
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen uptake in complexes related to [NiFeS]- and [NiFeSe]-hydrogenase active sites†
Xuemei
Yang
,
Lindy C.
Elrod
 ,
Joseph H.
Reibenspies
,
Michael B.
Hall
* and
Marcetta Y.
Darensbourg
*
,
Joseph H.
Reibenspies
,
Michael B.
Hall
* and
Marcetta Y.
Darensbourg
*
Texas A&M University, Department of Chemistry, College Station, TX 77843, USA. E-mail: marcetta@chem.tamu.edu
First published on 5th November 2018
Abstract
A biomimetic study for S/Se oxygenation in Ni(μ-EPh)(μ-SN2)Fe, (E = S or Se; SN2 = Me-diazacycloheptane-CH2CH2S); Fe = (η5-C5H5)FeII(CO) complexes related to the oxygen-damaged active sites of [NiFeS]/[NiFeSe]-H2ases is described. Mono- and di-oxygenates (major and minor species, respectively) of the chalcogens result from exposure of the heterobimetallics to O2; one was isolated and structurally characterized to have Ni–O–SePh–Fe–S connectivity within a 5-membered ring. A compositionally analogous mono-oxy species was implicated by ν(CO) IR spectroscopy to be the corresponding Ni–O–SPh–Fe–S complex; treatment with O-abstraction agents such as P(o-tolyl)3 or PMe3 remediated the O damage. Computational studies (DFT) found that the lowest energy isomers of mono-oxygen derivatives of Ni(μ-EPh)(μ-SN2)Fe complexes were those with O attachment to Ni rather than Fe, a result consonant with experimental findings, but at odds with oxygenates found in oxygen-damaged [NiFeS]/[NiFeSe]-H2ase structures. A computer-generated model based on substituting −SMe for the N-CH2CH2S− sulfur donor of the N2S suggested that constraint within the chelate hindered O-atom uptake at that sulfur site.
Introduction
Hydrogenases are metalloenzymes that catalyze reversible H2 production from protons and electrons. Likely originating in pre-biotic ages and under a reducing atmosphere, the active sites contain iron, nickel and sulfur, along with simple diatomic ligands, CO and CN, in optimal arrangements that produce superb biocatalysts found throughout nature.1 These structures offer guidance for design of molecular catalysts comprising earth abundant metals for application in electrolyzers and fuel cells.2,3 While the protein superstructures protect the biological redox centers from O2 as an oxidant that competes with protons, there is ample evidence, including protein crystal structures, of the detrimental effects of O2 invasion into the active site. Indeed the initial understanding of the [NiFeS]-H2ase was plagued with Ni-based EPR signals from various deactivated enzyme states damaged by partial oxidation or oxygenation. Vague terms such as “ready” and “unready” states, with recovery from oxygen damage on the order of seconds vs. hours, respectively, were adopted in attempts to express reductive (added H2 or H+ + e−) reactivation requirements.4,5As shown in Fig. 1 two subclasses exist for the [NiFe]-H2ase enzymes. The predominant form, [NiFeS]-H2ase, contains a set of four cysteines around Ni; two Cys-S bridge Ni to an FeII(CO)(CN)2 unit. A second form, [NiFeSe]-H2ase is identical to the [NiFeS]-H2ase except a terminal cysteine is replaced by selenocysteine. As the incorporation of selenocysteine requires an intricate dedicated biosynthetic machinery, as well as a high energetic cost, this form is found in few microorganisms; an example is Methanococcus voltae.6,7
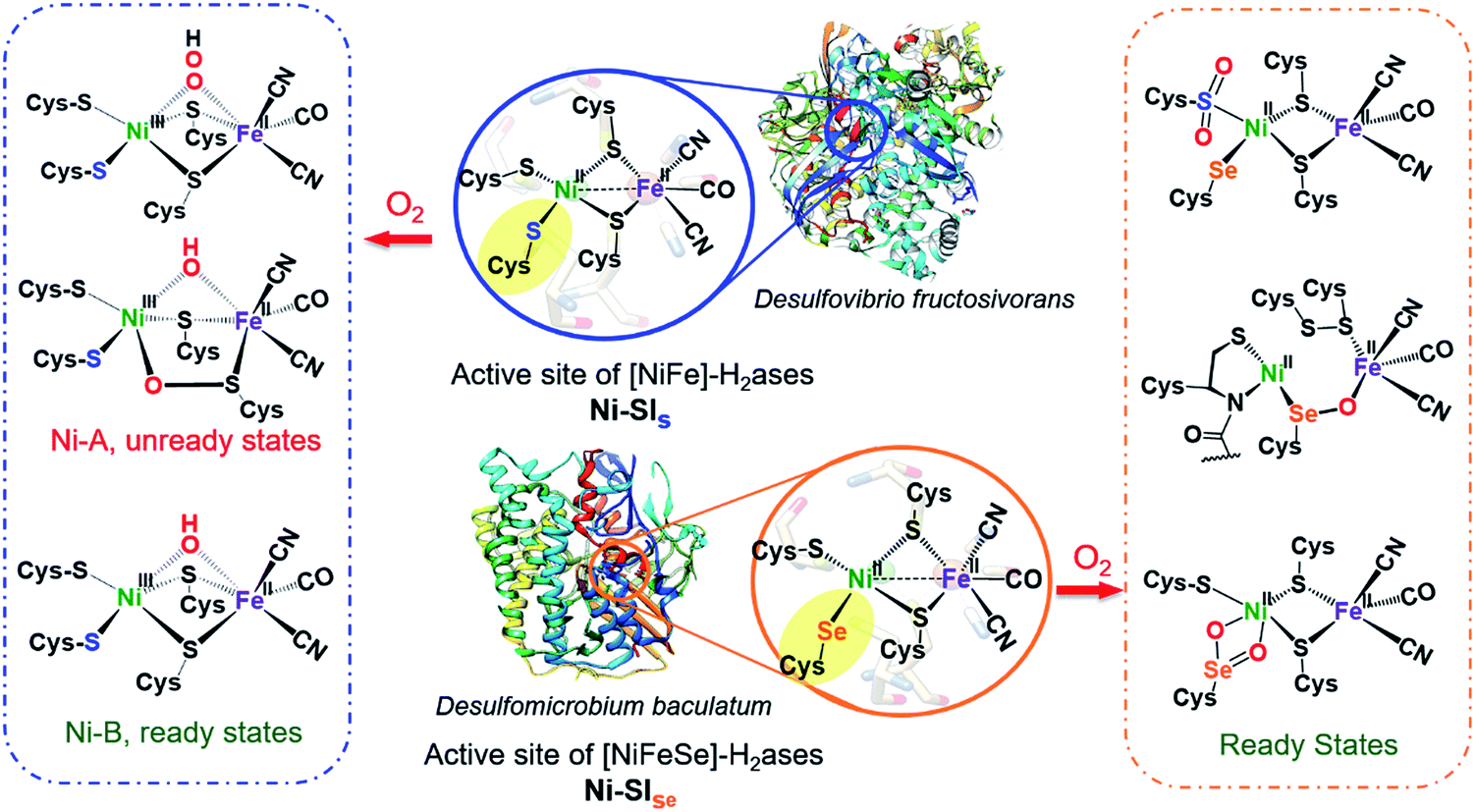 | ||
| Fig. 1 Reduced (middle) active sites of [NiFe]-H2ases and selected oxidized active sites of [NiFeS] (left) and [NiFeSe]-H2ases (right).8,9,15,16 | ||
Despite sharing similar active site structures, [NiFeSe]-H2ase is deemed superior to the all-sulfur, [NiFeS]-H2ase, with respect to H2 production and O2 tolerance, the latter defined as resistance to O2 and recovery of full activity on appropriate reductive treatment.4,8,9 Fundamental differences between sulfur and selenium include the higher acidity of Se-Cys, which might account for the proton reduction disparities, while the low redox potential and high nucleophilicity of selenium in selenolates relative to sulfur in thiolates might account for the higher oxygen-tolerance of [NiFeSe]-H2ase.7
Both the ready and unready states of [NiFeS]-H2ase are identified as containing NiIII; the ready state contains a hydroxide bridge between Ni and Fe, (Ni–B), while the unready state is proposed to contain a κ1-peroxide bridge or a sulfenate within a 5-membered Ni–O–Scys–Fe–Scys ring (Ni–A), as depicted in Fig. 1.10,11 However, in [NiFeSe]-H2ase, there are thus far revealed only rapid recovery or ready states comprised of oxygenates of Se- and S-cysteines; selected structures are shown in Fig. 1.9 Notably, multiple levels of oxidized chalcogenide sites, both S and Se, have been uncovered in [NiFeSe]-H2ase; nevertheless the basic NiFe core structure is maintained and reductive repair is possible. As these oxygenated chalcogenides play vital roles in the reversibility of oxygen damage, small molecule analogues are needed to explore aerobic damage and repair, both for insight into the enzyme as well as the design of robust synthetic catalysts. Examples are limited.
Ogo, et al. isolated a high valent iron(IV) peroxo complex on reacting solvent-coordinated complexes [NiIILFeII(RCN)(η5-C5Me5)]+ with O2.12–14 However, no oxygenated sulfur species were reported from their system. The first S-oxygenated [NiFe] complex was reported by Driess's lab as a model for sulfenate intermediates in O2-tolerant hydrogenase; it was synthesized from FeBr2 and the pre-formed sulfenato nickel complex17 rather than direct oxygenation.18
Herein, we describe a biomimetic study for S/Se oxygenation in complexes Ni(μ-EPh)(μ-SR′)Fe, (E = S or Se; R′ = N2S) with certain features of the [NiFeS]/[NiFeSe]-H2ases' active sites. We report the first XRD structure of an oxygenate of selenium within a Ni–Fe complex and use it as a reference point for DFT computational analyses and predictions.
Results and discussion
Synthesis and characterizations
Nickel dithiolate complexes such as NiN2S2 are well known to react as metalloligands by formation of Ni-(μ2-SR)2-M′ bridges.19 Adopting a [NiN2S]22+ bimetallic nickel dimer20 as platform for dimer cleavage reactions,19,21,22 we have derived monomeric Ni(N2S)(EPh) (E = S and Se, complexes A and B, respectively). These cis-dichalcogenides are subsequently used as metalloligands to an iron receiver unit, (η5-C5H5)FeII(CO)+, in analogy to well-known NiN2S2 derivatives.23 The synthetic scheme shown in Fig. 2 is further detailed (mass spectra, elemental analyses, 1H and 13C NMR spectra, and XRD crystal structures) in the ESI.† The lower value of ν(CO) (1934 cm−1) in the NiSePhFe+, complex 2, as contrasted to NiSPhFe+, complex 1 (1939 cm−1), is attributed to the better electron donor properties of Se over S,7 resulting in better pi-backbonding from FeII to the CO reporter ligand.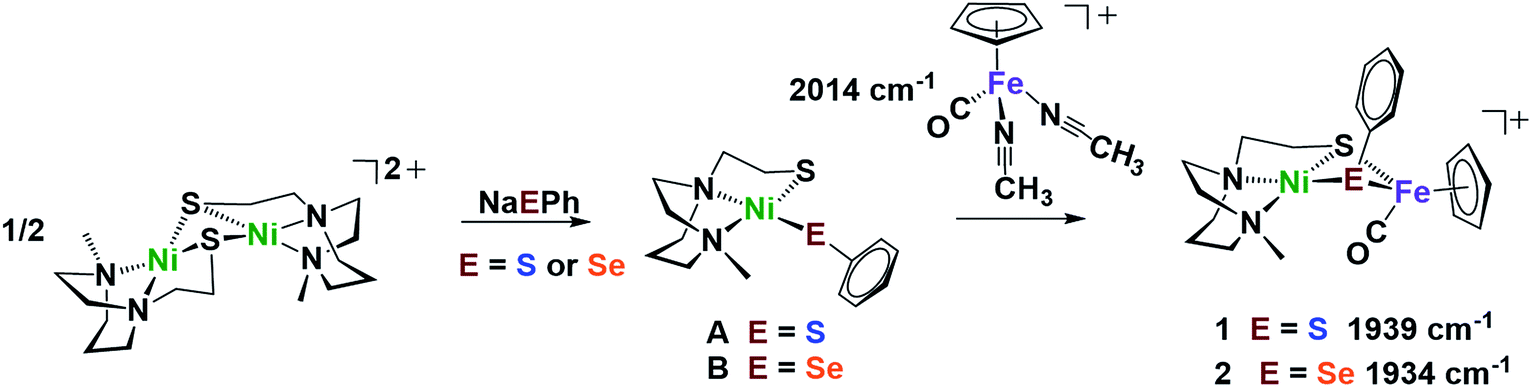 | ||
| Fig. 2 Synthetic scheme for NiEPhFe+ complexes 1 and 2. The ν(CO) IR values of the products recorded in CH2Cl2. | ||
X-ray quality crystals of monomeric Ni complex A were obtained by diethyl ether vapor transfer into a solution of CH3CN. Complex B and the Ni–Fe complexes (1, 2 and 2′), were crystallized from a pentane-layered CH2Cl2 solution at −35 °C. Polymorphs, 2 and 2′, are distinguished by the orientation of the SePh planes in the NiSePhFe+ complexes. Dark red needle crystals of 2 exist in a pbca space group; 2′, as dark red blocks, is in the P21/c space group.
Molecular structures determined by XRD are unexceptional, Fig. 3. The >3.0 Å distances between Ni and Fe are beyond bonding. The NiEPhFe+ complex structures find a typical piano-stool geometry exists about the [(η5-C5H5)Fe(CO)]+ unit, and, as usual for bridging dithiolates, butterfly-like cores are due to the lone pair-imposed steric requirement of the chalcogenide bridges. Hinge angles, defined as the intersection of the best N2SE plane with the SEFe plane, are ca. 140–145°. The selenium-containing complexes, 2 and 2′ contain hinge angles slightly larger than found in 1.
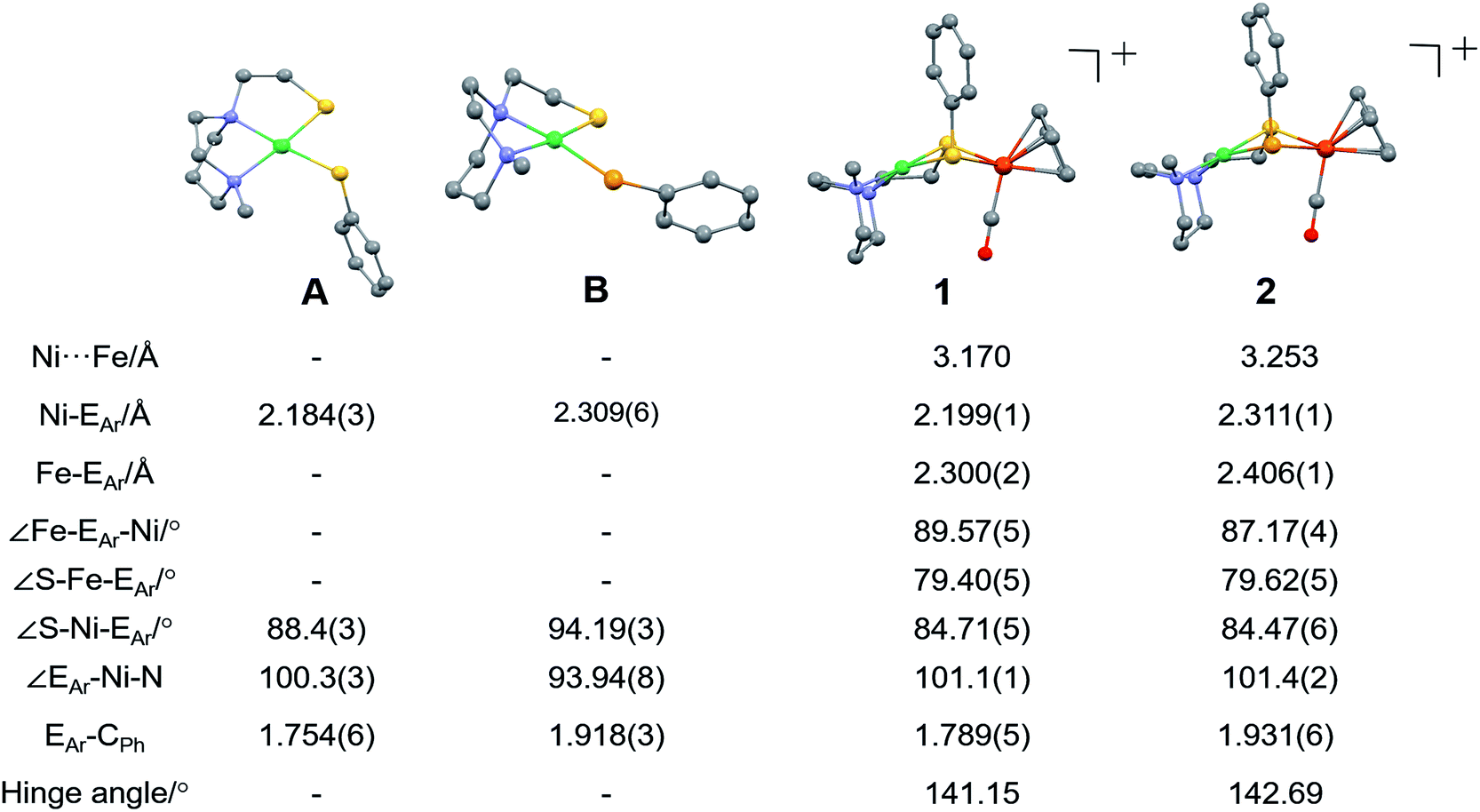 | ||
| Fig. 3 Molecular structures of A, B and 1 (NiSPhFe), 2 (NiSePhFe), determined by single-crystal XRD, with the BF4− ions and H atoms omitted. E in A and 1 is sulfur; E in B and 2 is selenium. | ||
Reactivity
The chemical responsiveness of the heterobimetallic complexes was illustrated by reaction with CO(g) which, in both the NiSPhFe+ and NiSePhFe+ complexes, resulted in Ni–Fe splitting by transferring the EPh to Fe, generating (η5-C5H5)Fe(CO)2EPh, and returned the Ni to its [NiN2S]22+ dimeric form. On mixing complex 1 with an excess of Na+SePh− in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2/MeOH, complex 2 is formed over the course of hours. The opposite, i.e., an attempt to replace the bridging SePh− in complex 2 by SPh−, was unsuccessful. These reactions emphasize the mobility of the EPh− unit in the NiEPhFe+ complex.
:1 CH2Cl2/MeOH, complex 2 is formed over the course of hours. The opposite, i.e., an attempt to replace the bridging SePh− in complex 2 by SPh−, was unsuccessful. These reactions emphasize the mobility of the EPh− unit in the NiEPhFe+ complex.
At 22 °C, O2 gas was bubbled through CH2Cl2 solutions of heterobimetallic complexes 1 and 2/2′; the reactions were monitored by FTIR spectroscopy. The clear reddish black solution of NiSePhFe+ developed into a slightly cloudy, dark red solution after 2 h, with ν(CO) shifting from a sharp band at 1934 cm−1 to a broader band at 1954 cm−1, designated as complex 2+O (Fig. 4). On filtration, the mother liquor mainly contained complex 2+O and, on removal of solvent and redissolving in CH2Cl2, a high resolution ESI-MS+ showed the parent molecular ion to have m/z 552.9634 (calc. for [2+O]+, 552.9661). This mono-oxygenate was isolated in ca. 50% yield (Fig. S20†). The uptake of two oxygen atoms was additionally indicated by a minor signal at m/z 568.9419 (calc. for [2+2O]+, 568.9611). The residue, which was soluble in CH3CN, was confirmed as the [NiN2S]22+ dimer by XRD with crystals obtained by ether diffusion; it was isolated in 35% yield. A remaining insoluble yellow solid was found to be an iron(III) oxide complex (see ESI†). Reactions performed in air instead of pure O2 yielded (more slowly) the same products.
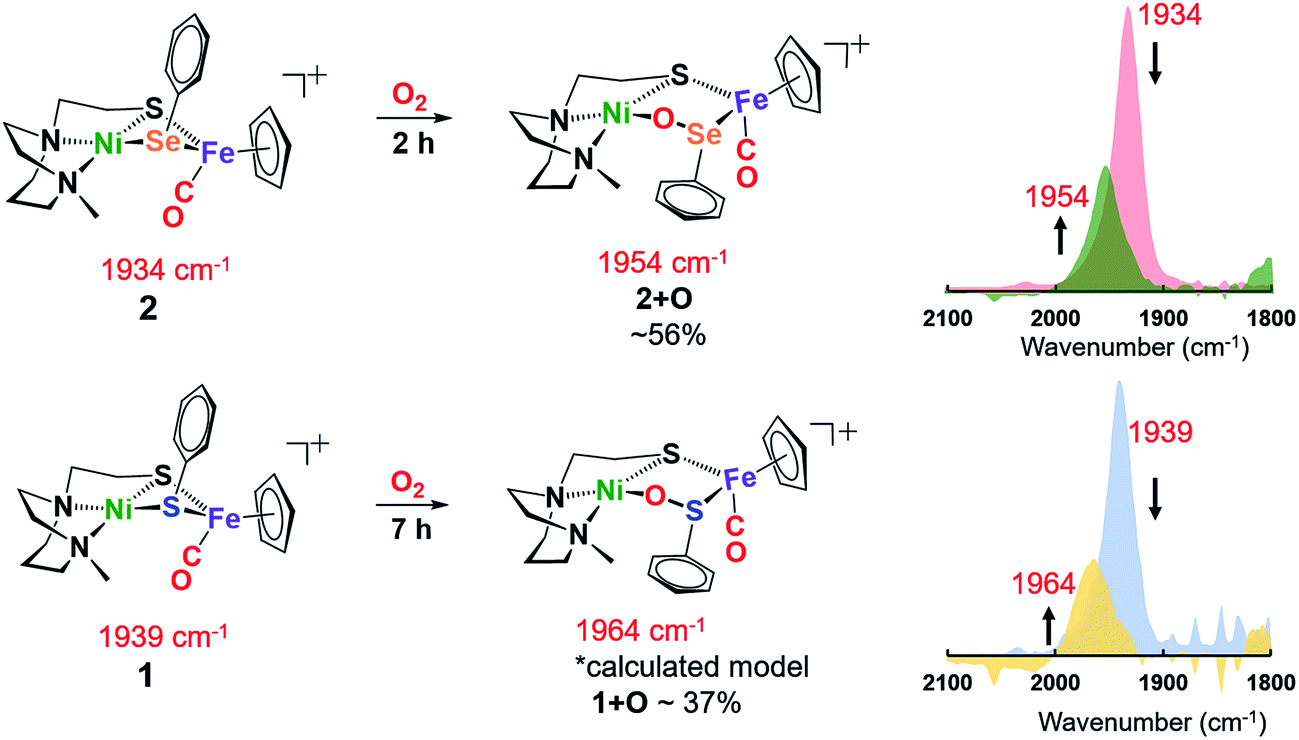 | ||
| Fig. 4 Reactions of 1 and 2 in the presence of 1 atm O2 in CH2Cl2 at 22 °C; conversions determined by ν(CO) analysis. | ||
The O2-bubbled CH2Cl2 solution of the all-sulfur analogue NiSPhFe+, 1, required 7 h to completion, indicated by the decrease in IR absorption at ν(CO) 1939 cm−1 as the oxidized species appeared as a broad band at ν(CO) 1964 cm−1 (Fig. 4). The reactivity (time) difference is consistent with the fact that S2− is oxidized with more difficulty than Se2−.7 The high resolution ESI-MS+ of products from 1 and O2 in CH3CN indicated the uptake of one, two, and three oxygens (Fig. S19†). Based on DFT calculations (vide infra), complex 1+O is of a similar structure as 2+O. Both 1+O and 2+O show sharp signals in their NMR spectra and are EPR-silent, indicating that Ni and Fe are low spin, +2 species (Fig. S30, S31 and S38†). To confirm that the 1+O and 2+O were derived from molecular O2, the same reactions were carried out with added 18O2. Mass spectral analysis indicated the uptake of labelled O-18, Fig. S43 and S44.†
Crystals of complex 2+O were obtained by layering pentane on DCM solutions of 2+O. As seen in Fig. 5, complex 2+O contains a 5-membered, puckered NiOSeFeS ring; the Ni(II) is the center of a square planar N2SO binding site. From XRD the bond distances of Ni–S and Fe–S in 2+O are largely the same as in the reduced form 2 (or 2′); the oxygen insertion into the Ni–Se bond results in an Ni–O bond at 1.870(2) Å. The Fe–Se bond length becomes slightly shorter in 2+O, 2.333(1) Å, as compared to 2, 2.406(1) Å. The distance between Ni and Fe in 2+O is 3.568 Å, ca. 0.3 Å longer than in the reduced form, reflecting the expansion of the ring. Note that the Ni–A form of the [NiFe]-H2ase enzyme active site with sulfenate also has a 5-membered NiOSFeS ring, Fig. 1.24 The Ni⋯O distance in Ni–A is 1.824 Å, while in 2+O it is 1.870(2) Å. In contrast, a minor fraction from [NiFeSe]-H2ase oxidation reported by Volbeda et al. has a different connectivity, NiSeOFe, Fig. 1.9 Complex 2+O is, to our knowledge, the first model of an oxygenated [NiFeSe]-H2ase synthetic analogue derived from direct reaction.
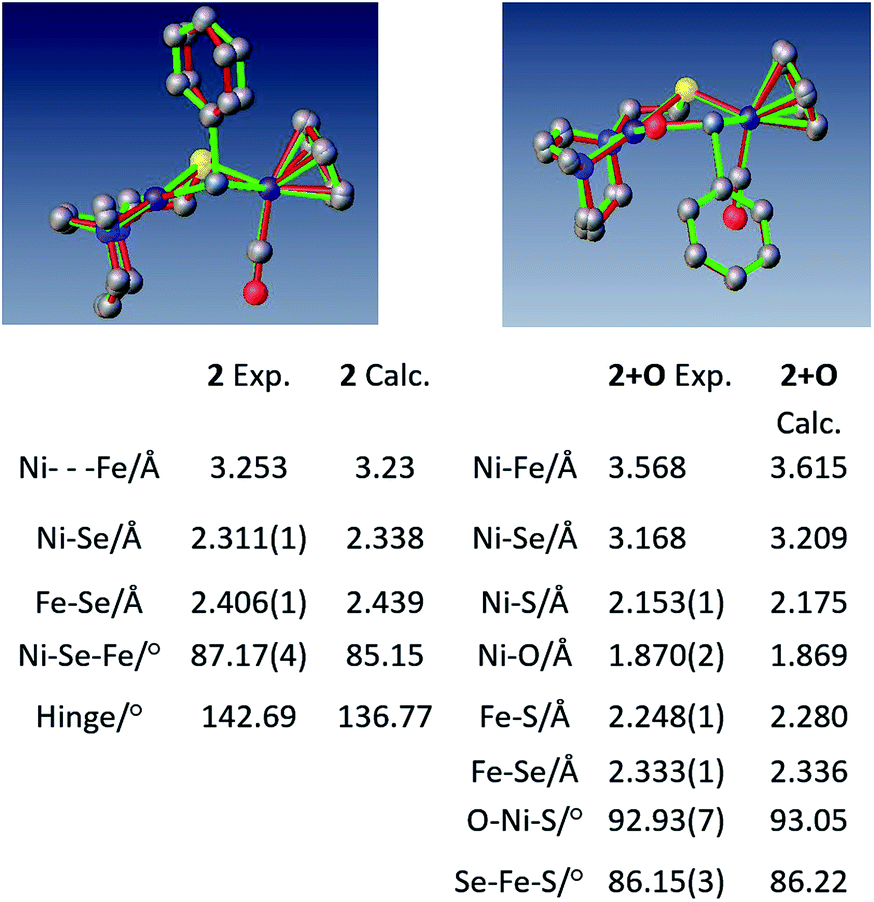 | ||
| Fig. 5 Overlay of experimental and computational structures of 2 and 2+O along with selected geometric parameters. Hydrogens deleted for clarity. | ||
As a comparison to the NiEPhFe complexes 1 and 2, constrained NiN2S2 derivatives, [NiN2S2·Fe(η5-C5H5)(CO)]+[BF4]− and [NiN2S2·Fe(η5-C5Me5)(CO)]+[BF4]− N2S2 = bismercaptoethanediazacycloheptane) were tested for reactivity with oxygen.25,26 After stirring their solutions in an O2 atmosphere for several days, they remain intact with no indication of reaction. We surmise that the CO which is bound to Fe prevents O2 activation at the Fe center and both sulfur and nickel are deactivated towards O2 reactivity in the rigid chelating N2S2 ligands. However, in complexes 1 and 2, the mobility at EPh (E = S or Se) provides a potential site for O2 attack on the Ni or Fe, consistent with the splitting of 1 and 2 by CO(g) (Fig. S33†). For comparison, elevated temperature (70 °C) and 11 bar CO(g) are required to break one Ni–S bond in [NiN2S2·Fe(η5-C5H5)(CO)]+[BF4]−.25
Computational section
The crystal structures of 2 and 2+O were used as geometric starting points for DFT calculations (TPSSTPSS functional with the 6-311++G(d,p) basis set on all nonmetal atoms and the 6-311+G basis set for Ni and Fe atoms) and then optimized using the Gaussian 09 program using the SMD implicit solvation model with acetonitrile as the solvent.28 Cartesian coordinates of calculated structures are given in the ESI.† The calculated structures for 2 and 2+O showed excellent agreement with the corresponding experimental structures, Fig. 5. Similar structures were found for the sulfur analogues, 1 and 1+O.The structure of 2+O was investigated to determine contributing factors to the thermodynamically favored oxygenation product. A less rigid model of 2+O was made by breaking the carbon–carbon bond linking the sulfur donor to the neighbouring nitrogen donor, changing the resulting methylene fragments into methyl groups, and replacing the phenyl group on Se-Ph with a methyl group. This bidentate N2SMeSeMe model, with unconnected SMe, is designated as 3 and is designed to create similar electronic, steric environments and constraints around the sulfur and selenium atoms.
Four oxygenated isomers, 3+Oa–d, as well as four corresponding isomers, 2+Oa–d, were calculated in which the location of the oxygen atom was varied according to the scheme described in Fig. 6. The isomer 2+Oa, which corresponds to the experimental structure, is indeed the lowest energy isomer of 2+O; the next lowest isomer, 2+Ob, is 8.6 kcal higher in energy. This contrasts with the N2SMeSeMe model in which the lowest energy isomer, 3d, has the oxygen atom inserted between the nickel and the sulfur atoms rather than the nickel and selenium. The corresponding isomer 2+Od is the highest energy isomer for the tridentate model and the oxygen atom appears in a position bridging the nickel sulfur bond rather than full insertion. This is likely due to the geometric restraints imposed by the tridentate N2S ligand that disfavor the addition of the oxygen atom into its rigid binding arrangement. This constraint is not found in the more open bidentate model with separated SMe donors. There it is found that formation of a sulfur–oxygen bond is thermodynamically favored, by 3.2 kcal mol−1, as compared to the formation of a selenium–oxygen bond.
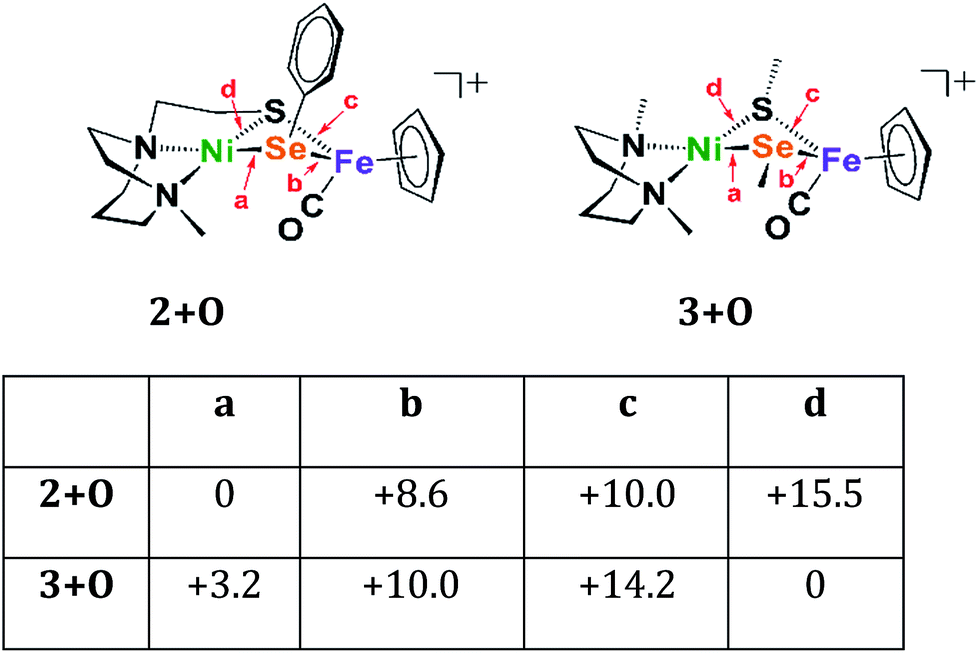 | ||
| Fig. 6 Relative energies of oxygenated isomers of N2SSePh model, 2+O, and the bidentate, separated donors in the N2SMeSeMe model, 3+O, in kcal mol−1. A range of functionals and basis sets were explored and found to elicit minor differences on energies of complexes but do not change the ordering of isomers' energies (Fig. S41†). | ||
O-atom removal results
As arbiters between O2 and the [NiFe] or [NiFeSe]-H2ase active sites, chalcogenides prevent complete degradation of the organometallic active sites of hydrogenases, and perform this role reversably.24,27 To explore the possibility of reversal of oxygenated selenate/sulfenate, O-abstracting agents, PR3 (R = Me or o-tolyl), were employed for O-atom removal (Fig. 7). The ν(CO) monitor of the reaction of 2+O and 1 equiv. of PMe3 in DCM showed a band shift from 1954 to 1934 cm−1. The latter absorption is made up from a mixture of complex 2 and the PMe3 species (η5-C5H5)Fe(SePh)(PMe3)(CO), which is confirmed by the +ESI-MS data (Fig. S37†). The reaction of a mixture of 1+O and 1+2O with 1 equiv. of PMe3 resulted in partial conversion of 1+O to 1, finding that 1+2O was unchanged, even in presence of excess PMe3. The +ESI-MS (Fig. S36†) indicates the formation of reduced complex 2, unreacted 1+2O and the PMe3 derivative (η5-C5H5)Fe(SPh)(PMe3)(CO). Removal of the O-atom from 1+O and 2+O by reducing agents containing H−, as well as electrochemically (e− + H+), have thus far been unsuccessful.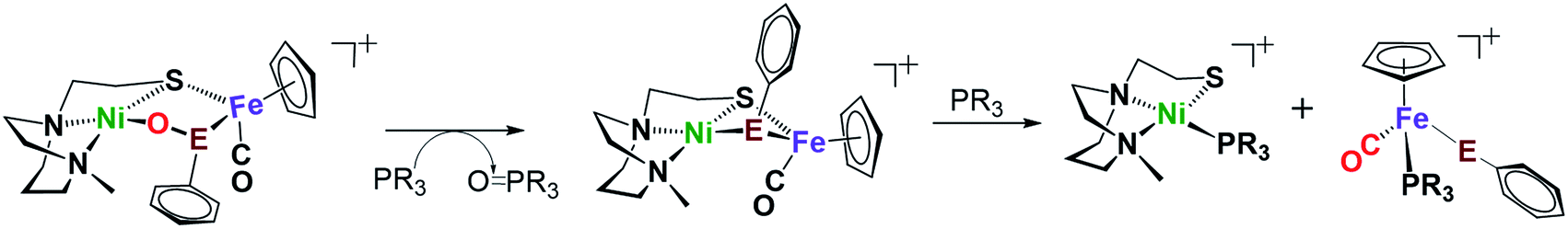 | ||
| Fig. 7 Reaction of complex 2+O (E = Se) or 1+O (E = S) with O-abstracting agents, PR3 (R = Me or o-tolyl). | ||
Conclusion
In summary, we have observed oxygenation and O-atom removal from two biomimetic complexes with features of the [NiFeS]/[NiFeSe]-H2ase active sites. Our study highlights the usefulness of the [NiN2S]22+ synthetic platform for generating NiFe biomimetic complexes. Clearly the NiEPhFe+ complex differs from the active site of [NiFeSe]-H2ase as the selenium in the model is in a bridging position, rather than terminal as nature has adopted. The relative reactivities however are consistent with what is found in nature. As compared to complex 1, the selenium-bridged complex 2 required a shorter time to generate oxygenated selenium, 2+O. The partial conversion of oxygenated chalcogenides (Se or S oxidation states of 0) back to reduced S/Se (oxidation state −2) in this study provides a foundation for the mechanism of reactivation of S/Se-oxygenated [NiFeE]-H2ases. Oxygen-uptake by S/Se in [NiFeS]- and [NiFeSe]-H2ases results in modification of the active site, but less severe than would result in irreparable degradation.27 During the oxygenation and O-atom removal from the chalcogen atom, we note that the mobility in the Ni–E bond plays a vital role.The variety of oxygenates thus far found in nature, Fig. 1, speak to the ability of the chalcogens to maintain a close attachment to the NiFe site. Detailed O2/H2ase active site reaction mechanisms remain obscure but of great interest.29,30 Our studies of model complexes point to the possibility of opening the Ni–E bond in the Ni–(μEPh)–Fe unit. Such bond cleavage exposes a reactive lone pair on sulfur or selenium that attracts the electrophilic O2 molecule concomitant with providing an open site on Ni2+ for assisted activation, leading to the product. A related strategy for protection of the active site from oxidative damage is seen in the MBH (membrane-bound respiratory [NiFeS]-hydrogenase) wherein the proximal [4Fe-3S] cluster donates two electrons and one proton for reduction of adventitious O2.31
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Foundation (CHE-1300787, CHE-1664866 to M. B. H. and CHE-1266097, CHE-1665258 to M. Y. D.) and the Robert A. Welch Foundation (A-0648 to M. B. H. and A-0924 to M. Y. D.).Notes and references
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081 CrossRef CAS PubMed.
- L. L. Beer, E. S. Boyd, J. W. Peters and M. C. Posewitz, Curr. Opin. Biotechnol., 2009, 20, 264 CrossRef CAS PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015 CrossRef CAS PubMed.
- C. S. A. Baltazar, M. C. Marques, C. M. Soares, A. M. DeLacey, I. A. C. Pereira and P. M. Matias, Eur. J. Inorg. Chem., 2011, 948 CrossRef CAS.
- D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2015, 8, 2283 RSC.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206 CrossRef CAS PubMed.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821 CrossRef CAS PubMed.
- M. C. Marques, R. Coelho, A. L. De Lacey, I. A. Pereira and P. M. Matias, J. Mol. Biol., 2010, 396, 893 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, M. Iannello, A. L. De Lacey, C. Cavazza and J. C. Fontecilla-Camps, Chem. Commun., 2013, 49, 7061 RSC.
- M. Horch, L. Lauterbach, M. A. Mroginski, P. Hildebrandt, O. Lenz and I. Zebger, J. Am. Chem. Soc., 2015, 137, 2555 CrossRef CAS PubMed.
- H. Ogata, W. Lubitz and Y. Higuchi, Dalton Trans., 2009, 7577 RSC.
- M. Isegawa, A. K. Sharma, S. Ogo and K. Morokuma, Organometallics, 2018, 37, 1534 CrossRef CAS.
- T. Kishima, T. Matsumoto, H. Nakai, S. Hayami, T. Ohta and S. Ogo, Angew. Chem., Int. Ed., 2016, 55, 724 CrossRef CAS PubMed.
- T. Matsumoto, T. Kishima, T. Yatabe, K.-S. Yoon and S. Ogo, Organometallics, 2017, 36, 3883 CrossRef CAS.
- E. Garcin, X. Vernede, E. Hatchikian, A. Volbeda, M. Frey and J. Fontecilla-Camps, Structure, 1999, 7, 557 CrossRef CAS PubMed.
- A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580 CrossRef CAS PubMed.
- C. A. Grapperhaus and M. Y. Darensbourg, Acc. Chem. Res., 1998, 31, 451 CrossRef CAS.
- N. J. Lindenmaier, S. Wahlefeld, E. Bill, T. Szilvasi, C. Eberle, S. Yao, P. Hildebrandt, M. Horch, I. Zebger and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 2208 CrossRef CAS PubMed.
- J. A. Denny and M. Y. Darensbourg, Chem. Rev., 2015, 115, 5248 CrossRef CAS PubMed.
- R. M. Jenkins, M. L. Singleton, L. A. Leamer, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2010, 49, 5503 CrossRef CAS PubMed.
- E. M. Gale, D. M. Cowart, R. A. Scott and T. C. Harrop, Inorg. Chem., 2011, 50, 10460 CrossRef CAS PubMed.
- D. Huang, L. Deng, J. Sun and R. H. Holm, Inorg. Chem., 2009, 48, 6159 CrossRef CAS PubMed.
- D. Brazzolotto, M. Gennari, N. Queyriaux, T. R. Simmons, J. Pecaut, S. Demeshko, F. Meyer, M. Orio, V. Artero and C. Duboc, Nat. Chem., 2016, 8, 1054 CrossRef CAS PubMed.
- A. Volbeda, L. Martin, E. Barbier, O. Gutiérrez-Sanz, A. L. D. Lacey, P.-P. Liebgott, S. b. Dementin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg Chem., 2014, 20, 11 CrossRef PubMed.
- S. Ding, P. Ghosh, A. M. Lunsford, N. Wang, N. Bhuvanesh, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2016, 138, 12920 CrossRef CAS PubMed.
- P. Ghosh, M. Quiroz, N. Wang, N. Bhuvanesh and M. Y. Darensbourg, Dalton Trans., 2017, 46, 5617 RSC.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani;V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. A. Montgomery Jr; J. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; J. M. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; O. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski; D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Ogo, Coord. Chem. Rev., 2017, 334, 43 CrossRef CAS.
- S. Ogo, Chem. Rec., 2014, 14, 397 CrossRef CAS PubMed.
- Y. Shomura, K. S. Yoon, H. Nishihara and Y. Higuchi, Nature, 2011, 479, 253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1861574, 1861577–1861580 and 1861890. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04436h |
| This journal is © The Royal Society of Chemistry 2019 |