP | >99 | 90/10 | 40 (S, aR) |
| 3 | (S)-SegPhos | >99 | >99/1 | 92 (S, aR) |
| 4 | (S)-DM-SegPhos | >99 | >99/1 | 80 (S, aR) |
| 5 | (S)-DTBM-SegPhos | >99 | >99/1 | 40 (S, aR) |
| 6 | (S)-MeO-BiPhep | >99 | >99/1 | 86 (S, aR) |
| 7 | (S)-MeO-DM-BiPhep | >99 | >99/1 | 53 (S, aR) |
| 8 | (S)-DifluorPhos | >99 | >99/1 | 94 (S, aR) |
| 9 | (S)-DifluorPhos | >99 | >99/1 (89) | 96 (S, aR) |
| 10e | (S)-DifluorPhos | >99 | >90/10 | 96 (S, aR) |
|
||||
Under the optimal reaction conditions, the substrate scope was then studied, as depicted in Table 3. We first examined methyl ketones with various substituents (Me, MeO, F, Cl, CF3, etc.) on both of the bridged benzene rings. In general, all tested substrates provided the corresponding enantioenriched dibenz[c,e]azepines in high yields and excellent ees (2b–2k, 87–94% yields, 89–97% ee), regardless of the nature and position of the substituents. Remarkably, pyridine-bridged substrate 1l is also applicable, yielding the corresponding product 2l in 89% yield and 84% ee. It is noteworthy that 2 equivalents of Ti(OiPr)4 are required to enable good conversion, which is possibly due to the competitive coordination of the N atom on the pyridine versus the N atom of imine. Disappointingly, the enantiocontrol is very susceptible to the steric bulk of the substituent R3. Ethyl-substituted substrates (1m) afforded the product with dramatically decreased enantioselectivity (60% ee for 2m).
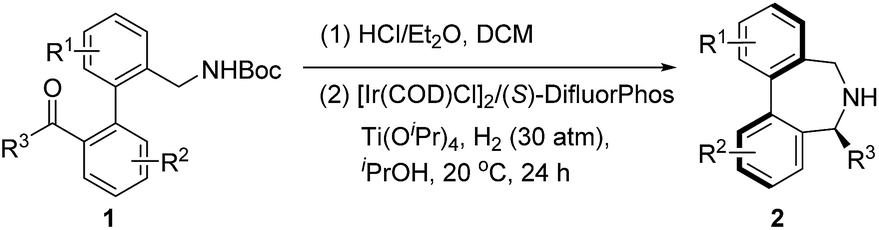

When we applied the above-mentioned standard reaction conditions to diarylketone-derived substrates, inferior performance was unfortunately observed in terms of both reactivity and enantiocontrol. After re-optimizing the reaction conditions with 1n, including the evaluation of the ligands, H2 pressure and the reaction time (see the ESI† for details), we were glad to discover that the desired reductive amination product 2n could be obtained in 72% yield and 89% ee in the presence of the [Ir(COD)Cl]2/(S)-SegPhos complex and 60 atm of H2 for 48 h. The moderate yield of 2n was caused by the low reactivity of the imine intermediate towards reduction.
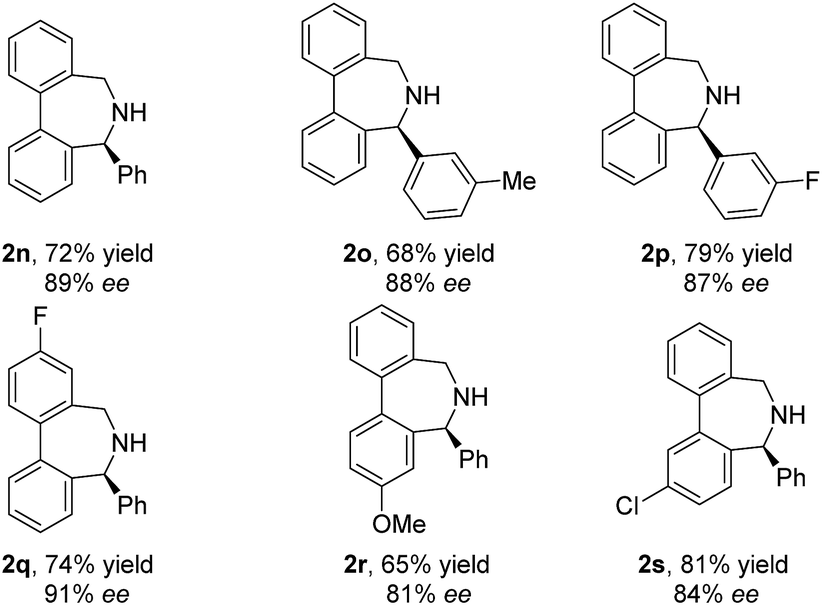
With the slightly revised reaction conditions, we found that several substrates (1o to 1s) contain either a Me, F, MeO or Cl substituent on each benzene ring, as summarized in Table 4. Gratifyingly, all examined substrates underwent the reaction smoothly to yield the corresponding products in decent to good yields with generally good ee (2o–2s, up to 81% yield and 91% ee).

To demonstrate the practical utility of this method, scale-up synthesis of 2a was performed on the 1.5 mmol scale, producing 2a in 88% yield and 96% ee. Dibenz[c,e]azepine B is an analogue of the alkaloid allocolchicine with bioactivity against antimicrotubules and was originally synthesized by Baudoin and co-workers14 from chiral-pool starting materials. We were curious if our catalytic asymmetric method could be applied to achieve the biologically active molecule B. To test our idea, imine hydrochloride salt 3 was prepared because direct ARA was unsuccessful. When 3 HCl was subjected to the standard hydrogenation conditions (DifluorPhos as the ligand), the desired product B was unfortunately obtained with poor enantiocontrol (25% ee). Nevertheless, 81% yield and 75% ee of B were achieved when ZhaoPhos was used as the ligand (Scheme 1).
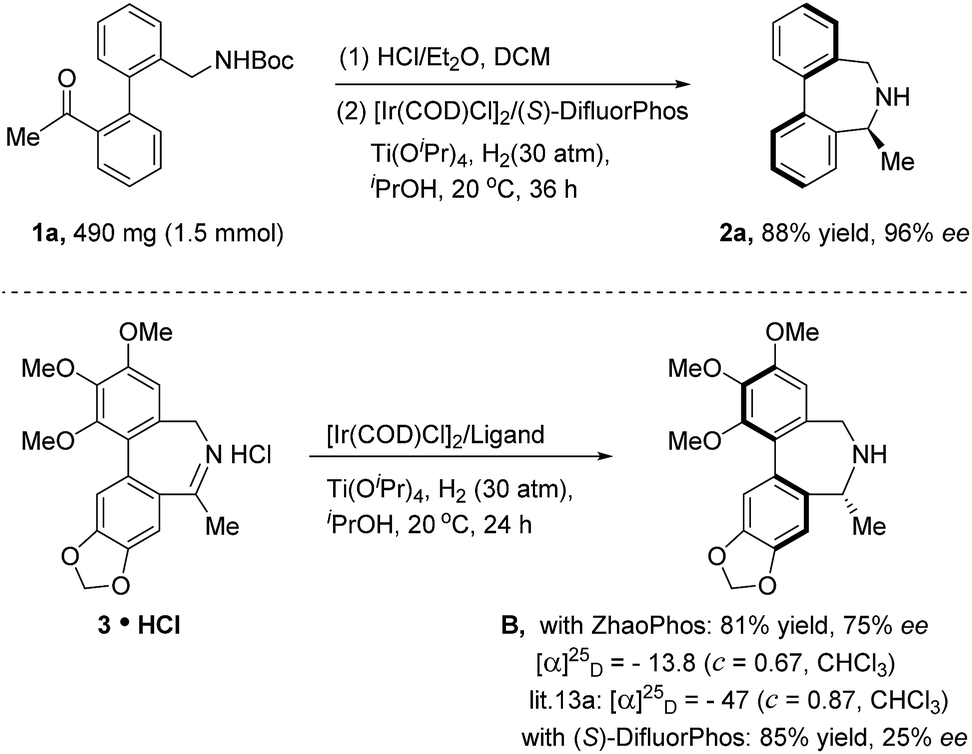 | ||
| Scheme 1 Scale-up synthesis of 2a and asymmetric hydrogenation towards bioactive compound B. | ||
The obtained enantioenriched axially chiral biaryl amines could be easily derivatized into the Kündig amine 4 with a procedure reported by Wallace.4a Likely, treatment of the N-Boc derivative 5, prepared from 2a, with 5 equivalents of Schlosser's “LIDAKOR” base15 at −78 °C, followed by a quenching operation with methyl chloroformate, provided 6 as a mixture of two rotamers.16 Subsequent removal of the Boc group using HCl/Et2O afforded enantioenriched seven-membered cyclic amino acid derivative 7 in a high yield (top, Scheme 2).
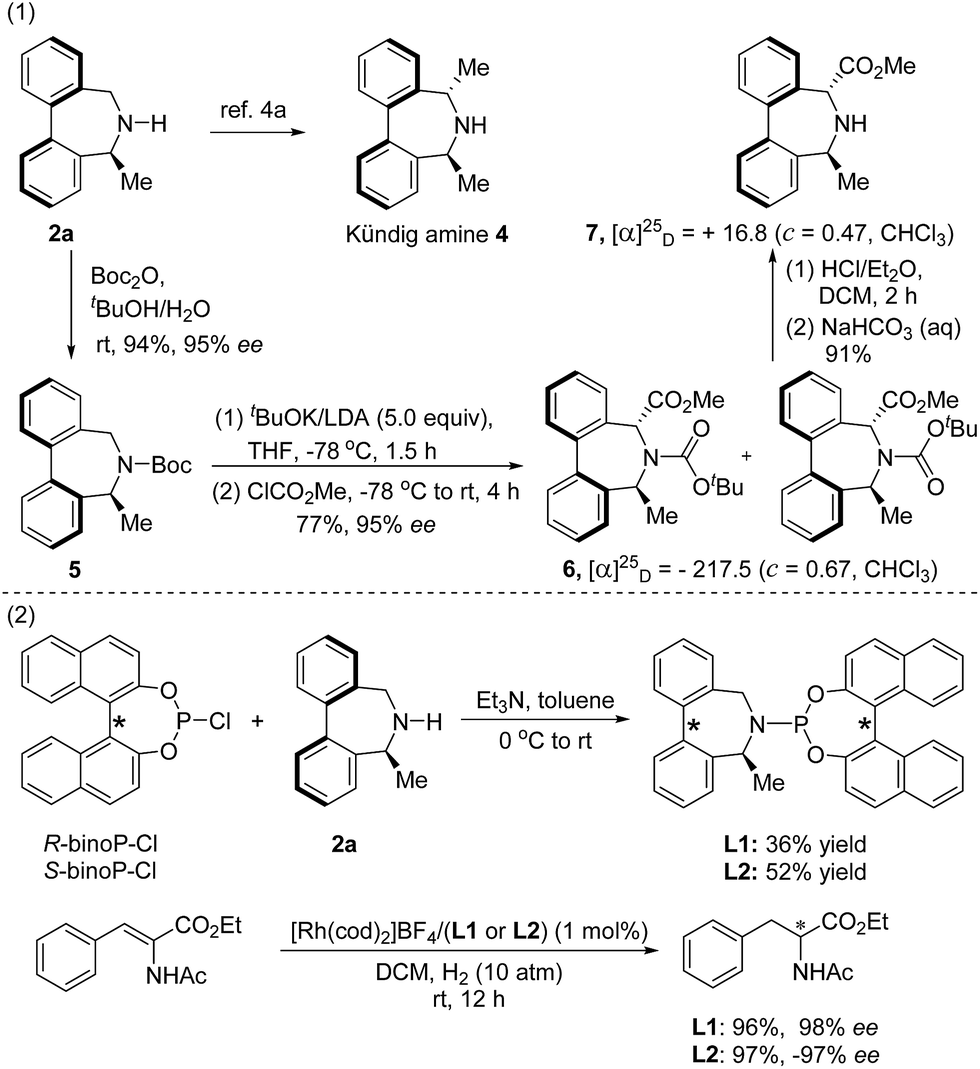 | ||
| Scheme 2 Synthetic applications of 2a. | ||
In addition, two chiral monodentate phosphorus ligands L1 and L2 were efficiently synthesized by condensation of the chiral amine 2a with (R) or (S)-(1,1′-binaphthyl-2,2′-diyl) chlorophosphite. The application of both ligands in rhodium-catalyzed asymmetric hydrogenation of α-dehydroamino acid derivatives17 afforded the hydrogenated product with excellent enantioselectivity (bottom, Scheme 2).
Conclusions
In summary, we have developed a highly enantioselective method towards chiral dibenz[c,e]azepines through a one pot N-Boc deprotection/intramolecular ARA sequence of bridged biaryl derivatives. Under optimal reaction conditions, dibenz[c,e]azepines containing both central and axial chiralities are conveniently obtained with generally excellent enantiocontrol (up to 97% ee). Our method represents an intriguing catalytic enantioselective synthesis of Ar-bridged axially cyclic amines. The synthetic utility of the developed method was exhibited by an unprecedented enantioselective synthesis of an analogue of allocolchicine. Derivatisation of the obtained product into more complicated and synthetically interesting amino acid derivatives and chiral ligands for transition metal catalysis was also performed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Southern University of Science and Technology, the Shenzhen Science and Technology Innovation Committee (No. KQTD20150717103157174, JSGG20160608140847864 and JCYJ20170817110055425), the Shenzhen Nobel Prize Scientists Laboratory Project (C17783101) and the National Natural Science Foundation of China (No. 21432007 and 21801119) for financial support.Notes and references
- (a) X. Gu, Z. Ren, X. Tang, H. Peng, Q. Zhao, Y. Lai, S. Peng and Y. Zhang, Eur. J. Med. Chem., 2012, 51, 137 CrossRef CAS PubMed; (b) S. A. Bright, A. BrinkØ, M. T. Larsen, S. Sinning, D. C. Williams and H. H. Jensen, Bioorg. Med. Chem. Lett., 2013, 23, 1220 CrossRef CAS PubMed; (c) S. Pache, C. Botuha, R. Franz, E. P. Kündig and J. Einhorn, Helv. Chim. Acta, 2000, 83, 2436 CrossRef CAS; (d) T. Kano, H. Sugimoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 18130 CrossRef CAS PubMed; (e) T. Kano, S. Song, Y. Kubota and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 1191 CrossRef CAS PubMed; (f) P. C. B. Page, J. C. Bartlett, Y. Chan, D. Day, P. Parker, B. R. Buckley, G. A. Rassias, A. M. Z. Slawin, S. M. Allin, J. Lacour and A. Pinto, J. Org. Chem., 2012, 77, 6128 CrossRef PubMed; (g) M. Gerdin, M. Penhoat, C. Pétermann and C. Moberg, J. Organomet. Chem., 2008, 693, 3519 CrossRef CAS.
- M. De Lera Ruiz, Y. M. Berlin, J. Zheng, R. G. Aslanian, K. D. McCormick and Q. Zeng, WO2010011653 A1, 2010.
- I. H. Hall, A. R. K. Murthy and S. D. Wyrick, J. Pharm. Sci., 1986, 75, 622 CrossRef CAS PubMed.
- (a) S. L. Pira, T. W. Wallace and J. P. Graham, Org. Lett., 2009, 11, 1663 CrossRef CAS PubMed; (b) S. Postikova, M. Sabbah, D. Wightman, I. T. Nguyen, M. Sanselme, T. Besson, J. F. Brière, S. Oudeyer and V. Levacher, J. Org. Chem., 2013, 78, 8191 CrossRef CAS PubMed; (c) P. C. Bulman Page, C. A. Pearce, Y. Chan, P. Parker, B. R. Buckley, G. A. Rassias and M. R. J. Elsegood, J. Org. Chem., 2015, 80, 8036 CrossRef CAS PubMed.
- (a) L. A. Saudan, G. Bernardinelli and E. P. Kündig, Synlett, 2000, 483 CAS; (b) C. A. Cheetham, R. S. Massey, S. L. Pira, R. G. Pritchard and T. W. Wallace, Org. Biomol. Chem., 2011, 9, 1831 RSC.
- S. P. France, G. A. Aleku, M. Sharma, J. Mangas-Sanchez, R. M. Howard, J. Steflik, R. Kumar, R. W. Adams, I. Slabu, R. Crook, G. Grogan, T. W. Wallace and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 15589 CrossRef CAS PubMed.
- For recent work on the synthesis of chiral dibenzo[b,d]azepines, see J. Liu, X. Yang, Z. Zuo, J. Nan, Y. Wang and X. Luan, Org. Lett., 2018, 20, 244 CrossRef CAS PubMed.
- For reviews and selected examples of TM-catalyzed ARA, see (a) T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753 CrossRef CAS; (b) C. Wang and J. Xiao, Top. Curr. Chem., 2014, 343, 261 CrossRef CAS PubMed; (c) H.-U. Blaser, H.-P. Buser, H.-P. Jalett, B. Pugin and F. Spindler, Synlett, 1999, 867 CrossRef CAS; (d) R. Kadyrov and T. H. Riermeier, Angew. Chem., Int. Ed., 2003, 42, 5472 CrossRef CAS PubMed; (e) R. Kadyrov, T. H. Riermeier, U. Dingerdissen, V. Tararov and A. Börner, J. Org. Chem., 2003, 68, 4067 CrossRef CAS PubMed; (f) L. Rubio-Pérez, F. J. PérezFlores, P. Sharma, L. Velasco and A. Cabrera, Org. Lett., 2009, 11, 265 CrossRef PubMed; (g) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967 CrossRef CAS PubMed; (h) S. Zhu, S. Fleischer, H. Jiao, K. Junge and M. Beller, Adv. Synth. Catal., 2014, 356, 3451 CrossRef; (i) H. Huang, X. Liu, L. Zhou, M. Chang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5309 CrossRef CAS PubMed; (j) D. Steinhuebel, Y. Sun, K. Matsumura, N. Sayo and T. Saito, J. Am. Chem. Soc., 2009, 131, 11316 CrossRef CAS PubMed; (k) X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 2024 CrossRef CAS PubMed; (l) J. Gallardo-Donaire, M. Hermsen, J. Wysocki, M. Ernst, F. Rominger, O. Trapp, A. S. K. Hashmi, A. Schäfer, P. Comba and T. Schaub, J. Am. Chem. Soc., 2018, 140, 355 CrossRef CAS PubMed; (m) P. Yang, L. H. Lim, P. Chuanprasit, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12083 CrossRef CAS PubMed.
- For selected recent reviews on TM-catalyzed asymmetric imine hydrogenation, see (a) J. Xie, S. Zhu and Q. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (b) W. Tang and J. Xiao, Synthesis, 2014, 46, 1297 CrossRef; (c) D. Wang, C. Hou, L. Chen, X. Liu, Q. An and X. Hu, Chin. J. Org. Chem., 2013, 33, 1355 CrossRef CAS.
- For TM-catalyzed intramolecular ARA, see (a) F. Chen, Z. Ding, J. Qin, T. Wang, Y. He and Q. Fan, Org. Lett., 2011, 13, 4348 CrossRef CAS PubMed; (b) Y. Zhang, Q. Yan, G. Zi and G. Hou, Org. Lett., 2017, 19, 4215 CrossRef CAS PubMed; (c) B. Song, C. Yu, Y. Ji, M. Chen and Y. Zhou, Chem. Commun., 2017, 53, 1704 RSC; (d) N. A. Strotman, C. A. Baxter, K. M. J. Brands, E. Cleator, S. W. Krska, R. A. Reamer, D. J. Wallace and T. J. Wright, J. Am. Chem. Soc., 2011, 133, 8362 CrossRef CAS PubMed; (e) G. D. Williams, R. A. Pike, C. E. Wade and M. Wills, Org. Lett., 2003, 5, 4227 CrossRef CAS PubMed; (f) H. Zhou, Y. Liu, S. Yang, L. Zhou and M. Chang, Angew. Chem., Int. Ed., 2017, 56, 2725 CrossRef CAS PubMed; (g) C. Yu, J. Wang and Y. Zhou, Org. Chem. Front., 2018, 5, 2805 RSC.
- T. Yang, Q. Yin, G. Gu and X. Zhang, Chem. Commun., 2018, 54, 7247 RSC.
- The real role of Ti(OiPr)4 is unclear at this stage. We also tested some other Lewis acids including AlCl3, InCl3, FeCl3, Zn(OTf)2, BF3·Et2O and Ti(OEt)4, and the results disclosed that only titanium Lewis acid could efficiently accelerate the transformation (see the ESI† for details).
- Deuterium-label experiments with D2 were conducted, which excluded i-PrOH as a potential hydrogen-transfer reagent and the possibility of involvement of hydrogenated enamine intermediates in this case (see the ESI† for details).
- (a) A. Joncour, A. Décor, S. Thoret, A. Chiaroni and O. Baudoin, Angew. Chem., Int. Ed., 2006, 45, 4149 CrossRef CAS PubMed; (b) A. Joncour, A. Décor, J.-M. Liu, M.-E. Tran Huu Dau and O. Baudoin, Chem.–Eur. J., 2007, 13, 5450 CrossRef CAS PubMed.
- S. Takagishi and M. Schlosser, Synlett, 1991, 119 CrossRef CAS.
- For studies on amide rotamers, see (a) W. E. Stewart and T. H. Slidell, Chem. Rev., 1970, 70, 517 CrossRef CAS; (b) M. Oki, The Chemistry of Rotational Iso-mers, Springer-Verlag, New York, 1993 CrossRef.
- Rh-catalyzed asymmetric hydrogenation of α-dehydroamino acid derivatives has been recognized as a model reaction to test the effectiveness of new chiral phosphoramidite ligands. See (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) Y. Fu, X. Guo, S. Zhu, A. Hu, J. Xie and Q. Zhou, J. Org. Chem., 2004, 69, 4648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04482a |
| This journal is © The Royal Society of Chemistry 2019 |