
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversibility and reactivity in an acid catalyzed cyclocondensation to give furanochromanes – a reaction at the ‘oxonium-Prins’ vs. ‘ortho-quinone methide cycloaddition’ mechanistic nexus†
Christian D.-T.
Nielsen
 a,
Wouter J.
Mooij
a,
David
Sale
b,
Henry S.
Rzepa
a,
Jordi
Burés
c and
Alan C.
Spivey
*a
a,
Wouter J.
Mooij
a,
David
Sale
b,
Henry S.
Rzepa
a,
Jordi
Burés
c and
Alan C.
Spivey
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: a.c.spivey@imperial.ac.uk
bProcess Studies Group, Syngenta, Jealott's Hill, Bracknell, Berkshire RG42 6EY, UK
cSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 19th October 2018
Abstract
Herein we report a combined experimental and computational investigation of the acid catalyzed cyclocondensation reaction between styrenyl homoallylic alcohols and salicylaldehyde to form furanochromanes. We disclose a previously unreported isomerisation of the ‘unnatural’ trans-fused products to the diastereomeric ‘natural’ cis-fused congeners. Notwithstanding the appeal of assuming this corresponds to endo to exo isomerisation of Diels–Alder (D–A) adducts via concerted retro-cycloaddition/cycloaddition reactions of an in situ generated ortho-quinone methide with the styrenyl alkene, our combined Hammett/DFT study reveals a stepwise Prins-like process via discrete benzylic carbocation intermediates for all but the most electron deficient styrenes. As these reactions fortuitously lie at the intersection of these two mechanistic manifolds, it allows us to propose an experimentally determined indicative ρ+ value of ca. −3 as marking this nexus between a stepwise Prins-type pathway and a concerted cycloaddition reaction. This value should prove useful for categorising other reactions formally involving ‘ortho-quinomethides’, without the need for the extensive computation performed here. Logical optimisation of the reaction based upon the mechanistic insight led to the use of HFIP as an additive which enables exclusive formation of ‘natural’ cis-fused products with a ∼100-fold reaction rate increase and improved scope.
Introduction
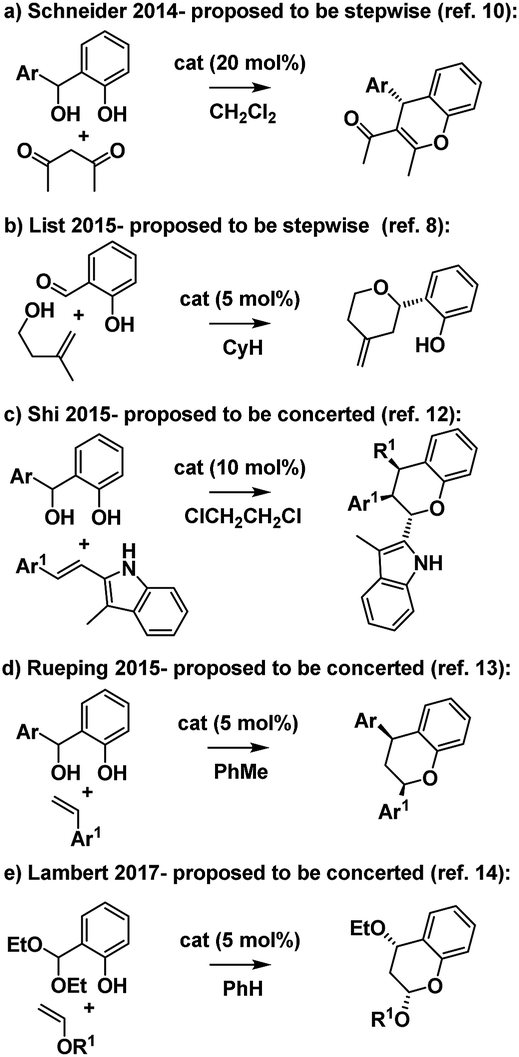
Ortho-quinone methides (oQMs) are useful intermediates in synthesis.1 Answering the call of Pettus in his 2002 review of their synthetic utility, these “underdeveloped and underutilized”2 intermediates have enjoyed a significant revival in the literature since then. In particular, legions of new acid catalyzed cyclocondensations have been reported as being oQM cycloaddition processes.3,4 A case in point is the reaction between salicylaldehyde and homoallylic alcohols, featuring C–C and C–O bond formation, to furnish furanochromanes (Scheme 1). | ||
| Scheme 1 Furanochromane forming reactions. | ||
Specifically, Brønsted acids have been shown to give trans-fused furanochromanes, putatively via either a protonated or neutral oQM cycloaddition process (Scheme 1a and b).5,6 Although List et al. had proposed a Prins-type mechanism for related reactions with other aldehydes (cf.Scheme 2b),7,8 in this case they cited the stereochemical fidelity by which (E)- and (Z)-4-phenylbut-3-en-1-ol (2e) converted to only 3a,4-trans or 3a,4-cis products 3e, respectively, as experimental support for a concerted mechanism. However, faithful translation of double bond stereochemistry could be an artefact arising from the rate of carbocation trapping exceeding the rate of C3a–C4 bond rotation rather than implicating a concerted oQM cycloaddition pathway. In our Lewis acid promoted reactions (Scheme 1c), we proposed a stepwise oxonium-Prins pathway in which a benzylic carbocation is trapped by the phenolic hydroxyl group, as supported experimentally by competitive trapping of the intermediate cation by bromine when using SnBr4 in the absence of the intramolecular nucleophile.9
 | ||
| Scheme 2 Related Brønsted acid catalyzed hydropyran forming reactions. | ||
Intrigued by these mechanistic discrepancies and cognizant of both the increasing number of related Brønsted acid catalyzed reactions and the ambiguity that remains between a concerted oQM cycloaddition pathway and a stepwise carbocation-mediated alternative for a variety of reactions (e.g.Scheme 2),2,10–14 we decided to investigate further.
Results and discussion
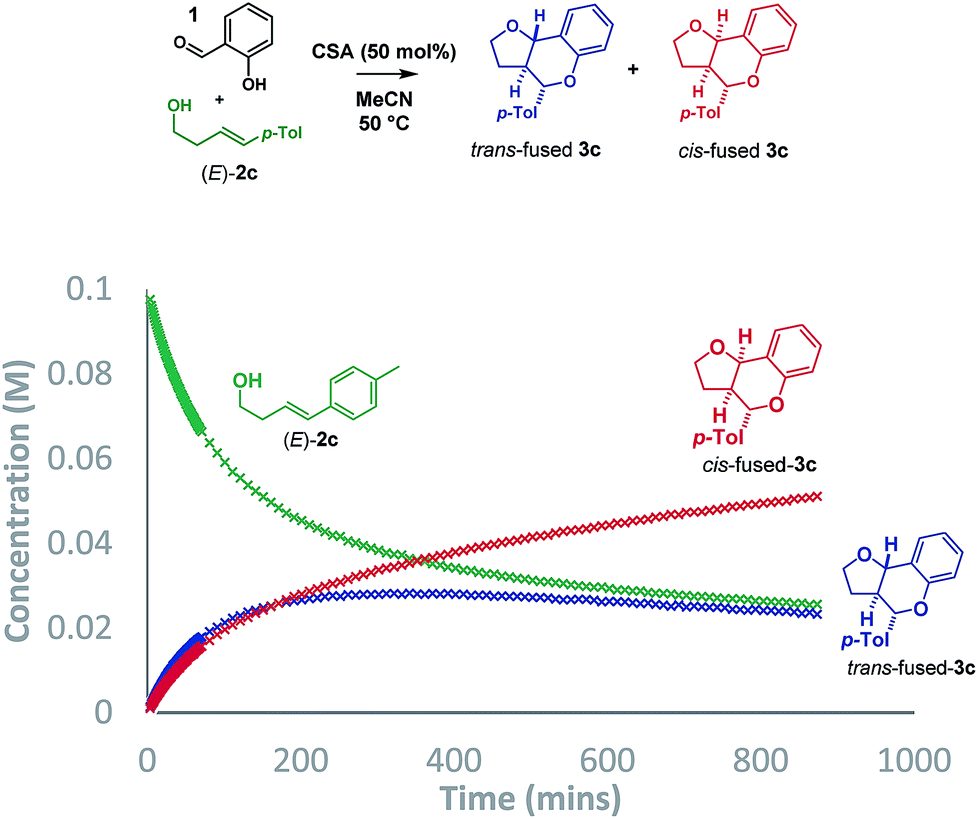
Initially, we planned to distinguish between the stepwise and concerted pathways by carrying out a Singleton 13C KIE at natural abundance experiment, recovering (E)-4-(4-tolyl)-but-3-en-1-ol [(E)-2c] from a reaction driven close to completion.15,16 However, DFT calculations revealed that the expected ratios for both pathways would be indistinguishable due to the highly asynchronous nature of the computed oQM cycloaddition transition state (TS). An analogous situation was reported by List when studying the chiral Brønsted acid catalyzed formation of dihydropyran from benzaldehydes and dienes in which the isotopic distribution in the product was inconclusive by virtue of being consistent with both stepwise carbocation-mediated and highly asynchronous cycloaddition pathways.17As such, we sought an alternative method to probe the mechanism and opted to collect reaction progress data by 1H NMR for the Brønsted acid promoted reaction of salicylaldehyde (1) with (E)-2c. The conditions described by List using p-TSA (10 mol%) in cyclohexane afforded a ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of trans:cis-fused isomers at the ring-junction as reported,5 but the conditions were inhomogeneous. Alternatively, a reaction mixture having (±)-camphorsulfonic acid (CSA, 50 mol%) in acetonitrile at 50 °C was homogeneous and allowed us to monitor the reaction progress by 1H NMR to 74% conversion in 14.5 h (Scheme 3).
:1 mixture of trans:cis-fused isomers at the ring-junction as reported,5 but the conditions were inhomogeneous. Alternatively, a reaction mixture having (±)-camphorsulfonic acid (CSA, 50 mol%) in acetonitrile at 50 °C was homogeneous and allowed us to monitor the reaction progress by 1H NMR to 74% conversion in 14.5 h (Scheme 3).
 | ||
| Scheme 3 Plot of reaction progress. | ||
Intriguingly, although the reaction was initially moderately trans-ring junction selective (cf. initial rates in the 0–50 min region), it was apparent that a thermodynamically driven trans to cis isomerization was taking place leading to progressive enrichment in the latter as the reaction progressed. DFT analysis (B3LYP+GD3BJ/Def2-TZVPP) suggested that cis-fused 3c is 3.9 kcal mol−1 lower in free energy than trans-fused 3c. This thermodynamic difference likely has a component attributable to a destabilizing 4 electron nO ↔ σC9b–H interaction in the trans-fused structure. Indeed, the prevalence of 5,6-cis-fused systems in bioactive natural products (e.g. flavonoids: lophirone H18 and cordigol;19 and pterocarpan isoflavonoids: medicarpin20 and phaseolin21) suggests their relative thermodynamic stability. Irrespective of the thermodynamic basis of the phenomenon, a reversible isomerization process renders Singleton analysis unsuitable for interrogation of the mechanism of this reaction.
We envisioned that isomerization was most likely occurring by reversion of the forwards reaction: i.e. either via a retro-Prins/Prins pathway or a retro-cycloaddition/cycloaddition pathway (path a, Scheme 4), following protonation of the tetrahydropyranyl oxygen. Fragmentation-recombination via a benzyne (path b, Scheme 4) from this same protonated species appeared unlikely given the poor nO → σ* orbital alignment for cleavage of the C9b–CAr bond, but could not be entirely discounted. Alternatively, protonation of the furan could facilitate formation of the cis-fused product via a retro-oxy-Michael/oxy-Michael pathway (path c, Scheme 4) or a Grob fragmentation-bicyclisation pathway (path d, Scheme 4).22
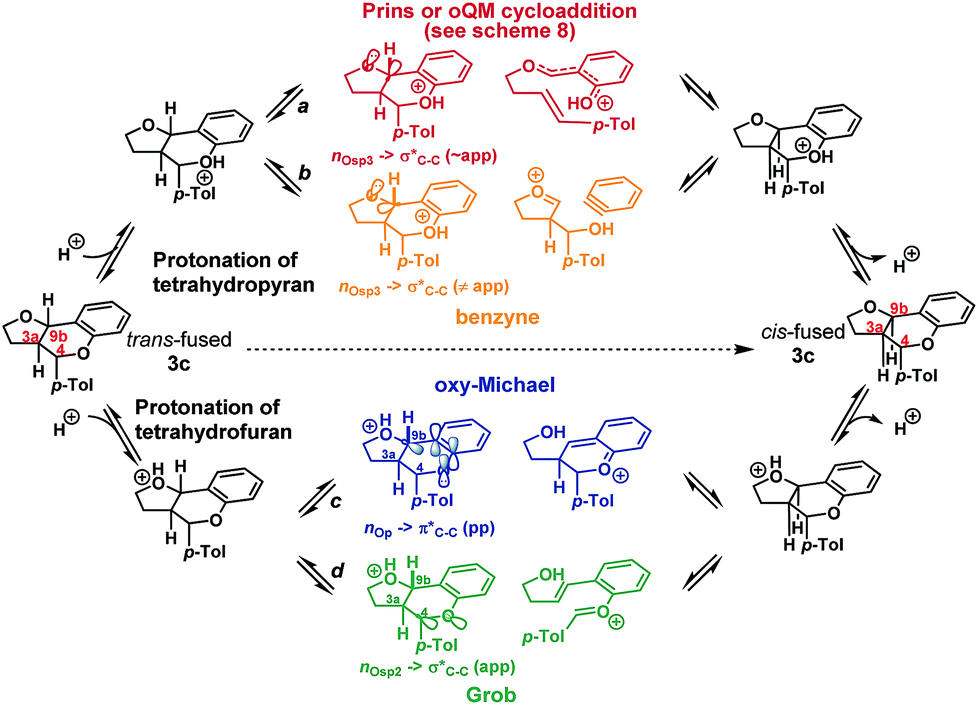 | ||
| Scheme 4 Potential pathways for isomerization. | ||
Inspired by the work of Rychnovsky,23 we anticipated that racemization would be a key indicator of mechanism as pathways a and d proceed via achiral intermediates, whereas pathways b and c proceed via intermediates which retain two stereocentres from the substrate. To test this, we isolated the enantiomerically pure trans-fused 3c and subjected it to the reaction conditions (Scheme 5).
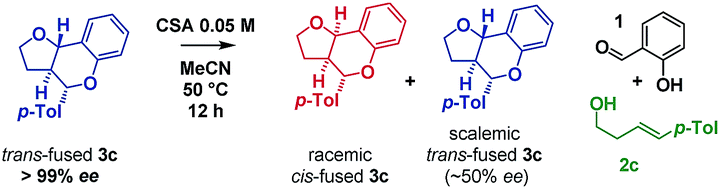 | ||
| Scheme 5 Isomerization study on enantiopure furanochromane monitored by CSP-HPLC. | ||
After 12 h, analysis revealed the formation of racemic cis-fused 3c as well as partially racemized recovered trans-fused 3c (∼50% ee). This indicated that the isomerization occurs via an achiral intermediate which can convert to the thermodynamically preferred cis-fused isomer but also revert to the trans-fused isomer and implicates the Prins/oQM cycloaddition and/or Grob mechanisms as operating (paths a and d).
Additionally, two new peaks were observed by CSP-HPLC; these were identified as the salicylaldehyde (1) and homoallylic alcohol (E)-2c. The formation of these compounds rules out the Grob fragmentation pathway (path d), for which adventitious hydrolysis of the intermediate oxonium ion would lead to p-tolualdehyde and 2-[(1E)-4-hydroxy-1-buten-1-yl]phenol (which were not observed). All evidence therefore pointed towards a mechanism of isomerization via either a retro-Prins/Prins pathway or a retro-cycloaddition/cycloaddition pathway (path a) as originally anticipated. To corroborate this, 1H NMR reaction progress analysis monitoring the isomerization of isolated trans-fused 3c22 was performed (Scheme 6).
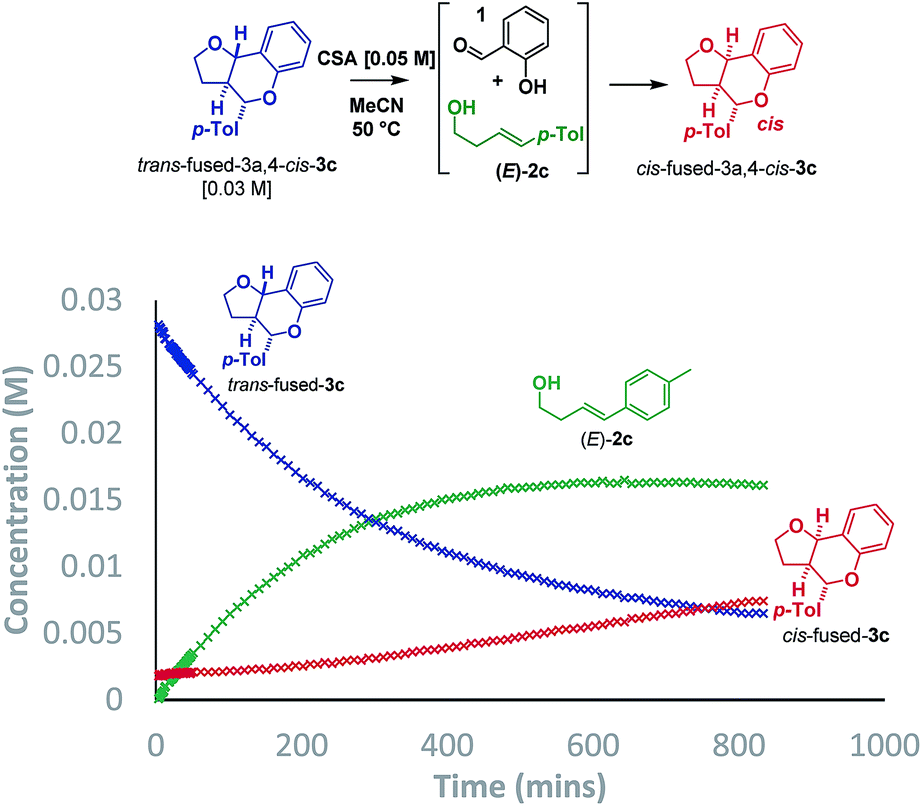 | ||
| Scheme 6 Isomerization study by 1H NMR. | ||
In agreement with the racemization study, a build-up of homoallylic alcohol (E)-2c was observed over a period of 15 h as the concentration of the trans-fused isomer reduced and that of the cis-fused isomer increased. It was also possible to isolate (E)-2c from a reaction mixture wherein the trans-fused diastereomer was partially isomerized.
Having established that the isomerization, like the furanochromane formation itself, takes place via either a Prins or oQM cycloaddition pathway, we set out to distinguish between these possibilities. Inspection of the oxonium ion/ortho-quinone methide conformations required to access the isomeric products via either mechanistic manifold reveals that the trans-fused products are formed from endo-like conformation A, whereas the cis fused products are formed from exo-like conformation B (Scheme 7).
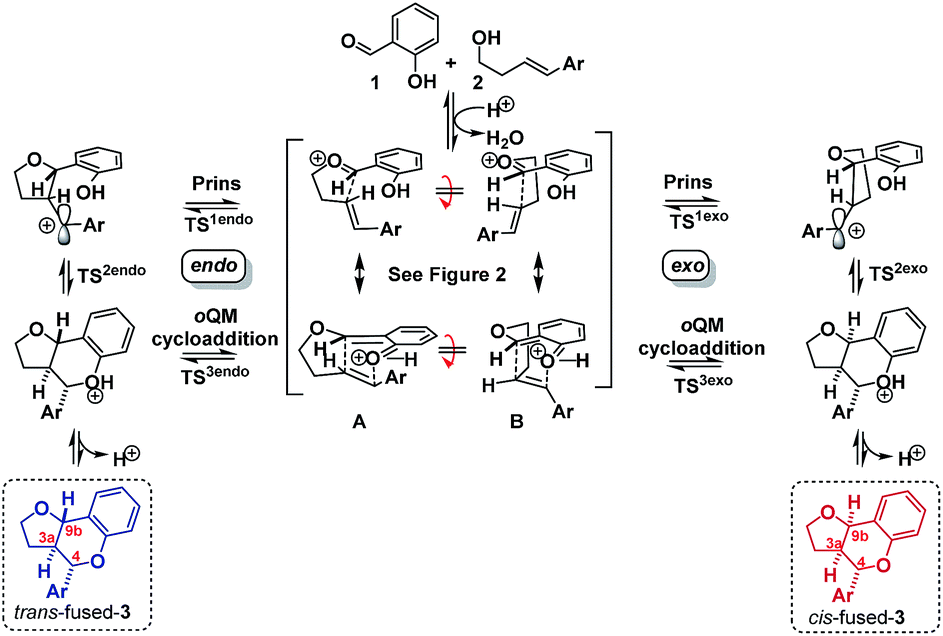 | ||
| Scheme 7 ‘Endo’ vs. ‘exo’ transition states. | ||
The endo conformer A appears poised for π–π stacking interactions whereas the exo conformer B is not, suggesting that ‘secondary orbital overlap’, as widely invoked for D–A reactions, could take place in the endo TSs, accounting for the kinetic preference.
Intrigued as to how this would manifest itself in experimental data, we set out to determine a Hammett correlation for the reaction.24–27 To this end, six homoallylic alcohols with para-substituents of varying electronic demand from p-OMe to p-Cl (2a–2f) were each subjected to the reaction conditions and the reactions monitored by 1H NMR. While traditional Hammett analysis utilizes initial rates,24,27 we tracked the reactions for an extended period of time to allow COPASI28 fitting of kinetic parameters. Use of these kinetic parameters would allow us to deconvolute the complexity arising from in situ isomerization in our subsequent Hammett analysis and obtain separate values for each isomer (Scheme 8).
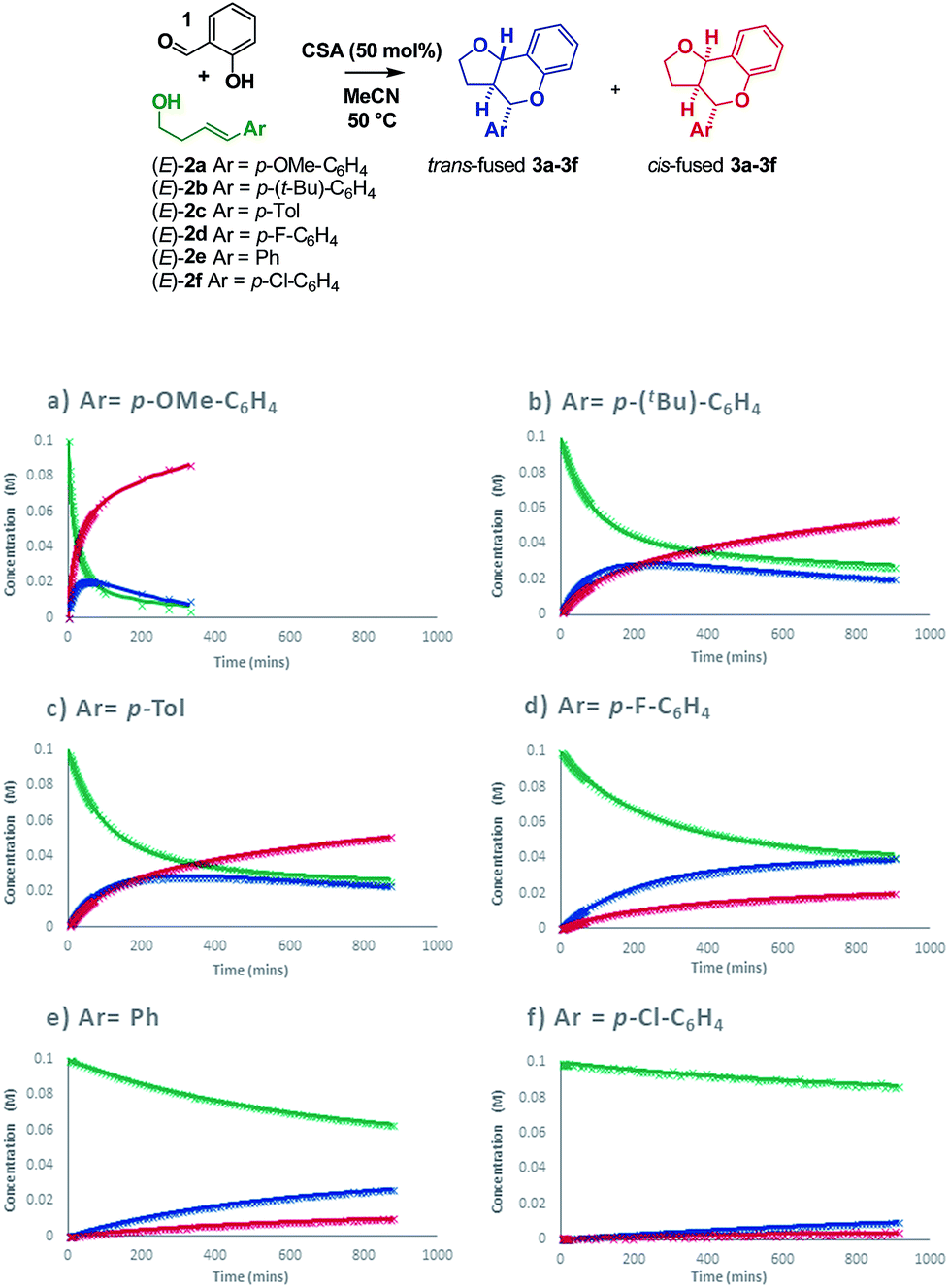 | ||
| Scheme 8 Reaction progress with varying homoallylic alcohols. | ||
Visual inspection of the graphs shows that the substituent has a profound effect on the rate of reaction. This was corroborated by the Hammett plots obtained from COPASI modelling of the data. The ρ+ value for formation of the trans-fused products was found to be less negative (ρ+ = −2.79, R2 = 0.87) than that for the formation of the cis-fused products (ρ+ = −3.69, R2 = 0.94), but the rate-determining TSs in both cases clearly have substantial carbocation character (Fig. 1).
 | ||
| Fig. 1 Hammett plot of COPASI fitted rate constants for formation of cis-fused (red) and trans-fused (blue) products vs. σ+. | ||
To better understand the origin of this disparity and to help interpret the observed data in terms of mechanism, we turned to DFT analysis at the B3LYP+GD3BJ/Def2-TZVPP level, using HCl as the acid and a continuum solvation model. The B3LYP functional with inclusion of GD3BJ dispersion terms has recently been shown to give suitably reliable activation energies.29 The TS activation free energies associated with the oQM cycloaddition pathway were significantly lowered (∼9 kcal mol−1) following protonation leading us to discount the neutral oQM mediated pathway (cf.Scheme 1b). Intrinsic reaction coordinate (IRC) analysis revealed that for all six substrates, an intermediate carbocation is formed en route to the kinetic trans-fused products. Moreover, the Hammett plot for this HCl catalyzed Prins pathway, constructed using computed activation free energies for the stepwise transition states TS1endo and TS2endo, correlates well with that from the CSA catalyzed experiments (ρcalc+ = −3.73, R2 = 0.96). IRC analysis suggests that for the cis-fused pathway, the substrates having electron releasing substituents (negative σ+ values) should proceed via a carbocation intermediate but that the substrates having electron withdrawing substituents (positive σ+ values) should follow a concerted asynchronous, acid catalyzed HDA pathway (i.e. one having no intermediate by IRC and hence no stationary point corresponding to TS2). As the switch of mechanism to a concerted process only applies to the p-Cl substituent in our experimental series (because more electron deficient substrates fail to react) the overall sensitivity to electronics as represented by the experimental ρ+, predominantly reflects that of the stepwise mechanism. Notwithstanding this, the computed Hammett ρ+ value for the cis-fused pathway (ρcalc+ = −4.34, R2 = 0.98), like the experimental one, is more negative than that for the trans-fused pathway.
DFT computation also supports the idea that intramolecular π–π stacking lowers the energies of the endo- relative to exo-TSs by a combination of dispersion and electrostatic attractive forces [i.e. noncovalent-interactions (NCIs)] attractive forces which are more significant in the former than the latter (cf.Fig. 2avs.2d).30
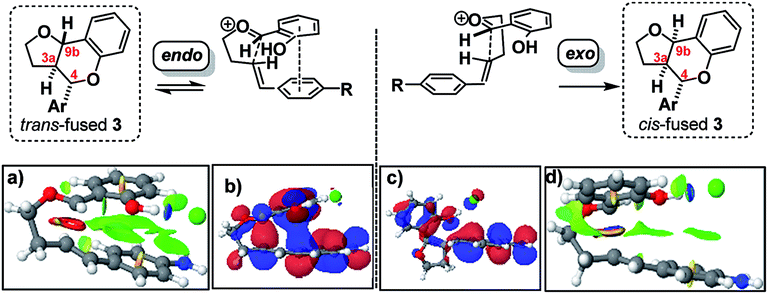 | ||
| Fig. 2 Analysis of endo and exo TS structures showing NCI surfaces (a and d)/and molecular orbitals (b and c). See FAIR data sub-collection https://doi.org/10.14469/hpc/4175 for 3D rotatable structures and relative transition state and product free energies. | ||
Quantitatively, the free energies of the endo TSs are computed to be lower by 6–8 kcal mol−1cf. the exo isomers. The difference between endo and exo TS energies more normally does not exceed 4 kcal mol−1,40 currently attributed to the accumulation of attractive dispersive and electrostatic (NCI) effects. The larger stabilization of the endo TS in our system may also be due to contributions from genuine orbital interactions38 (Fig. 2bvs.2c) reminiscent of those found in the π-complex-like TS involved in the benzidine rearrangement.31,32
Since these interactions are strongest for electron rich substrates, they increasingly compete for the electron density released by the para-substituent as σ+ becomes more negative. This dilutes the ability of the para substituent to stabilize the benzylic cation, explaining the aforementioned disparity in ρcalc+ values for formation of trans- vs. cis-fused products.
We conclude that under Brønsted acid conditions these furanochromane forming reactions proceed to the kinetic trans-fused products via an oxonium-Prins pathway, despite commonly being classed as oQM cycloadditions in the literature. However, as revealed by DFT IRC analyses, there is a continuum between these mechanistic extremes with the acid catalyzed cycloaddition pathway becoming competitive for the associated thermodynamically driven isomerization to give cis-fused products as the homoallylic alcohol-derived alkenes become more electron deficient. So, as suggested by our initial Singleton analysis, this reaction lies at the intersection of these two mechanistic manifolds. This situation provides an opportunity to use our findings to propose a qualitative benchmark for correlation of experimentally determined ρ+ values with likely mechanism for related acid catalyzed cyclocondensation reactions. Those systems with relatively low sensitivity to the electronics of the para substituents are likely concerted oQM cycloadditions, whereas those with relatively high sensitivity are likely stepwise Prins-type reactions with the ρ+ value corresponding to this nexus being ca. −3. We hope that this value will prove useful for indicative mechanistic categorisation of related reactions formally involving oQMs (cf.Scheme 2), particularly where Singleton analysis is inappropriate/inconclusive and extensive computation along the lines performed here has yet to be undertaken.
Having resolved these mechanistic questions, we sought to utilize this insight to optimize the conditions used for the practical synthesis of furanochromanes. Carbocations are known to be stabilized by hexafluoroisopropanol (HFIP)33–35 and recent calculations by Houk36 have illuminated the role of H-bonding by HFIP in accelerating the rate of certain inverse-electron demand D–A reactions by favoring a zwitterionic pathway over a concerted alternative. In light of this, we investigated the effect of HFIP in these reactions. HFIP alone did not furnish product but as an additive resulted in a substantial increase in reactivity upon addition of as little as 8 equivalents.22 Taking advantage of the fact that HFIP also improved the solubility of the CSA, we opted to use 12 equivalents of HFIP in chloroform at RT. Using these conditions (conditions A), formation of cis-fused 3c (Ar = p-Tol) from homoallylic alcohol (E)-2c was complete in just over 1 h giving an isolated yield of 91% (Scheme 9).
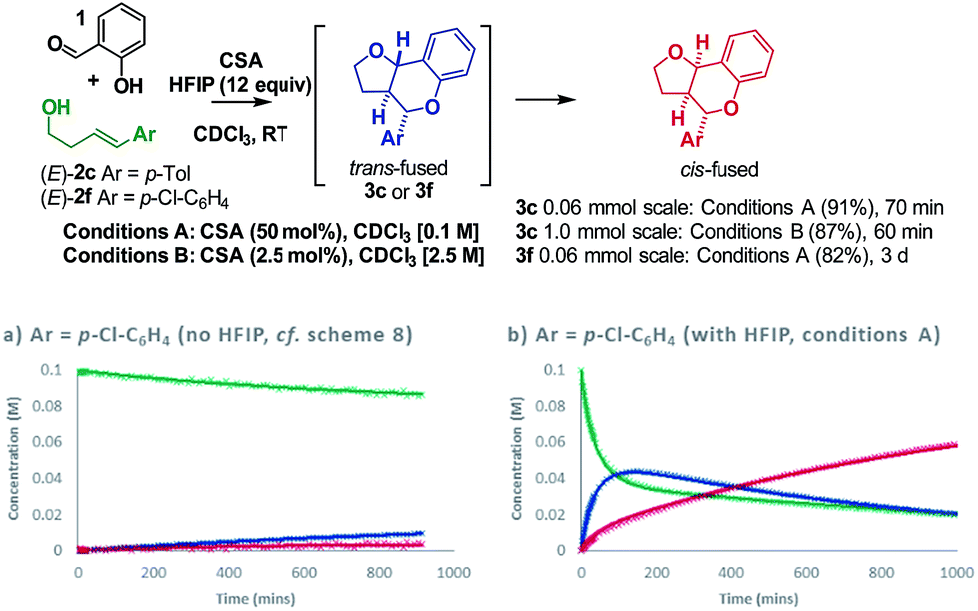 | ||
| Scheme 9 Use of HFIP as an additive. | ||
This result compares with a 70% conversion to a ∼2:1 cis:trans fused mixture of 3c after 15 h under the previous conditions at 50 °C. Similarly, the electron poor homoallylic alcohol (E)-2f, which progressed with <20% conversion to a ∼1:2. cis:trans-fused mixture of 3f after 15 h under the previous conditions, proceeded smoothly under condition A to furnish exclusively the cis-fused 3f product in an isolated yield of 82%. Comparison of reaction profiles illustrates this HFIP induced rate increase (Scheme 9avs.9b). Moreover, the COPASI fitted rate constants indicate a ∼100-fold rate enhancement. Increasing the concentration of the reaction and dropping the amount of CSA to 2.5 mol% allowed a practical preparative synthesis of 3c in just 1 h (conditions B, 87%, Scheme 9).
As HFIP is itself a Brønsted acid, we sought to confirm that the observed rate acceleration was due to its ability to stabilize the intermediate benzylic carbocation. To this end, we investigated the stereochemical fidelity with which the Z/E stereochemistry of homoallylic alcohol 2c was translated into cis/trans-3a,4-relative stereochemistry in the product 3c/7 (Scheme 10).
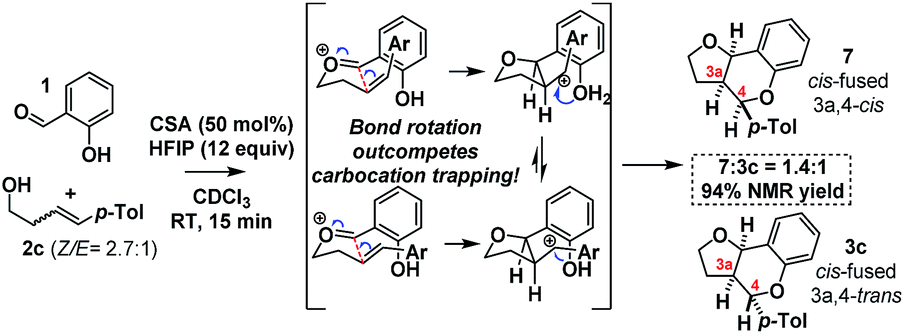 | ||
| Scheme 10 Stereochemistry scrambling with HFIP. | ||
It was found that in contrast to the complete fidelity displayed by this reaction when conducted in the absence of HFIP, a 2.7:1 mixture of (Z/E)-2c led to partially scrambled C3a–C4-stereochemistry in the product. This suggests that the acceleration is due to the stabilization of the intermediate benzylic carbocation. Recent calculations suggest carbocation lifetimes must exceed 1000 fs for loss of stereochemical information.37 In a control reaction, under the same conditions but in the absence of the salicylaldehyde (1), 2c was found to cyclize to cleanly furnish 2-tolyl-tetrahydrofuran. This reaction presumably occurs via protonation of the alkene, carbocation formation and then intramolecular cyclization.38
Looking to test the limits of the new protocol, we looked to deploy 2-aminobenzaldehyde (8) in place of salicylaldehyde (1) as a substrate. While this aniline has previously only been employed in Friedländer quinoline synthesis,39,40 and despite concerns regarding its polymerization and its basicity relative to phenol, we were delighted to find that the desired furanotetrahydroquinoline 9 could be isolated using the chloroform/HFIP conditions albeit promoted by a stoichiometric amount of CSA (Scheme 11).
 | ||
| Scheme 11 Tetrahydroquinoline formation with HFIP. | ||
Based on the foregoing studies we expect that this reaction proceeds via a stepwise Prins pathway involving the relatively rarely encountered trapping of a benzylic carbocation by an aniline nitrogen.41,42 Related reactions have however recently been suggested to proceed via oQM imine formation then cycloaddition (Scheme 12a and b).42–48
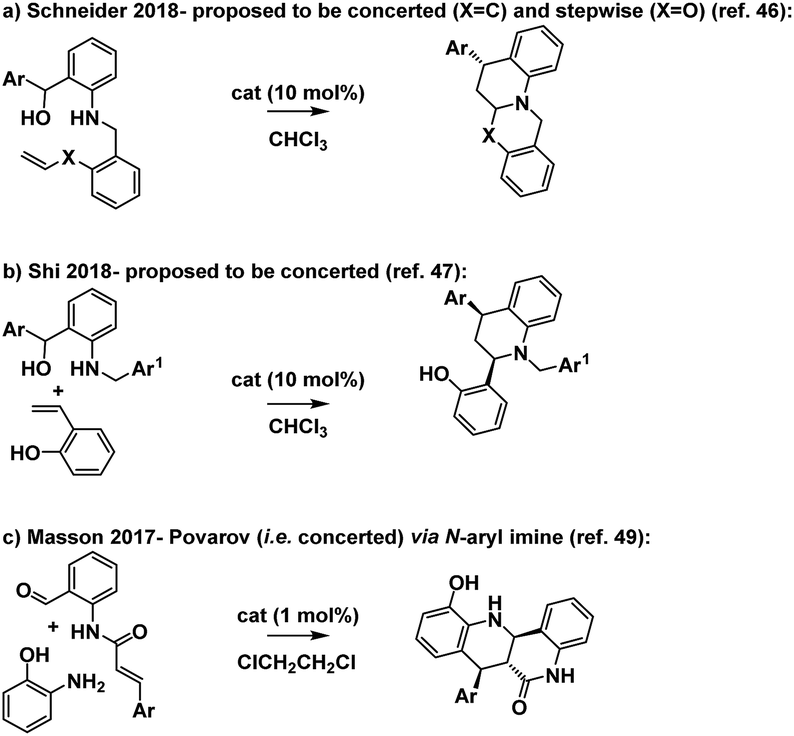 | ||
| Scheme 12 Tetrahydroquinoline forming reactions. | ||
The reaction is notable in providing access to a substituted tetrahydroquinoline via a disconnection which is complimentary to the more established hetero-D–A reaction of an N-aryl imine with an electron rich alkene (the Povarov reaction, e.g.Scheme 12c).49–51 The Povarov reaction has also been proposed to proceed via both stepwise and concerted pathways.52
Conclusion
In summary, we have conducted an experimental and computational investigation into the mechanism of formation of furanochromanes under Brønsted acid catalysis. Kinetic trans-fused products are all shown to be formed via a stepwise oxonium-Prins reaction. A new isomerization to furnish the thermodynamic and biologically active cis-fused congeners also follows this pathway for all but the most electron deficient substrates, which react slowly via an asynchronous acid catalyzed oQM cycloaddition pathway.25 Rational modification of the conditions by addition of HFIP achieved a ca. 100-fold rate enhancement to give cis-fused configured furanochromane products exclusively and allowed access to a furanotetrahydroquinoline by using 2-aminobenzaldehyde in place of salicylaldehyde.Beyond the mechanistic insight and practical scope extension the work has provided for this furanochromane-forming reaction, the study has illuminated a correlation between the Hammett ρ+ value and mechanism for this class of acid catalyzed cyclocondensation. High sensitivity to electronic factors (i.e. ρ+ value more negative than ca. −3) militates in favour of a concerted oQM cycloaddition pathway, particularly for electron withdrawing substituents, whereas low sensitivity (i.e. ρ+ value less negative than ca. −3) is indicative of a stepwise Prins-type pathway. Moreover, this sensitivity and hence mechanism of formation can differ between stereoisomeric products because the electronic communication between the Hammett para-substituent and the benzylic position is ‘intercepted’ by intramolecular π–π interactions in endo- but not exo-TSs – a phenomenon referred to as ‘secondary orbital overlap’ in D–A parlance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. D. T. N. thanks Syngenta (Pharmacat consortium) for a PhD studentship. W. J. M. thanks the People Programme (Marie Curie Actions) of the EU Seventh Framework Programme (FP7/2007–2013) under REA Grant Agreement No. 607466.Notes and references
- C. D.-T. Nielsen, H. Abas and A. C. Spivey, Synthesis, 2018, 50, 4008–4018 CrossRef CAS.
- R. W. Van De Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367–5405 CrossRef CAS.
- N. J. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160–9173 CrossRef CAS.
- A. A. Jaworski and K. A. Scheidt, J. Org. Chem., 2016, 81, 10145–10153 CrossRef CAS.
- Y. Xie and B. List, Angew. Chem., Int. Ed., 2017, 56, 4936–4940 CrossRef CAS.
- L. M. Zhao, A. L. Zhang, H. S. Gao and J. H. Zhang, J. Org. Chem., 2015, 80, 10353–10358 CrossRef CAS.
- Y. Xie, G. J. Cheng, S. Lee, P. S. J. Kaib, W. Thiel and B. List, J. Am. Chem. Soc., 2016, 138, 14538–14541 CrossRef CAS.
- G. C. Tsui, L. Liu and B. List, Angew. Chem., Int. Ed., 2015, 54, 7703–7706 CrossRef CAS.
- A. C. Spivey, L. Laraia, A. R. Bayly, H. S. Rzepa and A. J. P. White, Org. Lett., 2010, 12, 900–903 CrossRef CAS.
- O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923–7927 CrossRef CAS.
- W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910–1913 CrossRef CAS.
- J. J. Zhao, S. B. Sun, S. H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460–5464 CrossRef CAS.
- C. C. Hsiao, S. Raja, H. H. Liao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 5762–5765 CrossRef CAS.
- C. D. Gheewala, J. S. Hirschi, W.-H. Lee, D. W. Paley, M. J. Vetticatt and T. H. Lambert, J. Am. Chem. Soc., 2018, 140, 3523 CrossRef CAS.
- D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357–9358 CrossRef CAS.
- C. Wu, G. Yue, C. D.-T. Nielsen, K. Xu, H. Hirao and J. Zhou, J. Am. Chem. Soc., 2016, 138, 742–745 CrossRef CAS.
- L. Liu, H. Kim, Y. Xie, C. Farè, P. S. J. Kaib, R. Goddard and B. List, J. Am. Chem. Soc., 2017, 139, 13656–13659 CrossRef CAS.
- H. Abas, S. M. Linsdall, M. Mamboury, H. S. Rzepa and A. C. Spivey, Org. Lett., 2017, 19, 2486–2489 CrossRef CAS.
- M. Born, P.-A. Carrupt, R. Zini, F. Brée, J.-P. Tillement, K. Hostettmann and B. Testa, Helv. Chim. Acta, 1996, 79, 1147–1158 CrossRef CAS.
- Z.-G. Feng, W.-J. Bai and T. R. R. Pettus, Angew. Chem., Int. Ed., 2015, 54, 1864–1867 CrossRef CAS PubMed.
- T. Rukachaisirikul, P. Innok, N. Aroonrerk, W. Boonamnuaylap, S. Limrangsun, C. Boonyon, U. Woonjina and A. Suksamrarn, J. Ethnopharmacol., 2007, 110, 171–175 CrossRef CAS.
- (a) C. D.-T. Nielsen, W. J. Mooij, D. Sale, H. S. Rzepa, J. Burés and A. C. Spivey, FAIR data archives, Imperial College Research Computing Services data repository, 2018, DOI:10.14469/hpc/3943 and sub-collections therein; (b) Standard pdf ESI, 10.1039/c8sc04302g.
- R. Jasti and S. D. Rychnovsky, J. Am. Chem. Soc., 2006, 128, 13640–13648 CrossRef CAS.
- F. A. Carroll, Perspectives on Structure and Mechanism in Organic Chemistry, 1998 Search PubMed.
- X. Creary, B. D. O'Donnell and M. Vervaeke, J. Org. Chem., 2007, 72, 3360–3368 CrossRef CAS.
- J. M. Wurst, G. Liu and D. S. Tan, J. Am. Chem. Soc., 2011, 133, 7916–7925 CrossRef CAS.
- C. K. Hazra, J. Jeong, H. Kim, M.-H. Baik, S. Park and S. Chang, Angew. Chem., Int. Ed., 2018, 57, 2692–2696 CrossRef CAS PubMed.
- S. Hoops, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- E. E. Kwan, Y. Zeng, H. A. Besser and E. N. Jacobsen, Nat. Chem., 2018, 10, 917 CrossRef CAS PubMed.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed.
- S. Yamabe, H. Nakata and S. Yamazaki, Org. Biomol. Chem., 2009, 7, 4631–4640 RSC.
- G. Ghigo, S. Osella, A. Maranzana and G. Tonachini, Eur. J. Org. Chem., 2011, 2326–2333 CrossRef CAS.
- M. C. DiPoto, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2015, 137, 14861–14864 CrossRef CAS PubMed.
- A. Acharya, D. Anumandla and C. S. Jeffrey, J. Am. Chem. Soc., 2015, 137, 14858–14860 CrossRef CAS.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS.
- Y. F. Yang, P. Yu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 18213–18221 CrossRef CAS PubMed.
- J. S. J. Tan, V. Hirvonen and R. S. Paton, Org. Lett., 2018, 20, 2821–2825 CrossRef CAS PubMed.
- N. Tsuji, J. L. Kennemur, T. Buyck, S. Lee, S. Prévost, P. S. J. Kaib, D. Bykov, C. Farès and B. List, Science, 2018, 1505, 1501–1505 CrossRef PubMed.
- V. K. Rai, F. Verma, G. P. Sahu, M. Singh and A. Rai, Eur. J. Org. Chem., 2018, 2018, 537–544 CrossRef CAS.
- D. S. Deshmukh and B. M. Bhanage, Synlett, 2018, 29, 979–985 CrossRef CAS.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS PubMed.
- J. Zhou and H. Xie, Org. Biomol. Chem., 2018, 16, 380–383 RSC.
- H.-H. Liao, A. Chatupheeraphat, C.-C. Hsiao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 15540–15544 CrossRef CAS.
- A. Chatupheeraphat, H. H. Liao, S. Mader, M. Sako, H. Sasai, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 4803–4807 CrossRef CAS.
- M. Kretzschmar, T. Hodík and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 9788–9792 CrossRef CAS PubMed.
- T. Hodík and C. Schneider, Org. Biomol. Chem., 2017, 15, 3706–3716 RSC.
- C. Schneider, M. Kretzschmar, F. Hofmann and D. Moock, Angew. Chem., Int. Ed., 2018, 57, 4774 CrossRef.
- L. Z. Li, C. S. Wang, W. F. Guo, G. J. Mei and F. Shi, J. Org. Chem., 2018, 83, 614–623 CrossRef CAS PubMed.
- L. He, M. Bekkaye, P. Retailleau and G. Masson, Org. Lett., 2012, 14, 3158–3161 CrossRef CAS PubMed.
- L. Jarrige, F. Blanchard and G. Masson, Angew. Chem., Int. Ed., 2017, 56, 10573–10576 CrossRef CAS PubMed.
- X. L. Yu, L. Kuang, S. Chen, X. L. Zhu, Z. L. Li, B. Tan and X. Y. Liu, ACS Catal., 2016, 6, 6182–6190 CrossRef CAS.
- G. Masson, C. Lalli, M. Benohoud and G. Dagousset, Chem. Soc. Rev., 2013, 42, 902–923 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: C. D.-T. Nielsen, W. J. Mooij, D. Sale, H. S. Rzepa, J. Burés and A. C. Spivey, FAIR data archives, Imperial College Research Computing Services data repository, 2018, DOI: 10.14469/hpc/3943 and sub-collections therein. See DOI: 10.1039/c8sc04302g |
| This journal is © The Royal Society of Chemistry 2019 |