
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free visible light photoredox enables generation of carbyne equivalents via phosphonium ylide C–H activation†
Mrinmoy
Das‡
,
Minh Duy
Vu‡
,
Qi
Zhang
and
Xue-Wei
Liu
 *
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: xuewei@ntu.edu.sg
First published on 4th December 2018
Abstract
Carbyne, an interesting synthetic intermediate, has recently been generated from hypervalent iodine precursors via photoredox catalysis. Given the underexplored chemistry of carbyne, due to the paucity of carbyne sources, we are intrigued to discover a new source for this reactive species from classical reagents – phosphonium ylides. Our novel strategy employing phosphonium ylides in an olefin hydrocarbonation reaction features a facile approach for constructing carbon–carbon bonds through metal-free and benign reaction conditions. Moreover, the hydrocarbonation products were delivered in a highly regioselective manner.
Introduction
Conventionally, in organic structures, carbon atoms with a lack of valences are categorized into 4 common classes, namely, carbocations, radicals, carbenes and carbanions. Since 1967, the interesting carbyne intermediate, which would belong to the 5th class, was discovered by Strausz in a decomposition reaction of diethyl mercurybisdiazoacetate.1–3 This species contains 3 non-bonding electrons and appears to be so reactive that its chemistry has not been widely established. Until earlier this year, Suero and co-workers reported convenient access to a diazomethyl radical, which is a direct equivalent to the carbyne species, via the use of hypervalent iodine and photoredox chemistry (Fig. 1a).4 In line with our interest in utilizing ylides in single electron transfer processes, we proposed that mono-substituted stabilized phosphonium ylides could undergo C–H activation to form ylide radicals, which would be new synthetic equivalents to carbyne. In this article, we report the reaction between the ylides and activated olefins to form a hydrocarbonation-like product under this novel mechanism.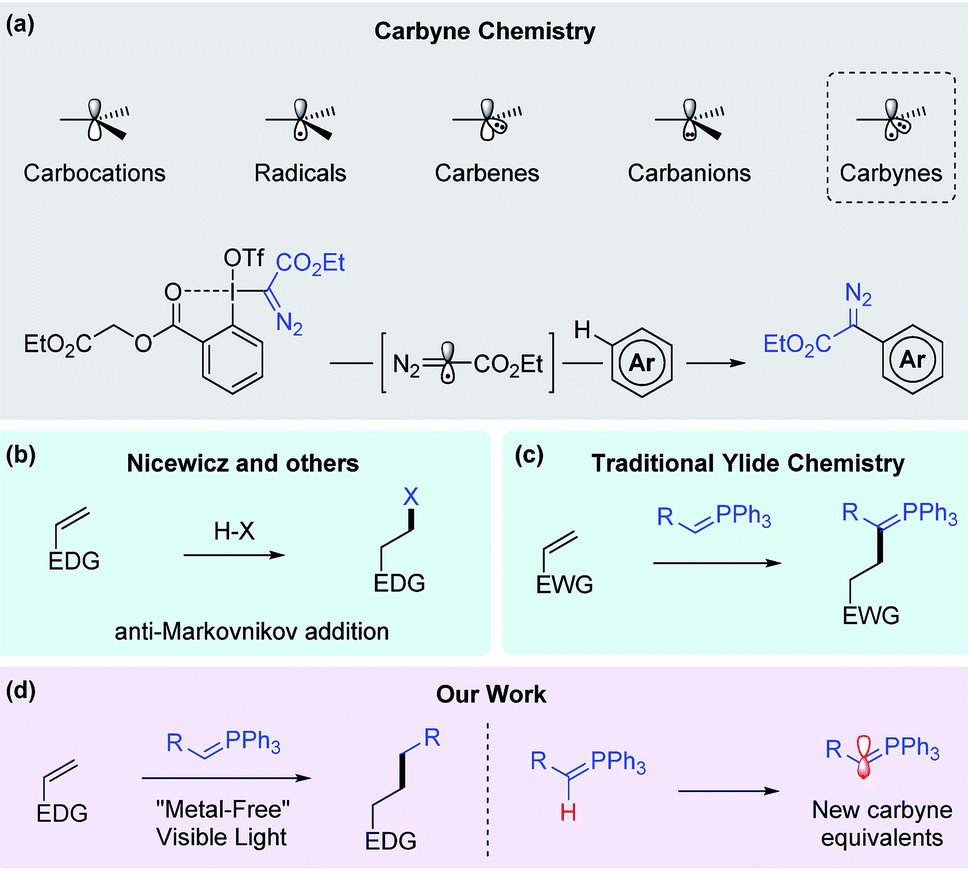 | ||
| Fig. 1 Overview of reported carbyne chemistry, anti-Markovnikov HX addition, ylide chemistry and our strategy. | ||
In recent years, olefins have been employed as valuable synthons for synthesis due to their ubiquitous occurrence in nature and the chemical industry. The atom transfer radical addition (ATRA) reaction is a classical choice to forge new carbon–carbon bonds from alkenes. Seminal work done by Stephenson,5 Reiser,6,7 Melchiorre,8 and Martin9 on photoredox catalysis together with Severin’s work10–12 on ruthenium catalysis has greatly advanced ATRA. Liu’s work on reductive olefin hydrocarbonation via nickel catalysis also offers a highly practical alternative for carbon–carbon bond formation from olefins.13 Additionally, since 2014, independent work done by Baran and Shenvi has revealed the possibility of radical formation from electron rich olefins via silane reduction with iron, manganese or cobalt catalysts.14–19 Furthermore, the new trend of methodology development using inactivated olefins as substrates for carbon–carbon bond formation has been blooming over the past few years thanks to great contributions from other groups.20–25
With the rise of visible light photoredox catalysis,26–28 its application to functionalize alkenes has been demonstrated by several groups. Nicewicz and co-workers have pioneered the anti-Markovnikov addition of olefins by utilizing acridinium photosensitizers since 2012 (Fig. 1b).29–32 The catalyst’s high oxidation potential (>2.0 V vs. SCE) is sufficient for the single electron oxidation process of several alkenes, leading to the attack of various nucleophiles such as alcohols,33 amines,34 carboxylates35 and halides.36 The work was subsequently extended to olefin anti-Markovnikov hydration by Lei and co-workers.37 Very recently, Huang et al. also demonstrated [4 + 2] cycloaddition of styrene using similar acridinium photosensitizers.38
Initially, we questioned whether the cationic radical intermediate resulting from olefin single electron oxidation would be vulnerable to carbon nucleophiles. In that case, useful carbon–carbon bond formation could be developed. Our tremendous efforts turned out unsuccessful while employing “hard” nucleophiles such as enolates or other organometallic reagents, presumably due to the low stability of the organophotocatalyst under the reaction conditions. When we turned our attention to phosphonium ylides, the classical carbon nucleophiles,39,40 our initial trials achieved remarkable success. In previous literature, the attack of ylides on electron deficient olefins was reported to form new elongated ylides through trivial Michael type addition (Fig. 1c).41 In our reaction, with the carbyne radical involvement subsequently disclosed, the final product is no longer a phosphorane due to phosphine cleavage via in situ reduction (Fig. 1d). To the best of our knowledge, there is only one recent example of a reaction between ester-stabilized ylides and olefin under photoredox conditions, as reported by Miura and Murakami.42 It was proposed that the formation of an alkyl radical from the phosphonium salt using an iridium photocatalyst is the key step in the mechanistic pathway.
Results and discussion
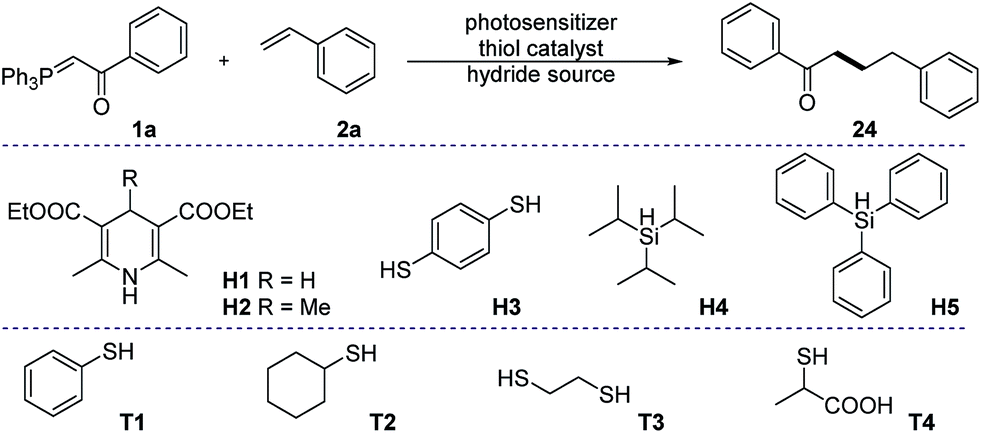
Prime investigations with model substrates 1a and 2a revealed successful carbon–carbon bond formation when using thiol as a hydrogen atom transfer (HAT) catalyst and the Hantzsch ester as a hydride donor. A wide range of aromatic as well as aliphatic thiols was found to catalyse the reaction (entries 1–4). Among them, thiophenol was chosen as the best catalyst due to its simplicity and high efficiency. Subsequently, some other hydride sources were evaluated in place of the Hantzsch ester. However, diminished yields were obtained in all of these cases (entries 5–8). Solvent screening (see ESI†) showed that chlorinated non-polar solvents, such as DCM or DCE, are the best candidates. The conversion of the phosphonium ylide 1a substrate was almost complete in the majority of trials and the frequent side product observed results from the reduction of 1a to acetophenone.43,44 We were pleased to discover that further diluted reaction concentrations slightly help to improve the yield of the desired product (entry 13). Control experiments (entries 9–12) confirmed the necessity of all parameters in the optimized conditions.With the optimized reaction conditions in hand, we proceeded to evaluate the substrate scope of both olefins and phosphonium ylides (Fig. 2). Generally, in most of the examples, moderate to excellent yields were observed with absolute regioselectivity as determined by crude 1H-NMR (the linear form is predominant). Ester stabilized phosphonium ylides (4–7) worked slightly better than their ketone counterparts (8–23). This is probably due to slow undesirable Wittig olefination of the expected product with the ylide starting materials. Aliphatic (8–10 and 18–20) and aromatic substituted ketone stabilized ylides (11–17 and 21–23) worked equally well. The electronic effect of substituents on the aromatic ring does not significantly affect the reaction efficiency (Table 1).
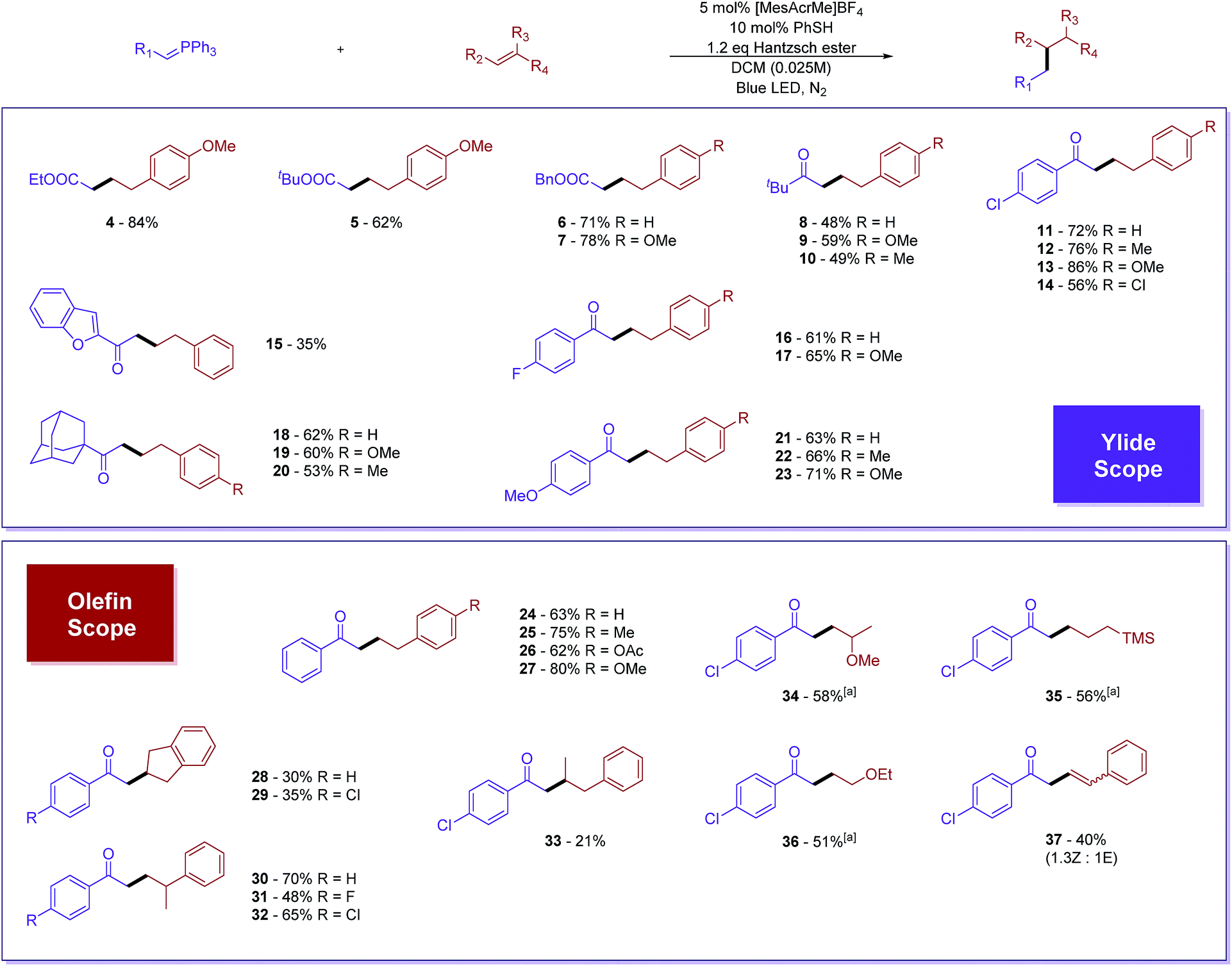 | ||
| Fig. 2 Phosphonium ylide and olefin substrate evaluation. All reactions were carried out under an inert atmosphere, with 0.1 mmol of the phosphonium ylides and 0.3 mmol of the corresponding alkenes in dry DCM (0.025 M). 9-Mesityl-10-methylacridinium tetrafluoroborate (5 mol%) was used as the photosensitizer in all experiments. Isolated yields were reported. a10 equivalents of alkenes were used instead. The isomeric ratio was determined by analysis of crude 1H-NMR. | ||
|
|
|||
|---|---|---|---|
| Entry | Thiol catalyst (10 mol%) | Hydride source (1.2 equiv.) | Resultsb |
| a All reactions were carried out under an inert atmosphere, with 0.1 mmol of 1a and 0.3 mmol of 2a in dry DCM (0.1 M). 9-Mesityl-10-methylacridinium tetrafluoroborate (5 mol%) was used as the photosensitizer in all experiments. b Yields were determined from the crude reaction mixture by 1H-NMR using 1,3,5-trimethoxy benzene as an internal standard. c Without photosensitizer. d Without light irradiation. e Diluted concentration: 0.025 M of 1a, isolated yield was reported. NR. no reaction. ND. not determined, complex mixture. | |||
| 1 | T1 | H1 | 57% |
| 2 | T2 | H1 | 51% |
| 3 | T3 | H1 | 20% |
| 4 | T4 | H1 | 24% |
| 5 | T1 | H2 | NR. |
| 6 | — | H3 | 49% |
| 7 | T1 | H4 | ND. |
| 8 | T1 | H5 | ND. |
| 9 | — | H1 | NR. |
| 10 | T1 | — | NR. |
| 11c | T1 | H1 | NR. |
| 12d | T1 | H1 | NR. |
| 13e | T1 | H1 | 63% |
In terms of olefin substrates, para-substituted styrenes gave reasonably good yields despite electronic effect differences (24–27). Additionally, α- and β-disubstituted styrene showed positive reactivity, although a low yield was obtained for the β-isomer presumably due to steric effects (30–33). Cyclic alkenes, such as indene, could be viable substrates for the reaction as well (28 and 29). Besides styrene derivatives, vinyl ether (34 and 36) and allylic trimethylsilane (35) could also work to a certain extent. Interestingly, terminal alkynes such as phenyl acetylene successfully reacted, furnishing the formation of disubstituted alkenes in decent yields with a mixture of (Z) and (E) isomers (37). It is worth noting that the alkene product in this case did not undergo further addition.
After measuring the redox potential, we realized that ylide 1a actually has a considerably lower oxidation potential (Eox = 1.12 V vs. SCE) than its alkene partner 2a (Eox = 1.97 V vs. SCE).45 The results from a fluorescence quenching plot (Fig. 3) also suggest that among all reaction components, ylide 1a is the most efficient quencher. We therefore propose that 1a might first undergo single electron oxidation by the excited photocatalyst, forming the cationic radical (A) (Fig. 4). This is in contrast with the mechanism for anti-Markovnikov addition for C–X bond formation by Nicewicz. To date, the reactivity of intermediate (A) has not been reported. We rationalize that it might undergo 3 different pathways (Fig. 4). Its cationic character could facilitate nucleophilic addition of an alkene while its radical character could enable radical addition to an alkene. Either pathway will result in a new radical cation intermediate with the ylide hydrogen intact. To our surprise, starting from deuterated ylide 1b, the obtained hydrocarbonation product does not contain any deuterium (verified by NMR and MS). We hence postulate that the hydrogen in (A) is acidic and possibly undergoes fast proton exchange with the medium. In other words, deprotonation of (A) leading to a neutral carbyne radical could happen. Further confirmation by a trapping reaction with the radical scavenger TEMPO showed traces of unstable trapping product (C) in the MS analysis of the reaction mixture. In addition, new peaks corresponding to compound (D) were also observed in the MS analysis. This explains well the formation of radical intermediate (B) which may undergo a HAT process with the TEMPO radical (MS peaks for (D) were not observed in the normal reaction without TEMPO). Together with recent work conducted by Suero and coworkers,4 this constitutes one of the rare examples of the formation and reaction of a carbyne intermediate equivalent.
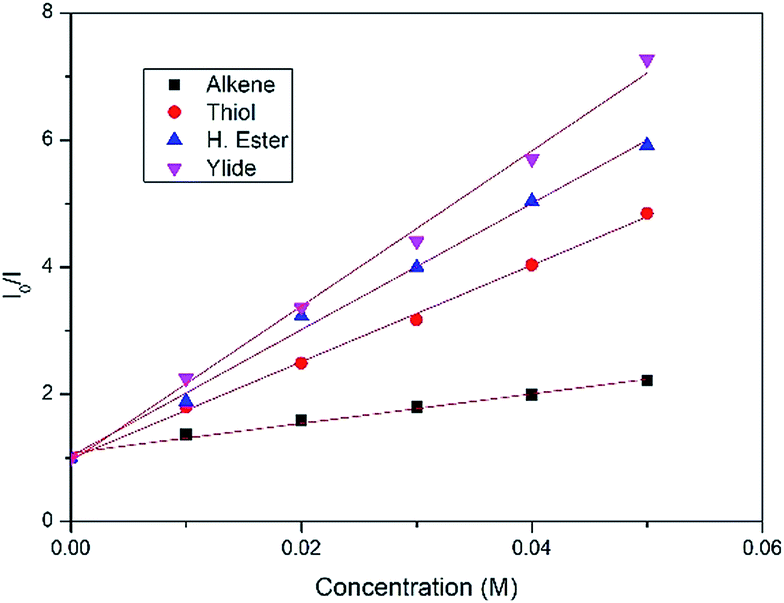 | ||
| Fig. 3 Stern–Volmer plot for fluorescence quenching of each component. | ||
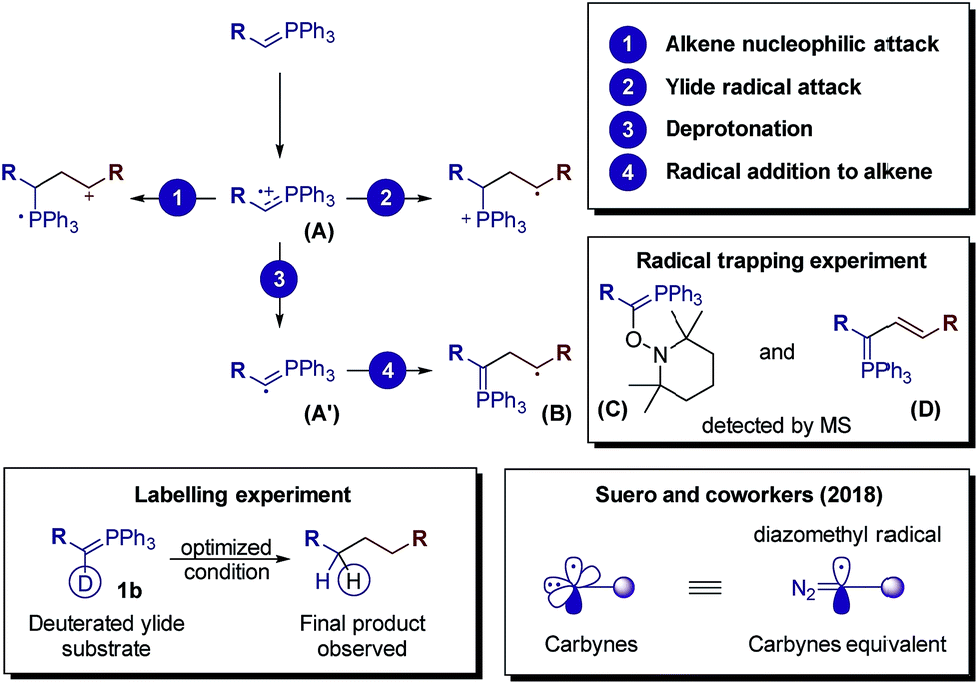 | ||
| Fig. 4 The fate of radical cation (A) resulting from single electron oxidation of the phosphonium ylide. | ||
To gain more mechanistic insight, we carried out other deuterium labelling experiments (Fig. 5a) using different deuterated Hantzsch esters. The results showed that deuterium only ends up at C2 of product 24d, despite its origin in the Hantzsch ester. This interesting phenomenon could explain that the source of hydrogen at C4 is from the thiol catalyst through HAT, and the deuterium could be incorporated into the final product via reduction with a Hantzsch ester, even though a detailed mechanism of the reduction is unclear. We also tested the probability of photoinduced reduction between the Hantzsch ester and ylide 1b, and complete reduction to acetophenone was observed after 12 hours (Fig. 5b).
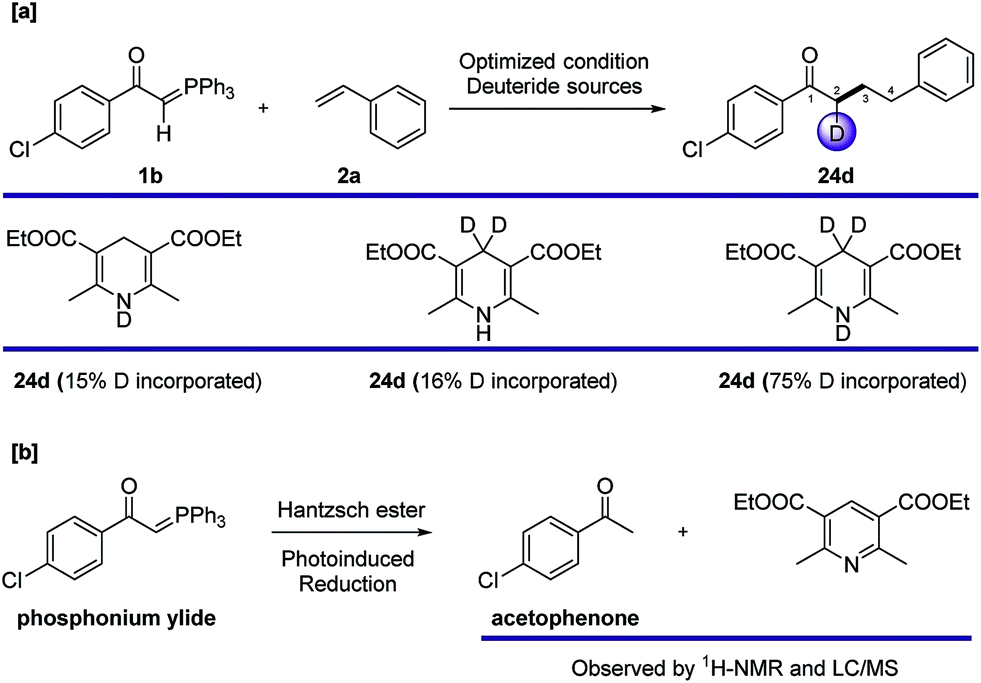 | ||
| Fig. 5 Other labelling experiments with deuterated Hantzsch esters and a photoinduced reduction test on the phosphonium ylide. | ||
With all of the information gathered so far, we have proposed a mechanism for our hydrocarbonation protocol, as shown in Fig. 6. The key step that determines regioselectivity in our protocol is the addition of the carbyne radical (A′) to the olefin. A linear chain isomer was obtained in all cases resulting from the stability of secondary radical (B).
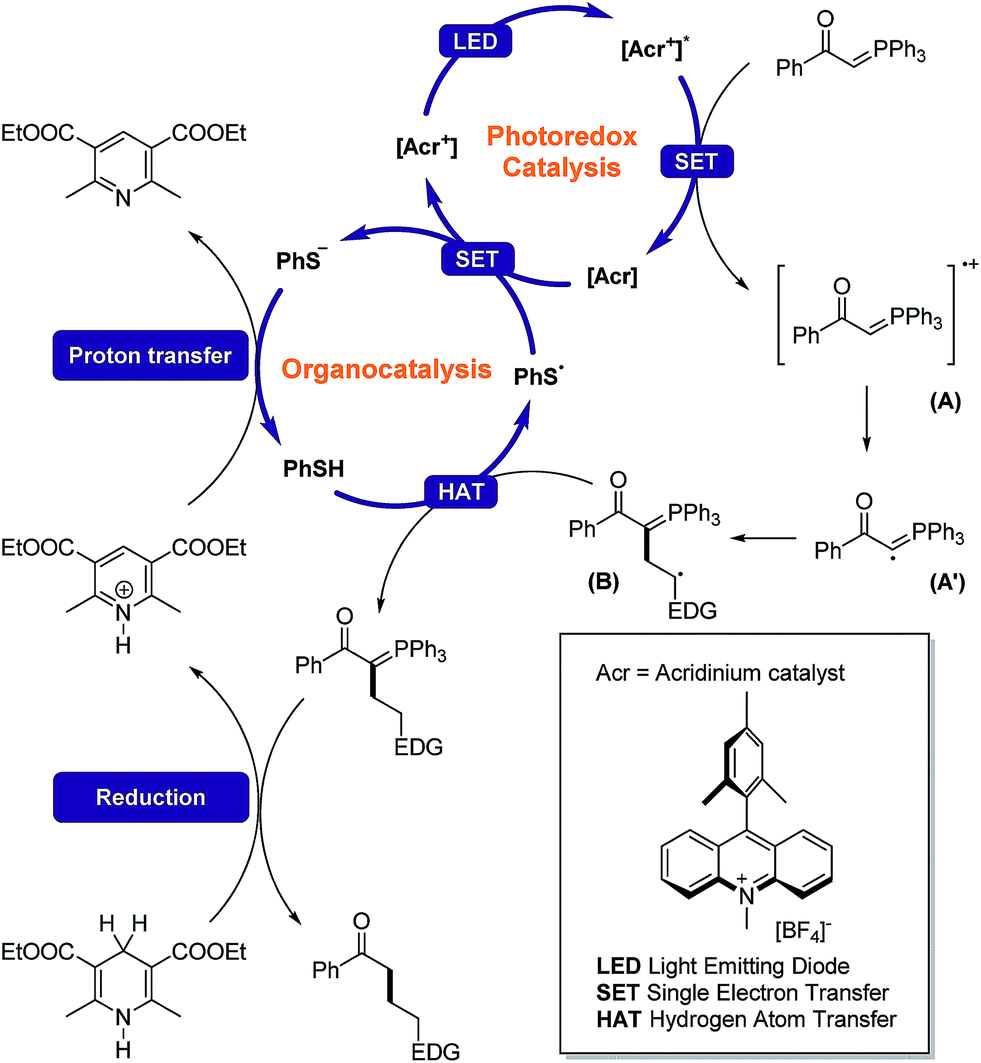 | ||
| Fig. 6 Our hypothesized mechanism. | ||
Conclusions
In conclusion, a benign method for the addition of phosphonium ylides to electron rich alkenes was developed. The reaction conditions are completely metal-free, can be carried out at room temperature, and utilize only visible light as an activation source. Our reactions gave absolute predictable regioselectivity and synthetically useful yields. Preliminary studies suggest a novel mechanism involving the recently introduced carbyne radical intermediate. The outlook of this study would be the employment of phosphonium ylides as carbyne equivalents in multi-functionalization reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Research Foundation (NRF2016NRF-NSFC002-005) and the Ministry of Education (MOE 2013-T3-1-002), Singapore for the financial support of this research.Notes and references
- M. Thap Do, H. E. Gunning and O. P. Strausz, J. Am. Chem. Soc., 1967, 89, 6785–6787 CrossRef.
- O. P. Strausz, D. M. Thap and J. Font, J. Am. Chem. Soc., 1968, 90, 1930–1931 CrossRef CAS.
- O. P. Strausz, G. J. A. Kennepohl, F. X. Garneau, D. Thap, B. Kim, S. Valenty and P. S. Skell, J. Am. Chem. Soc., 1974, 96, 5723–5732 CrossRef CAS.
- Z. Wang, A. G. Herraiz, A. M. del Hoyo and M. G. Suero, Nature, 2018, 554, 86–91 CrossRef CAS PubMed.
- C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- M. Knorn, T. Rawner, R. Czerwieniec and O. Reiser, ACS Catal., 2015, 5, 5186–5193 CrossRef CAS.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem.–Eur. J., 2012, 18, 7336–7340 CrossRef CAS PubMed.
- E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed.
- Y. Shen, J. Cornella, F. Juliá-Hernández and R. Martin, ACS Catal., 2017, 7, 409–412 CrossRef CAS.
- L. Quebatte, K. Thommes and K. Severin, J. Am. Chem. Soc., 2006, 128, 7440–7441 CrossRef CAS PubMed.
- K. Thommes, B. Içli, R. Scopelliti and K. Severin, Chem.–Eur. J., 2007, 13, 6899–6907 CrossRef CAS PubMed.
- M. A. Fernández-Zúmel, K. Thommes, G. Kiefer, A. Sienkiewicz, K. Pierzchala and K. Severin, Chem.–Eur. J., 2009, 15, 11601–11607 CrossRef PubMed.
- X. Lu, B. Xiao, Z. Zhang, T. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129–11137 CrossRef PubMed.
- J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS PubMed.
- J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS PubMed.
- S. A. Green, J. L. M. Matos, A. Yagi and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 12779–12782 CrossRef CAS PubMed.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- C. Obradors, R. M. Martinez and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 4962–4971 CrossRef CAS PubMed.
- X. Dong, J. Cui, J. Song, Y. Han, Q. Liu, Y. Dong and H. Liu, Chem. Commun., 2017, 53, 4903–4906 RSC.
- Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS PubMed.
- S. Bordi and J. T. Starr, Org. Lett., 2017, 19, 2290–2293 CrossRef CAS PubMed.
- Y. Shen, B. Huang, J. Zheng, C. Lin, Y. Liu and S. Cui, Org. Lett., 2017, 19, 1744–1747 CrossRef CAS PubMed.
- K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS PubMed.
- S. Sumino, A. Fusano and I. Ryu, Org. Lett., 2013, 15, 2826–2829 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- D. A. Nicewicz and D. S. Hamilton, Synlett, 2014, 25, 1191–1196 CrossRef PubMed.
- K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 CrossRef CAS PubMed.
- D. J. Wilger, M. GrandjeanJean-Marc, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 CrossRef CAS PubMed.
- X. Hu, G. Zhang, F. Bu and A. Lei, ACS Catal., 2017, 7, 1432–1437 CrossRef CAS.
- L. Wang, F. Wu, J. Chen, D. A. Nicewicz and Y. Huang, Angew. Chem., Int. Ed., 2017, 56, 6896–6900 CrossRef CAS PubMed.
- O. I. Kolodiazhnyi, in Phosphorus Ylides, Wiley-VCH Verlag GmbH, 2007, pp. 1–8, DOI:10.1002/9783527613908.ch01.
- R. Appel, R. Loos and H. Mayr, J. Am. Chem. Soc., 2009, 131, 704–714 CrossRef CAS PubMed.
- X.-L. Sun and Y. Tang, Acc. Chem. Res., 2008, 41, 937–948 CrossRef CAS PubMed.
- T. Miura, Y. Funakoshi, J. Nakahashi, D. Moriyama and M. Murakami, Angew. Chem., Int. Ed., 2018, 57, 15455–15459 CrossRef CAS PubMed.
- T. J. Van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 44, 4953–4962 CrossRef CAS.
- D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04195d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |