
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hollow nanoreactors for Pd-catalyzed Suzuki–Miyaura coupling and O-propargyl cleavage reactions in bio-relevant aqueous media†
Paolo
Destito‡
 a,
Ana
Sousa-Castillo‡
b,
José R.
Couceiro
a,
Fernando
López
*ac,
Miguel A.
Correa-Duarte
*b and
José L.
Mascareñas
*a
a,
Ana
Sousa-Castillo‡
b,
José R.
Couceiro
a,
Fernando
López
*ac,
Miguel A.
Correa-Duarte
*b and
José L.
Mascareñas
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidad de Santiago de Compostela, 15782, Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es
bDepartment of Physical Chemistry, Center for Biomedical Research (CINBIO), Southern Galicia Institute of Health Research (IISGS), Biomedical Research Networking Center for Mental Health (CIBERSAM), Universidade de Vigo, 36310 Vigo, Spain. E-mail: macorrea@uvigo.es
cInstituto de Química Orgánica General CSIC, Juan de la Cierva 3, 28006, Madrid, Spain. E-mail: fernando.lopez@csic.es
First published on 26th December 2018
Abstract
We describe the fabrication of hollow microspheres consisting of mesoporous silica nanoshells decorated with an inner layer of palladium nanoparticles and their use as Pd-nanoreactors in aqueous media. These palladium-equipped capsules can be used to promote the uncaging of propargyl-protected phenols, as well as Suzuki–Miyaura cross-coupling, in water and at physiologically compatible temperatures. Importantly, the depropargylation reaction can be accomplished in a bioorthogonal manner in the presence of relatively high concentrations of biomolecular components and even in the presence of mammalian cells.
Introduction
During the last decade, bioorthogonal reactions have emerged as prominent tools in chemical biology and have facilitated the study and rational modification of different biological processes.1–3 Among these reactions, Cu-catalyzed azide–alkyne cycloaddition (CuAAC) remains as a main reference in the field.4–7 However, the coordination and oxidation lability of Cu(I) complexes and the toxicity associated with this metal have prompted the development of alternative, metal-free reactions (e.g., strain promoted azide–alkyne cycloaddition, inverse electron demand Diels–Alder reactions, photoclick cycloaddition, among others).8–17While these transformations represent important tools in chemical biology, bioorthogonal reactions catalyzed by metals are especially attractive owing to the additional control possibilities offered by the presence of the catalyst. However, achieving such metal-mediated processes in aqueous biological environments is far from obvious. Indeed, only recently several bioorthogonal metal-promoted reactions that take place in aqueous media, and even in cellular settings, have been developed.18–24 Among the different metals that have been explored, palladium occupies a prominent position owing to its well-known transformative potential in organic solvents.18–27 Although palladium complexes can be deactivated in aqueous solvents, Davis and Lin have independently demonstrated that in the presence of specific additives it is possible to achieve Pd-mediated protein Suzuki–Miyaura and Sonogashira cross-coupling reactions in an aqueous milieu.28–32 Chen has reported the use of Pd salts, including [Pd(allyl)Cl]2, to promote depropargylation and deallenylation reactions in proteins containing caged lysines and tyrosines.33–35 However, recent studies suggest that achieving these deallylation or depropargylation reactions in complex aqueous media, such as standard tissue cultures (HBSS and MEM), requires the use of more elaborated, specifically designed phosphine–palladium complexes.36–38
An alternative to small-sized, well-defined palladium reagents is the use of Pd nanoparticles (Pd-NPs). Indeed, a number of Pd-NP-promoted processes, including Suzuki–Miyaura cross coupling, have been achieved in aqueous media. However, most of them require the addition of bases and temperatures higher than 50 °C, which represent significant limitations in terms of translating this type of reactivity to biological media.39–45 Moreover, and importantly, catalytic Pd-NPs tend to aggregate and/or can be readily passivated when used in aqueous mixtures containing biological components. Chen and coworkers have shown that Pd-NPs can promote the uncaging of propargyloxy protected amines in cell-surface glycans;46 however they indicated that the nanostructures need to be immediately used after their generation (within 30 s). Unciti-Broceta and Bradley have devised an elegant method to protect the NPs from the media, by embedding them within polystyrene microspheres, which allowed their use in different biological set-ups.47–51 While these constructs have found interesting applications, the difficulties in controlling the architecture of the resulting structures, especially with regard to the distribution of Pd-NPs, might represent a limitation in terms of obtaining improved designs.
Therefore, the development of alternative palladium-based nanocatalytic constructions that work in biological media represents a major current challenge. In this context, we envisioned a new strategy to achieve bioorthogonal palladium heterogeneous catalysis based on the use of hollow nanoreactors. Specifically, we reasoned that porous hollow microspheres equipped with palladium nanoparticles at the internal surface might work as biocompatible, smart reactive chambers.52–54 The thickness and porosity of the chamber shell could be rationally tuned to allow an effective flow of low molecular weight reactants and products, while avoiding both metal leakage and the entrance of large biomolecules that could passivate the catalytic surface (Fig. 1).
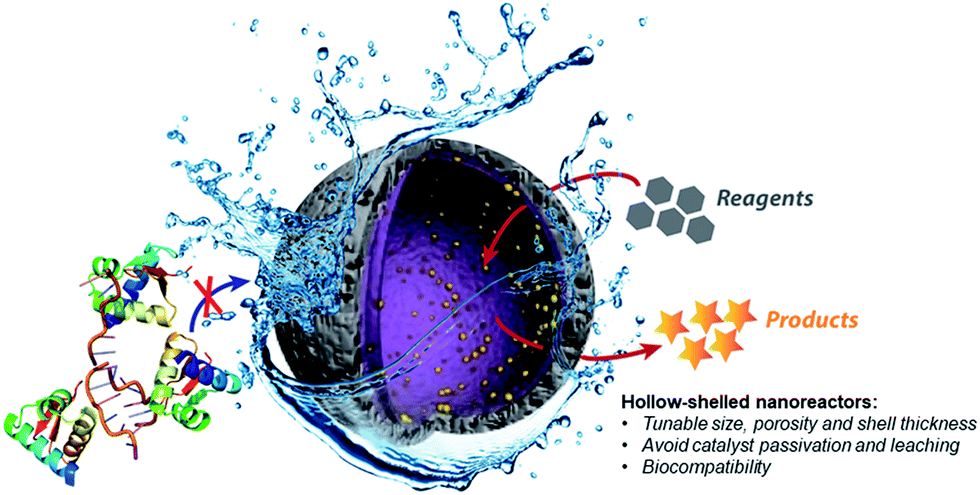 | ||
| Fig. 1 Prototype reactive chambers for metal-catalyzed biocompatible processes. | ||
While there are some precedents for the use of yolk@shell nanoparticles to carry out Pd-promoted processes (Suzuki–Miyaura coupling), the reactions have been performed in alcoholic rather than aqueous solvents, at non-physiological temperatures, and required high concentrations of the reactants.55–59
Herein we demonstrate that hollow microspheres consisting of mesoporous silica nanoshells equipped with Pd-NPs at their inner side behave as efficient Pd-nanoreactors for O-depropargylation reactions and Suzuki–Miyaura cross-couplings in aqueous buffer solutions (pH = 7.2). Importantly, while discrete palladium complexes like [Pd(allyl)Cl]2 failed to promote the propargyl-uncaging process when proteins like BSA are added to the reaction mixture (vide infra), our hollow nanoreactors remain active. Indeed, the capsules also work in cell culture medium and even in the presence of living cells.
Results and discussion
The fabrication of the hollow-structured mesoporous systems equipped with palladium nanostructures at their inner cavity was achieved according to a protocol that ensures the tuning of the silica shell (Scheme 1).56,60–62 Namely, homogeneous polystyrene beads used as sacrificial templates are coated with polyallylamine hydrochloride (PAH). Then, freshly prepared Pd nanoparticles (3.8 ± 0.5 nm, Fig. S1†) are deposited onto these polystyrene particles and subsequently coated with a homogeneous mesoporous silica layer using a tetraethoxysilane solution in ethanol (5%). The use of a polystyrene core facilitated its removal under soft conditions (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF/H2O), avoiding alternative calcination processes that could sinter and reduce the catalytic potential of the Pd-NPs (see the ESI for details, Fig. S2†).
:1 THF/H2O), avoiding alternative calcination processes that could sinter and reduce the catalytic potential of the Pd-NPs (see the ESI for details, Fig. S2†).
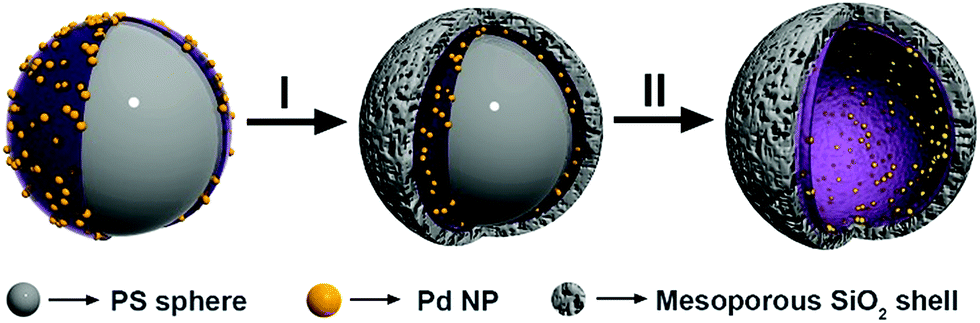 | ||
| Scheme 1 Strategy for the synthesis of hollow spheres equipped with Pd-NPs at their inner cavity. | ||
Fig. 2 shows typical transmission electron microscopy (TEM) images of the resulting nanocapsules exemplified by Pd-Cap1 with a homogeneous average size of 500 nm and a mesoporous silica shell thickness of 25 nm.60–62 The Pd-NPs are homogeneously distributed through the inner surface of this mesoporous shell as confirmed by STEM and EDS analyses (Fig. 2b and d and S3†). The content of Pd in these capsules, measured by ICP-MS, turned out to be 0.61 μg mg−1.
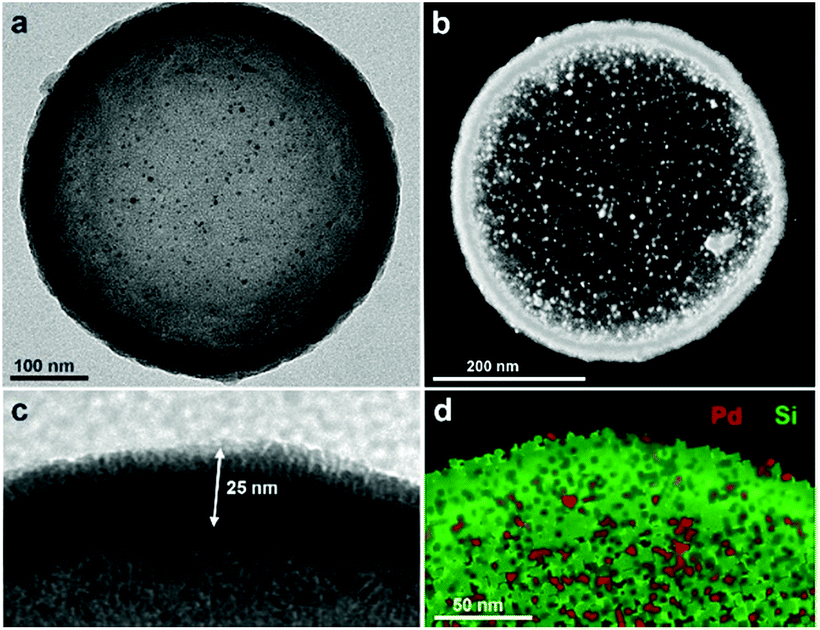 | ||
| Fig. 2 Characterization of Pd-Cap1. (a) Representative TEM image (500 nm diameter); (b) STEM image; (c) zoom-in view showing the vertically oriented mesopores in the silica shell with a thickness of 25 nm; (d) XEDS mapping showing the elemental distribution of Pd and Si. | ||
Prior to the reactivity tests, we checked the stability of the Pd nanocapsules (Pd-Cap1) in water at 37 °C. Gratifyingly, the structural features of the spheres and the size of the internalized Pd-NPs remained unaffected even after 30 days, confirming the protective role of the mesoporous shells in the characteristics of the Pd-NPs. The catalytic activity was preliminarily tested in the uncaging of propargyl phenol derivative 1, a pro-fluorogenic substrate which upon deprotection provides a fluorescent benzothiazole (2, Scheme 2, eqn (1)). The green fluorescence emission of this product (λexc = 460 nm and λem = 535 nm), which arises from an excited state intramolecular proton transfer (ESIPT) process, enables an efficient quantification of reaction yields at low μM concentrations.63 Treatment of 1 (50 μM) with Pd-Cap1 (5 mol% Pd content) in a 2:8 mixture of DMSO:water provided, after 3 h at 37 °C, the desired product (2) in 32% yield (the remaining starting material (1) accounts for the rest of the mass balance). Control experiments confirmed that the reaction does not proceed in the absence of Pd-Cap1.64 The yield could be improved to 53% by doubling the concentration of Pd-Cap1 (10 mol% of Pd content) and running the reaction for 24 h. Importantly, control experiments with non-encapsulated Pd-NPs, stabilized with polyvinylpyrrolidone (PVP, Pd-NP1), under otherwise identical reaction conditions, provided yields below 10% after 24 h, confirming that confinement of the Pd-NPs in the nanoreactor has a pivotal influence on their catalytic activity.64
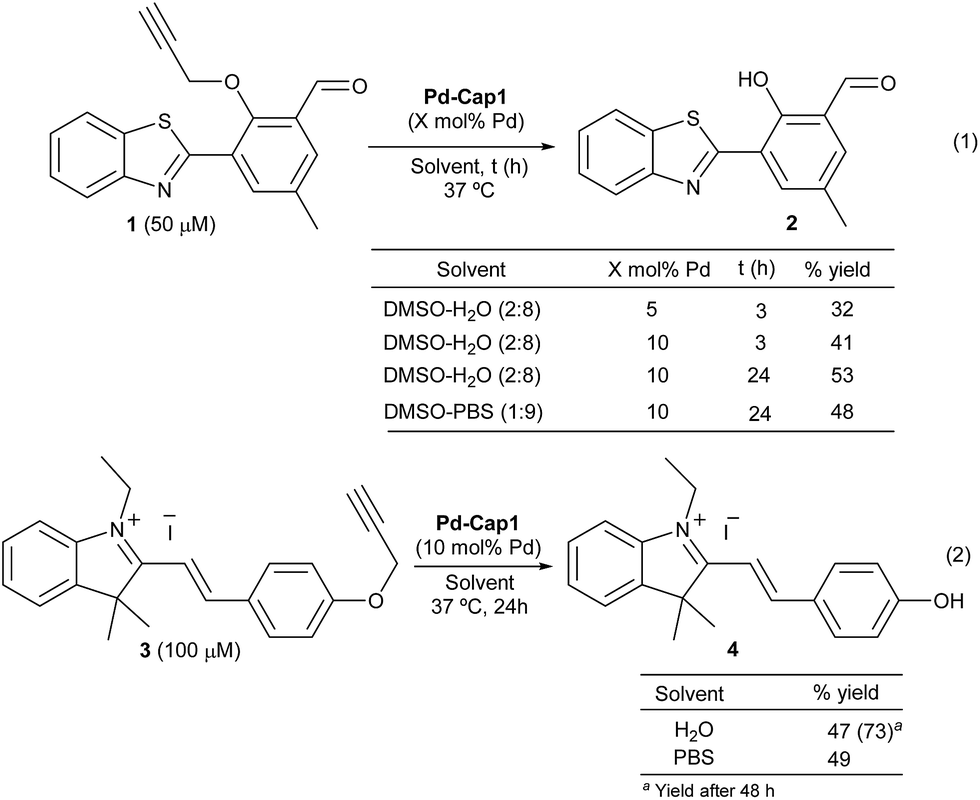 | ||
| Scheme 2
Pd-Cap1-promoted uncaging of 1 and 3. Conditions: a suspension of Pd-Cap1 (0.2 mL, 8.33 mg mL−1) in EtOH was centrifuged (8000 rpm) for 10 min. The supernatant was removed and a mixture of H2O:DMSO (4:1) was added, followed by addition of 1 or 3. The mixture was stirred at 37 °C, under air, for the indicated time and centrifuged (8000 rpm) prior to quantification by fluorescence analysis (for 2) or by HPLC (for 4). | ||
Remarkably, the reaction promoted by Pd-Cap1 could also be carried out in PBS, instead of water, with similar results (48% yield). To avoid the use of DMSO as the co-solvent, we checked the reactivity of the styryl indolinium probe 3 (Scheme 2, eqn (2)), which presents excellent water solubility in the μM range. Interestingly, the reaction outcome can be easily judged by naked-eye detection of the colorimetric change, and yields are easily quantified by HPLC-MS.64 Treatment of a water solution of 3 with Pd-Cap1 (10 mol% of Pd) provided, after 24 h at 37 °C, the depropargylated product 4 in 47% yield, while a significantly better value of 73% could be obtained if the reaction was carried out for 48 h (Scheme 2, eqn (2)). A similar yield of 4 after 24 h was obtained when the reaction was carried out in PBS (Scheme 2, eqn (2)). Overall, despite the yields and reaction rates are moderate, these results validate the potential of the palladium nanoreactors in water, at low temperatures and under high dilution conditions.
To check whether these Pd-nanoreactors could be applied to more challenging bimolecular processes, we tested their effectivity in Suzuki–Miyaura coupling between 2-thiophenyl boronic acid (5) and p-iodo benzaldehyde (6) (Scheme 3). The expected product, 4-(thiophen-2-yl)benzaldehyde (7), shows a strong blue fluorescence (λexc = 330 nm, λem = 434 nm, 10 μM in EtOH:H2O 1:1),65 facilitating reaction monitoring. Gratifyingly, when a water suspension of Pd-Cap1 (10 mol% Pd content), 5 (150 μM), 6 (100 μM) and K2CO3 (1 mM) was mixed at 37 °C for 3 h, the desired product 7 was obtained in 55% yield. Moreover, by running the reaction for 24 h, the yield of 7 could be further increased up to 78%. Not surprisingly, in the absence of a base the reaction did not occur. However, we were pleased to find that the reaction could be carried out using PBS 10× (pH 7.2) as solvent, without any additional base. In this case, a 65% yield of 7 was obtained after 3 h at 37 °C, while after 24 h the yield was 86%. Notably, this reaction represents the first application of hollow palladium nanocapsules in Suzuki–Miyaura cross coupling in an aqueous solvent and at physiological temperatures.
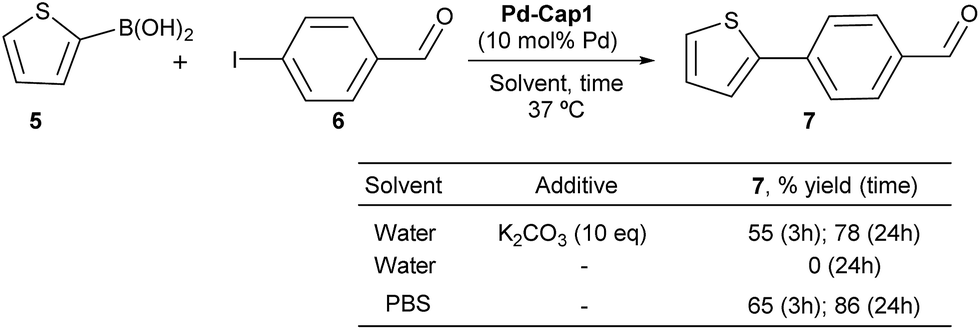 | ||
| Scheme 3 Suzuki–Miyaura cross-couplings of 5 and 6. | ||
Although it was not the main goal of this research, it is worth noting that these nanoreactors can be easily recovered by centrifugation after reaction completion, and preliminary experiments demonstrate that they can be reused, at least three times, without an important loss of catalytic activity (Fig. S16†).64 On the other hand, leaching was insignificant, as determined by ICP-MS analysis of the supernatants. Accordingly, mother liquids obtained by centrifugation after reaction did not show catalytic activity.64
We next analyzed the influence of the structural properties of the nanostructures on the reactivity. Thus, two additional Pd-nanocapsules of different sizes and shell thicknesses were prepared.64 Capsules Pd-Cap2, which are significantly smaller (280 nm) but of equal shell thickness (25 nm), were prepared from polystyrene beads of 230 nm (see Scheme 4 for the TEM image). On the other hand, Pd-Cap3, comprising a thicker mesoporous shell (50 nm), with an overall diameter of 500 nm were obtained following a slightly modified procedure.64 As can be deduced from the results of Scheme 4, Pd-Cap2 showed a very similar behavior in the cross-coupling between 5 and 6, providing an 85% yield of 7 after 24 h at 37 °C. On the other hand, the efficiency of Pd-Cap3, which features a 50 nm shell width, turned out to be slightly lower (7, 71% yield), indicating that increasing the shell thickness disrupts the catalytic activity, likely because of a less efficient flow of reactants and products throughout the shell. These results confirm that tuning the thickness of the surface allows extra control over the reaction kinetics.
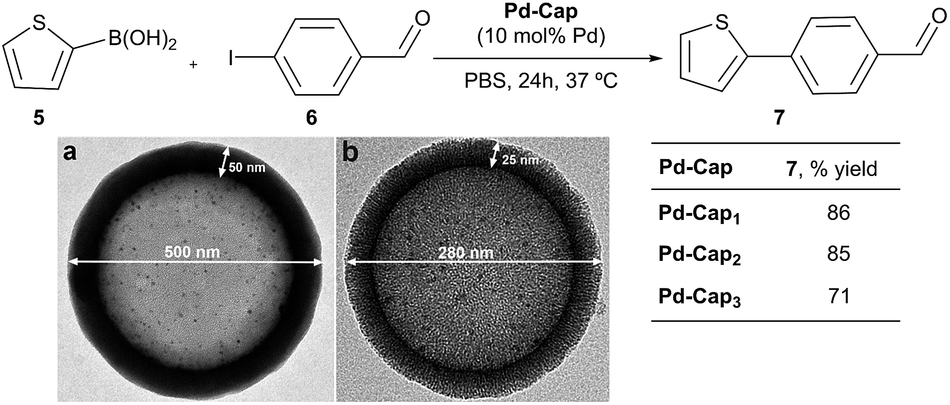 | ||
| Scheme 4 Reactivity of Pd-capsules with different diameters and thicknesses. (a) TEM image of Pd-Cap3; (b) TEM image of Pd-Cap2. | ||
At this point, we explored the viability of translating the above reactivities to aqueous media containing additional biomolecules, in order to test the bioorthogonality of the approach. Gratifyingly, the Suzuki–Miyaura coupling between 5 and 6, promoted by Pd-Cap1, proceeds with similar yields in the presence of one equivalent (with respect to the Pd content) of carbohydrates, such as glucose, or different amino acids like glycine, tyrosine or valine (Fig. 3). We later found that it also tolerates larger excesses of the additives (Fig. S11†). However, the reaction was inhibited by biorelevant thiols such as glutathione and did not proceed in cell lysates or in the presence of Vero cells. This inactivation is likely associated with the mechanistic complexity of the bimolecular process and the lateral reactivity of transient palladium intermediates with some of the components of the mixture (especially with thiols).
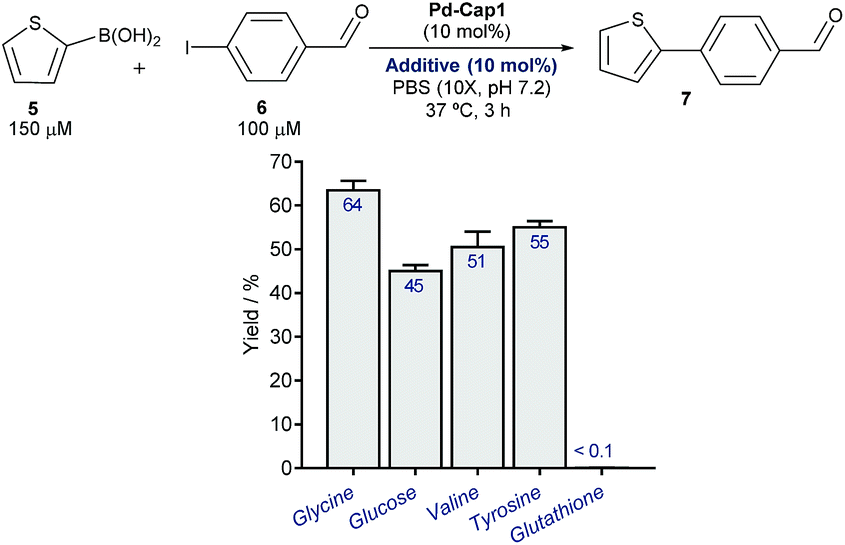 | ||
| Fig. 3 Pd-Cap1-promoted Suzuki–Miyaura reaction of 5 and 6 (top, mean ± s.e.m., n = 2), in the presence of additives. | ||
However, to our delight, the depropargylation of probe3proceeded in the presence of added glucose and different types of amino acids and even in the presence of glutathione (Fig. 4a and S12†). Interestingly, the reaction can also be performed at acidic pHs of 5.3 and 4.5 (45% and 46% yield, respectively). Moreover, the reaction is also effective in the presence of significant amounts of proteins like BSA (up to 600 μM, Fig. 4b). Importantly, under these conditions, discrete Pd complexes that had been previously used in lysine uncaging reactions, like [Pd(allyl)Cl]2,33–35 turned out to be completely ineffective, even at BSA concentrations as low as 100 μM. These results confirm the superiority of the Pd-nanocapsules, which prevents an otherwise rapid passivation.
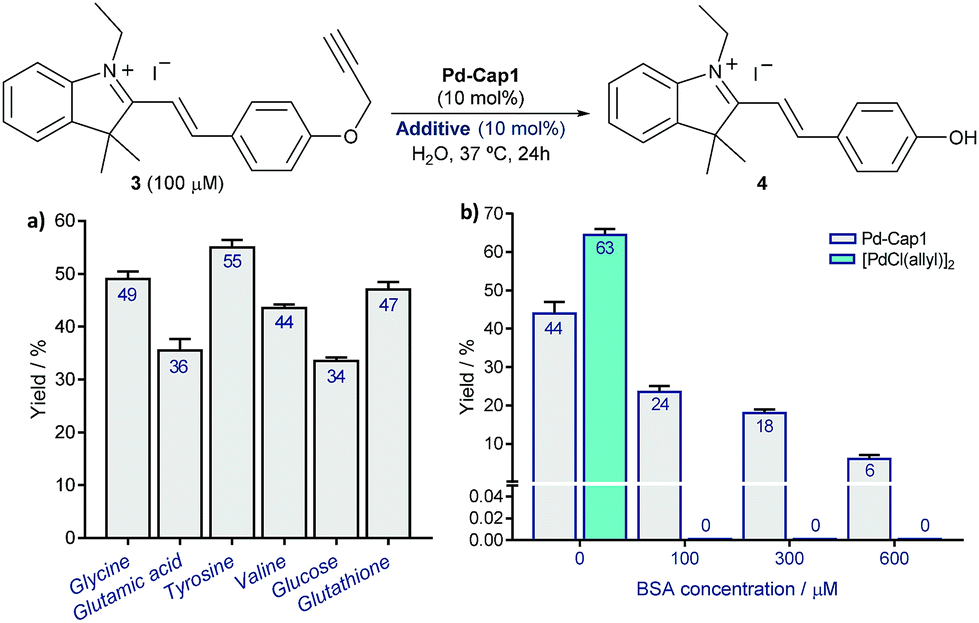 | ||
| Fig. 4 Pd-Cap1-promoted depropargylation of 3 (mean ± s.e.m., n = 2) in the presence of different additives, 1.0 equiv. with respect to Pd (a), and of increasing concentrations of BSA (b). The reaction is also effective in the presence of higher amounts of the additives as indicated in (a) (see Fig. S12).† | ||
Finally, a preliminary analysis of the toxicity in Vero cells revealed a superior biocompatibility of the nanoreactor Pd-Cap1, compared to [Pd(allyl)Cl]2. Indeed, while at low concentrations (2.5 μM), none of the Pd-systems were significantly toxic, at a concentration of 20 μM a significant toxicity of [Pd(allyl)Cl]2 was observed (85% of cells were dead after 24 h), whereas the Pd-nanocapsules still provided a good cell viability > 75% (Fig. S14†).64
Given their low toxicity, we studied their catalytic activity in cellular environments. With this purpose, we performed the depropargylation of the probe HBTPQ (8)66 in the presence of Vero mammalian cells.
This probe does not emit red fluorescence when irradiated with UV light but the cleavage of its propargyl moiety generates the red fluorescent compound HBTP (9, λexc = 330 nm, λem = 635 nm). In this experiment, the extracellular medium was removed and substituted with a solution of 50 μM HBTPQ (8) in DMEM or in PBS. Pd-Cap1 was added to reach a final 5 μM concentration of Pd, and the resulting cellular fluorescence was monitored by confocal microscopy. Gratifyingly, red fluorescence spots arising from the intracellular presence of the product were observed in the cells incubated with probe 8 (Fig. 5).
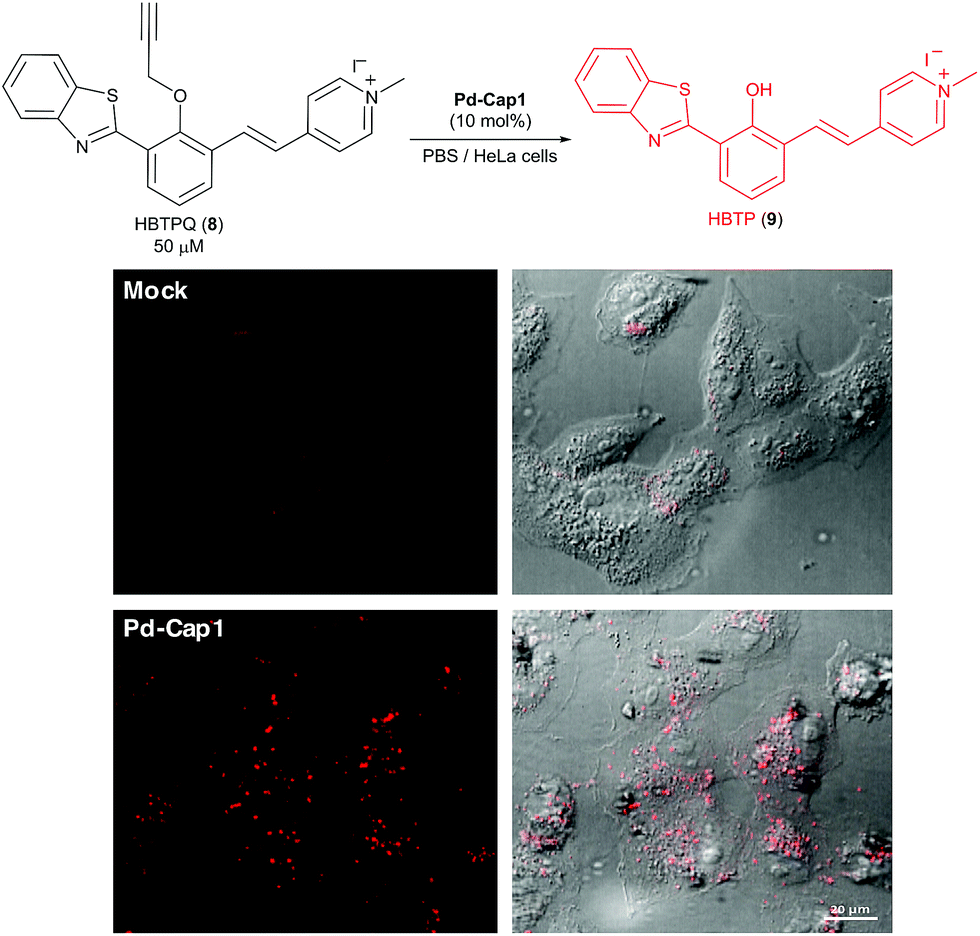 | ||
| Fig. 5 Micrographs of Vero cells incubated for 30 min in DMEM containing Pd-Cap1 (≈5 μM Pd) and 50 μM HBTPQ (8), washed and analyzed. | ||
Conclusions
To sum up, we have demonstrated that hollow microspheres consisting of mesoporous silica nanoshells with Pd-NPs at their inner face behave as efficient nanoreactors for Pd-catalyzed depropargylation and Suzuki–Miyaura intermolecular cross-coupling under physiologically compatible aqueous conditions. The aqueous stability, monodispersity, well-defined architecture and fabrication reproducibility of the capsules represent important advantages compared to other polymer-based structures. Moreover, their low toxicities to cells owing to an inert external surface and the bioorthogonality of the depropargylation augur well for further developments with hollow nanoreactors that contain well-defined metal catalysts inside the capsule. The development of this type of biocompatible reactor might allow the implementation of new therapies based on prodrug activation67 and/or contribute to the establishment of biocompatible, non-natural metabolic networks. Work in this direction is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received financial support from Spanish grants (SAF2016-76689-R, CTQ2017-84767-P, CTM2014-58481-R, CTM2017-84050-R and Orfeo-Cinqa network CTQ2016-81797-REDC), the Consellería de Cultura, Educación e Ordenación Universitaria (2015-CP082, ED431C/2017119 and Centro Singular de Investigación de Galicia accreditation 2016–2019, ED431G/09), the European Union (European Regional Development Fund-ERDF corresponding to the multiannual financial framework 2014–2020), and the European Research Council (Advanced Grant No. 340055).Notes and references
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007 RSC.
- M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS PubMed.
- L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef PubMed.
- J. Miguel-Ávila, M. Tomás-Gamasa, A. Ólmos, P. J. Pérez and J. L. Mascareñas, Chem. Sci., 2018, 9, 1947 RSC.
- For a recent Ru-based version, see: P. Destito, J. R. Couceiro, H. Faustino, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2017, 56, 10766 CrossRef CAS PubMed.
- Cycloadditions in Bioorthogonal Chemistry, ed. M. Vrabel and T. Carell, Springer Cham, Switzerland, 2016, pp. 1–157 Search PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895 RSC.
- M. Merkel, K. Peewasan, S. Arndt, D. Ploschik and H.-A. Wagenknecht, ChemBioChem, 2015, 16, 1541 CrossRef CAS.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307 RSC.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Top. Curr. Chem., 2015, 374, 3 CrossRef.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592 CrossRef CAS PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Acc. Chem. Res., 2011, 44, 828 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811 RSC.
- M. Martínez-Calvo and J. L. Mascareñas, Coord. Chem. Rev., 2018, 359, 57 CrossRef.
- J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106 CrossRef CAS PubMed.
- M. Yang, Y. Yang and P. R. Chen, Top. Curr. Chem., 2016, 374, 2 CrossRef PubMed.
- T. Völker and E. Meggers, Curr. Opin. Chem. Biol., 2015, 25, 48 CrossRef PubMed.
- M. Martínez-Calvo and J. L. Mascareñas, Chimia, 2018, 72, 791 CrossRef PubMed.
- C. Vidal, M. Tomás, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1913 CrossRef PubMed.
- M. Jbara, S. K. Maity and A. Brik, Angew. Chem., Int. Ed., 2017, 56, 10644 CrossRef CAS PubMed.
- S. V. Chankeshwara, E. Indrigo and M. Bradley, Curr. Opin. Chem. Biol., 2014, 21, 128 CrossRef CAS PubMed.
- J. Li and P. R. Chen, ChemBioChem, 2012, 13, 1728 CrossRef CAS PubMed.
- J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Chem. Commun., 2011, 47, 1698 RSC.
- N. Li, R. K. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316 CrossRef CAS PubMed.
- C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800 CrossRef CAS PubMed.
- Z. Gao, V. Gouverneur and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 13612 CrossRef CAS PubMed.
- J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352 CrossRef CAS PubMed.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118 CrossRef CAS PubMed.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055 CrossRef PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- For a related strategy, see: G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597 CrossRef CAS PubMed.
- For selected examples see: M. C. Hong, M. C. Choi, Y. M. Chang, Y. Lee, J. Kim and H. Rhee, Adv. Synth. Catal., 2012, 354, 1257 CrossRef CAS.
- P.-P. Fang, A. Jutand, Z.-Q. Tian and C. Amatore, Angew. Chem., Int. Ed., 2011, 50, 12184 CrossRef CAS PubMed.
- Y. Yu, T. Hu, X. Chen, K. Xu, J. Zhang and J. Huang, Chem. Commun., 2011, 47, 3592 RSC.
- G. Park, S. Lee, S. J. Son and S. Shin, Green Chem., 2013, 15, 3468 RSC.
- J. Yang, D. Wang, W. Liu, X. Zhang, F. Bian and W. Yu, Green Chem., 2013, 15, 3429 RSC.
- M. Charbonneau, G. Addoumieh, P. Oguadinma and A. R. Schmitzer, Organometallics, 2014, 33, 6544 CrossRef CAS.
- V. W. Faria, D. G. M. Oliveira, M. H. S. Kurz, F. F. Goncalves, C. W. Scheeren and G. R. Rosa, RSC Adv., 2014, 4, 13446 RSC.
- J. Wang, B. Cheng, J. Li, Z. Zhang, W. Hong, X. Chen and P. R. Chen, Angew. Chem., Int. Ed., 2015, 54, 5364 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239 CrossRef CAS PubMed.
- A. Unciti-Broceta, E. M. V. Johansson, R. M. Yusop, R. M. Sánchez-Martín and M. Bradley, Nat. Protoc., 2012, 7, 1207 CrossRef CAS PubMed.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864 CrossRef CAS PubMed.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212 RSC.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983 CrossRef CAS PubMed.
- J. Lee, S. M. Kim and I. S. Lee, Nano Today, 2014, 9, 631 CrossRef CAS.
- M. Pérez-Lorenzo, B. Vaz, V. Salgueiriño and M. A. Correa-Duarte, Chem.–Eur. J., 2013, 19, 12196 CrossRef PubMed.
- S. Sadjadi and M. M. Heravi, RSC Adv., 2016, 6, 88588 RSC.
- Z. Chen, Z.-M. Cui, F. Niu, L. Jiang and W.-G. Song, Chem. Commun., 2010, 46, 6524 RSC.
- F. Wei, C. Cao, Y. Sun, S. Yang, P. Huang and W. Song, ChemCatChem, 2015, 7, 2475 CrossRef CAS.
- S.-W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS PubMed.
- J. Niu, M. Liu, P. Wang, Y. Long, M. Xie, R. Li and J. Ma, New J. Chem., 2014, 38, 1471 RSC.
- M. Sanles-Sobrido, W. Exner, L. Rodríguez-Lorenzo, B. Rodríguez-González, M. A. Correa-Duarte, R. A. Álvarez-Puebla and L. M. Liz-Marzán, J. Am. Chem. Soc., 2009, 131, 2699 CrossRef CAS PubMed.
- M. Sanlés-Sobrido, M. Pérez-Lorenzo, B. Rodríguez-González, V. Salgueiriño and M. A. Correa-Duarte, Angew. Chem., Int. Ed., 2012, 51, 3877 CrossRef PubMed.
- C. Vázquez-Vázquez, B. Vaz, V. Giannini, M. Pérez-Lorenzo, R. A. Álvarez-Puebla and M. A. Correa-Duarte, J. Am. Chem. Soc., 2013, 135, 13616 CrossRef PubMed.
- B. Liu, H. Wang, T. Wang, Y. Bao, F. Du, J. Tian, Q. Li and R. Bai, Chem. Commun., 2012, 48, 2867 RSC.
- See the ESI for further details.†.
- For the photophysical properties of this compound see: K. Guo, Z. Gao, J. Cheng, Y. Shao, X. Lu and H. Wang, Dyes Pigm., 2015, 115, 166 CrossRef CAS.
- T. Gao, P. Xu, M. Liu, A. Bi, P. Hu, B. Ye, W. Wang and W. Zeng, Chem.–Asian J., 2015, 10, 1142 CrossRef CAS PubMed.
- C. Adam, A. M. Pérez-López, L. Hamilton, B. Rubio-Ruiz, T. L. Bray, D. Sieger, P. M. Brennan and A. Unciti-Broceta, Chem.–Eur. J., 2018, 24, 16783 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04390f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |