DOI:
10.1039/C8SC04061C
(Edge Article)
Chem. Sci., 2019,
10, 569-575
A divergent synthetic pathway for pyrimidine-embedded medium-sized azacycles through an N-quaternizing strategy†
Received
12th September 2018
, Accepted 18th October 2018
First published on 18th October 2018
Abstract
Medium-sized heterocycles have recently received significant attention because of their potential roles as modulators of protein–protein interactions, but their molecular diversity and synthetic availability are still inadequate to meet the demand. To address these issues, we developed a new divergent synthetic pathway for skeletally distinct pyrimidine-containing medium-sized azacycles. We introduced N-quaternized pyrimidine-containing polyheterocycles as novel key intermediates for diversity-generating reactions via selective bond cleavages or migrations and prepared 14 discrete core skeletons in an efficient manner. The skeletal diversity of the resulting molecular frameworks was confirmed by chemoinformatic analysis.
Introduction
The identification of novel bioactive small-molecules is an essential research element in drug discovery and chemical biology. Small-molecule collections with high three-dimensional (3D) skeletal diversity and complexity are invaluable resources in the discovery of new chemical probes or therapeutic agents, especially for regulating protein–protein interactions.1–3 In this regard, the emergence of diversity-oriented synthesis (DOS) has provided access to unprecedented molecular frameworks with maximized skeletal and stereochemical diversity, which enables an unbiased screening of compounds and discovery of their interactions with diverse biological targets.4–6 Recently, a variety of divergent synthetic strategies to create discrete core skeletons have been developed.7 Along with its structural diversity, the biological relevancy of a chemical library is another important consideration for targeting the bioactive chemical space. To satisfy the two criteria simultaneously, we devised a privileged substructure-based DOS (pDOS) strategy based on the assumption that privileged structural motifs could be effective “chemical navigators” to access unexploited biologically relevant chemical space.8,9 The pDOS strategy focuses on the assembly of discrete heterocyclic moieties around the privileged substructures through divergent synthesis in an efficient manner. In the last decade, the effectiveness of the pDOS strategy has been validated by the discovery of novel small-molecule modulators for various biological activities including anti-neuroinflammatory effects,10 chondrogenesis-inducing activity,11 and the inhibition of protein–miRNA interactions.12
As a continuation of our endeavour to develop new pDOS strategies, we envisioned that pyrimidines would be excellent navigators toward bioactive chemical space because they have been frequently found in bioactive natural products and extensively studied in medicinal chemistry as nucleoside analogues.13 Therefore, we developed pDOS pathways, which afforded diverse pyrimidine-embedded 6-14 and 7-membered15 azacyclic core skeletons through a variety of pairing strategies based on the synthetic versatility of pyrimidine. From this pyrimidine-containing pDOS library, we identified novel bioactive small molecules that modulated the cellular contents of lipid droplets16 or inhibited the protein–protein interaction between leucyl-tRNA synthetase (LRS) and Ras-related GTP-binding protein D (RagD).15
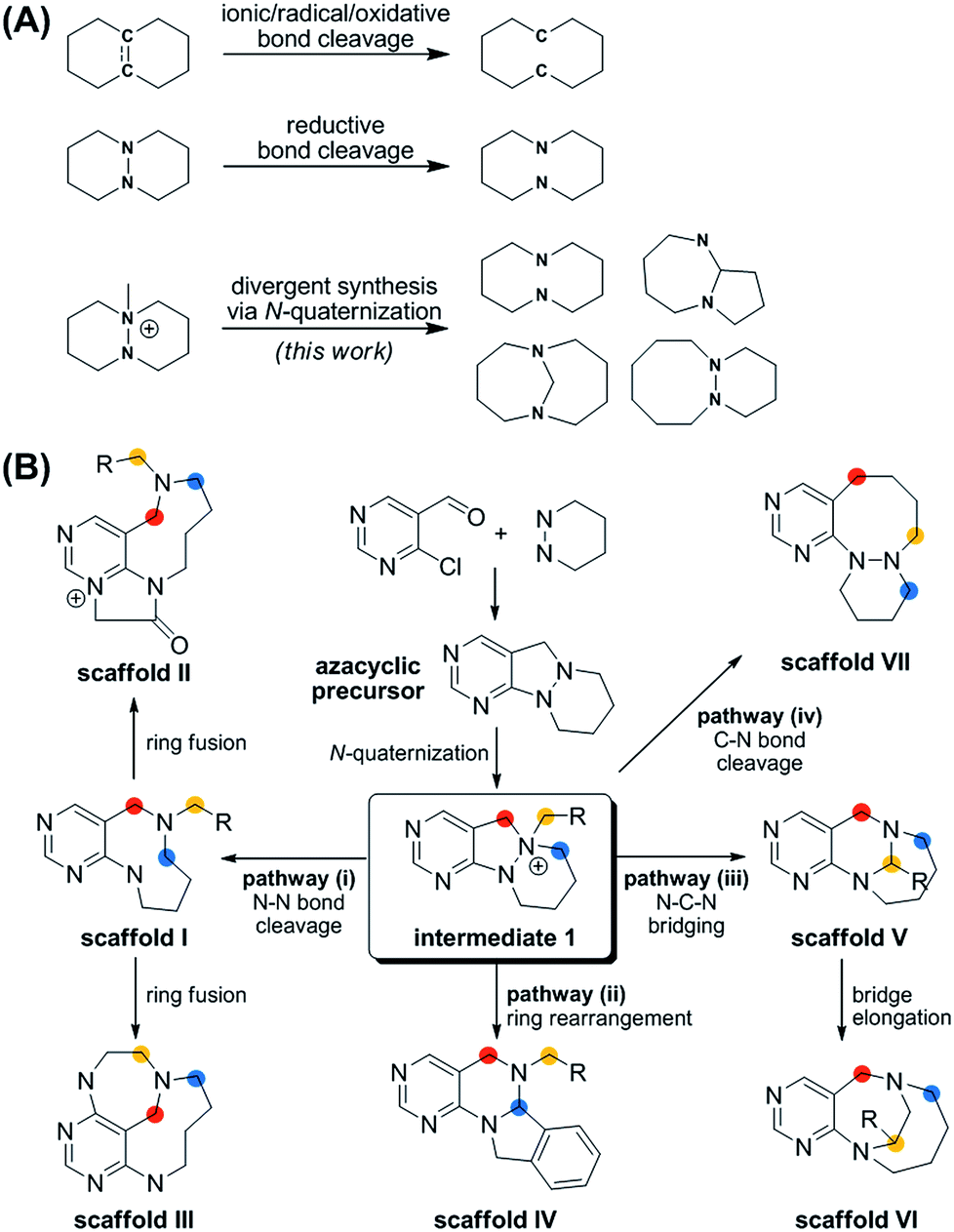
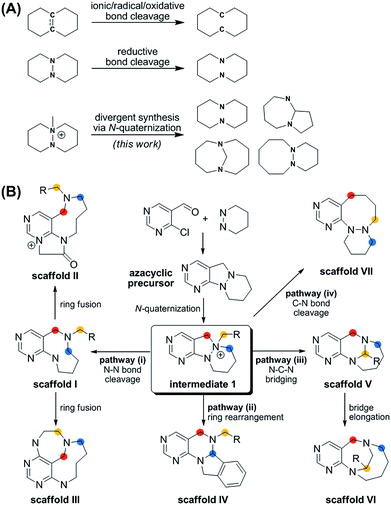
Herein, we describe a novel pDOS pathway for pyrimidine-fused medium-sized rings with high skeletal diversity and biological relevancy. Although 8- to 11-membered cyclic motifs have been observed in various bioactive natural products, these molecular frameworks are hard to find in the current list of top-selling drugs because of their limited synthetic accessibility and consequent underexposure in drug discovery screening exercises.17 The classical head-to-tail cyclization of linear precursors to access medium-sized rings is much more difficult than the formation of 5- or 6-membered rings because of entropic and enthalpic factors. In this regard, the selective cleavage of a central C–C single bond or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to generate a medium-sized carbocycle from a fused bicyclic precursor has been widely studied as an alternative method to access medium-sized rings (Fig. 1A).18 Recently, a series of DOS approaches for medium-sized molecular frameworks using the cleavage of C–C or CC zero bridges as the main strategy have been reported.19–21 However, there are limited studies22–24 on the cleavage of N–N bonds to afford the medium-sized heterocycles, especially those with the potential to interact with biopolymers differently than the analogous carbocycles.
C double bond to generate a medium-sized carbocycle from a fused bicyclic precursor has been widely studied as an alternative method to access medium-sized rings (Fig. 1A).18 Recently, a series of DOS approaches for medium-sized molecular frameworks using the cleavage of C–C or CC zero bridges as the main strategy have been reported.19–21 However, there are limited studies22–24 on the cleavage of N–N bonds to afford the medium-sized heterocycles, especially those with the potential to interact with biopolymers differently than the analogous carbocycles.
 |
| | Fig. 1 (A) Synthetic strategies for medium-sized rings via ring cleavage. (B) Divergent synthetic pathway for pyrimidine-containing medium-sized azacycles through N-quaternizing strategy. | |
More importantly, the reported methods for the reductive cleavage of N–N bonds are not suitable for diversity-generating reactions. To address this issue, we designed a divergent synthetic pathway for the facile construction of distinct pyrimidine-embedded medium-sized azacycles through chemoselective N–N bond cleavages or rearrangements from N-quaternized key intermediates (Fig. 1B). Fourteen discrete pyrimidine-containing medium-sized azacycles were synthesized and their 3D structural diversity was confirmed by in silico analysis.
Results and discussion
Design of diversity-generating pathways for pyrimidine-containing medium-sized azacycles
To prepare diverse pyrimidine-containing medium-sized rings, we first designed key intermediates 1, prepared by selective N-quaternization of azacyclic precursors derived from the reactions of functionalized pyrimidine moieties with cyclic hydrazines (Fig. 1B). As the polyheterocyclic precursors were designed to have a single tertiary amine whose nucleophilicity would be stronger than those of the other anilinic nitrogen, selective N-quaternization using a variety of alkyl halides was possible. Regarding the quaternized nitrogen as a reaction center, base- and/or hydride-mediated orthogonal transformations into diverse medium-sized azacycles were devised. In pathway (i), co-treatment with base and hydride allowed the selective cleavage of the N–N zero bridge, forming pyrimidine-fused medium-sized diazacycles (scaffold I). The core skeleton was further diversified to rigid tricyclic scaffolds II and III through differentiated ring fusions utilizing the functional groups of scaffold I. Base treatment of intermediate 1 triggered cleavage of the N–N bond followed by intramolecular aminal formation to give ring-rearranged scaffold IV via pathway (ii) or N–C–N bridged scaffold V via pathway (iii). Successive ring expansion from scaffold V allowed the formation of ethano-bridged scaffold VI. Finally, selective C–N bond cleavage without dissociation of the N–N bond afforded medium-sized polyheterocycles (scaffold VII) via pathway (iv). Consequently, distinct pyrimidine-embedded medium-sized azacycles with high skeletal diversity were successfully established under precisely controlled reaction conditions.
Synthesis of pyrimidine-containing key intermediates
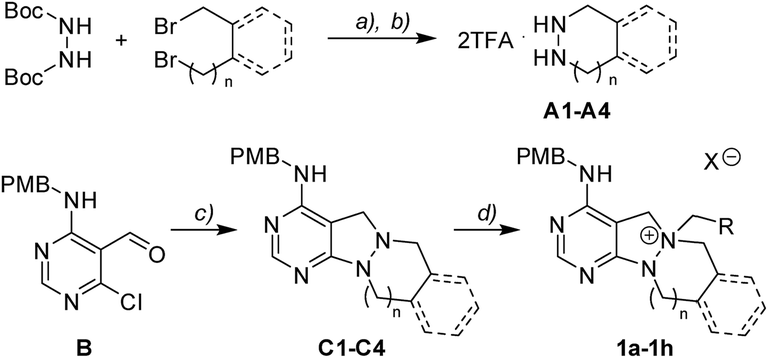
To investigate the designed transformations, we prepared pyrimidine-containing azacyclic precursors having a central N–N bond (Scheme 1). First, cyclic hydrazines (A1–A4) were synthesized from the coupling of di-tert-butyl hydrazodiformate with different alkyl dibromides via double SN2 reaction and the subsequent deprotection of the tert-butyloxycarbonyl (Boc) groups in the presence of trifluoroacetic acid (TFA). The sequential cyclization and reduction of functionalized pyrimidine (B) with the prepared cyclic hydrazines (A1–A4) efficiently afforded azacyclic precursors (C1–C4). Finally, the key intermediates (1a–1h) were prepared through selective N-quaternization of C1–C4 with various alkyl halides.
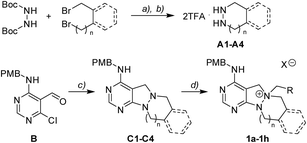 |
| | Scheme 1 Synthetic scheme for pyrimidine-containing azacyclic key intermediates 1a–1h. Reagents and conditions: (a) tetraethylammonium bromide (TEAB), 50% aq. NaOH, toluene/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), 100 °C; (b) TFA, DCM, r.t.; (c) cyclic hydrazine (A1–A4), TEA, EtOH, 80 °C, then NaBH4, EtOH, r.t.; (d) RCH2X, ACN, 40–80 °C. :1 v/v), 100 °C; (b) TFA, DCM, r.t.; (c) cyclic hydrazine (A1–A4), TEA, EtOH, 80 °C, then NaBH4, EtOH, r.t.; (d) RCH2X, ACN, 40–80 °C. | |
Scaffold differentiation studies
Pathway (i).
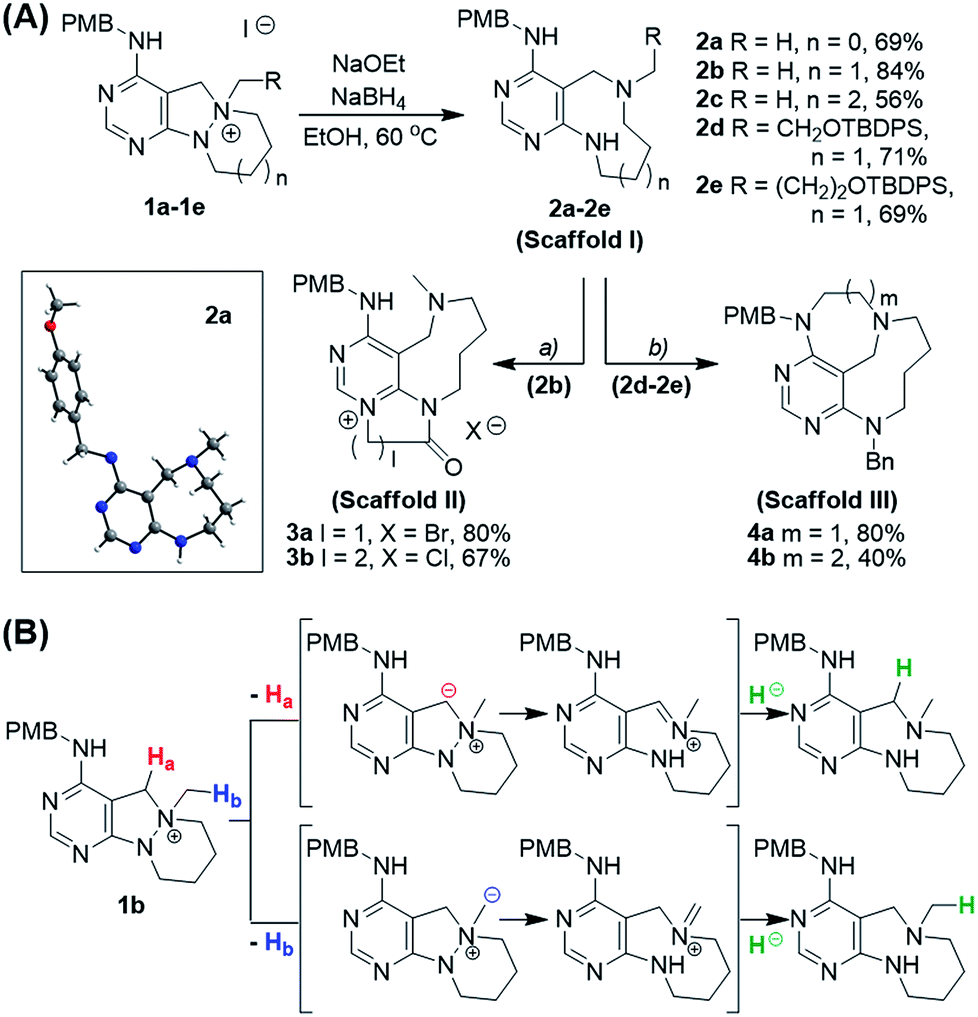
We initiated our investigation of the N–N bond cleavage reaction using 1b as a substrate. A previous report on the cleavage of an N–N zero bridge through a Hofmann-type elimination from an N-quaternized precursor using NaOMe24 served as our starting point for optimizing the reaction conditions. After screening a wide range of base, hydride source, and solvent combinations (see ESI† for optimization table), we found that treatment with NaOEt and NaBH4 in EtOH media smoothly transformed the N-quaternized key intermediate 1b into 9-membered diazonane-fused pyrimidine 2b in high yield (84%, Scheme 2A). To test the scope of this transformation for further diversification, other intermediates were examined under the optimized conditions. For instance, 8-membered diazocane- and 10-membered diazecane-fused pyrimidines 2a and 2c were successfully synthesized from 1a and 1c, respectively. The formation of medium-sized azacycles via N–N cleavage was clearly confirmed by the structural elucidation of 2a using X-ray crystallography. Moreover, it is worth mentioning that the N–N bond cleavage reaction conditions were sufficiently mild to produce pyrimidine-fused diazonanes 2d and 2e containing functionalized substituents (silyl-protected hydroxyethyl or hydroxypropyl moieties, respectively) for late-stage elaboration.
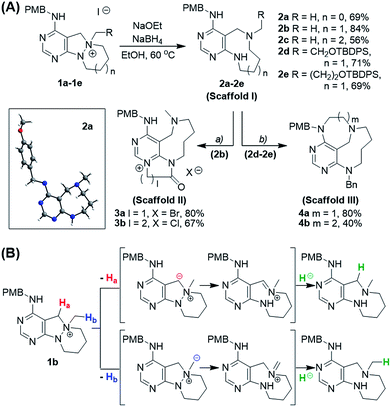 |
| | Scheme 2 (A) Exploration of N–N bond cleavage pathway (i) and subsequent ring fusions for scaffolds I–III. Reagents and conditions: (a) bromoacetyl bromide, ACN, 80 °C (for 3a) or 3-chloropropionyl chloride, DMF, r.t. to 120 °C (for 3b); (b) BnBr, ACN, then HF/pyridine/THF, then MsCl, DCM, then NaH, DMF, r.t. (B) Proposed mechanism of N–N bond cleavage reaction. | |
Although pleased with the outcome of these reactions, we were not confident that the cleavage of the N–N bond occurred via the previously reported Hofmann-type β-elimination because the reaction did not occur when using only NaOEt. Moreover, the presence of benzylic protons (Ha), which are more acidic than β-protons, brought the reaction mechanism into question. Therefore, we considered that the N–N bond could be cleaved through the base-triggered formation of an iminium species and its subsequent neutralization by hydride addition (Scheme 2B). To understand the reaction mechanism, we performed a deuterium-labelling experiment with 1b using NaBD4 to find the site of nucleophilic attack. According to the 1H NMR data, deuterium was introduced into the methyl position (Hb) predominantly compared to the benzylic position (Ha) (see ESI†). Additionally, when NaCN was used as a nucleophile instead of a hydride source, the cyanide-added product in the Hb position was obtained as a major product. On the basis of these results, we proposed a new reaction mechanism for N–N bond cleavage through base-promoted iminium formation followed by hydride-mediated neutralization as shown in Scheme 2B.
Along with the efficient cleavage of the N–N bond, further transformations of scaffold I were demonstrated by utilizing both pre-embedded and newly introduced functionalities (Scheme 2A). The 9-membered diazonane 2b was treated with dielectrophiles such as bromoacetyl bromide or 3-chloropropionyl chloride, which selectively acylated the newly generated aniline moiety and subsequently cyclized with the pyrimidyl nitrogen to afford scaffold II (3a and 3b) containing a pyrimidinium moiety. In the cases of functionalized diazonanes 2d and 2e, we successfully synthesized the conformationally restricted bridged scaffold III (4a and 4b) through an intramolecular substitution reaction using the pre-embedded aniline moiety.
Pathway (ii).
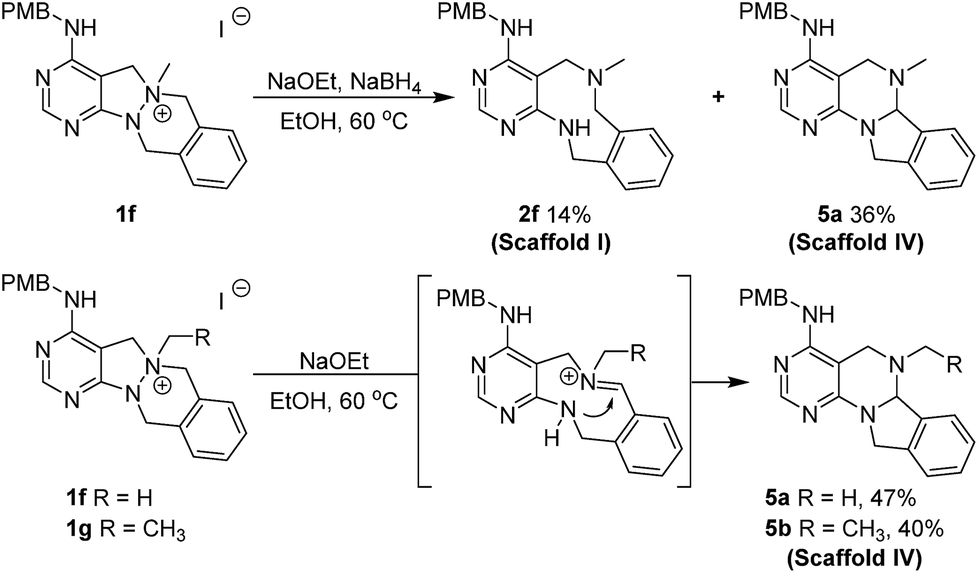
In the case of the tetracyclic N-quaternized intermediate 1f, which has more than one benzylic carbon adjacent to the quaternized nitrogen, the ring-rearranged tetracyclic product 5a was predominantly obtained compared to the N–N bond cleaved tricyclic product 2f under the usual reaction conditions for the N–N bond cleavage reaction (Scheme 3). This ring transformation is likely caused by base-promoted iminium formation followed by intramolecular nucleophilic attack of the anilinic nitrogen to form the energetically favorable 6-membered ring, which is preferred compared to the intermolecular hydride attack.25 Therefore, a new 6/6/5/6-tetracyclic scaffold IV (5a and 5b) could be accessed by the simple base treatment of 6/5/6/6-tetracyclic intermediates (1f and 1g).
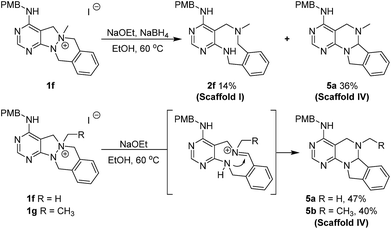 |
| | Scheme 3 Exploration of ring-rearrangement pathway (ii) for scaffold IV. | |
Pathway (iii).
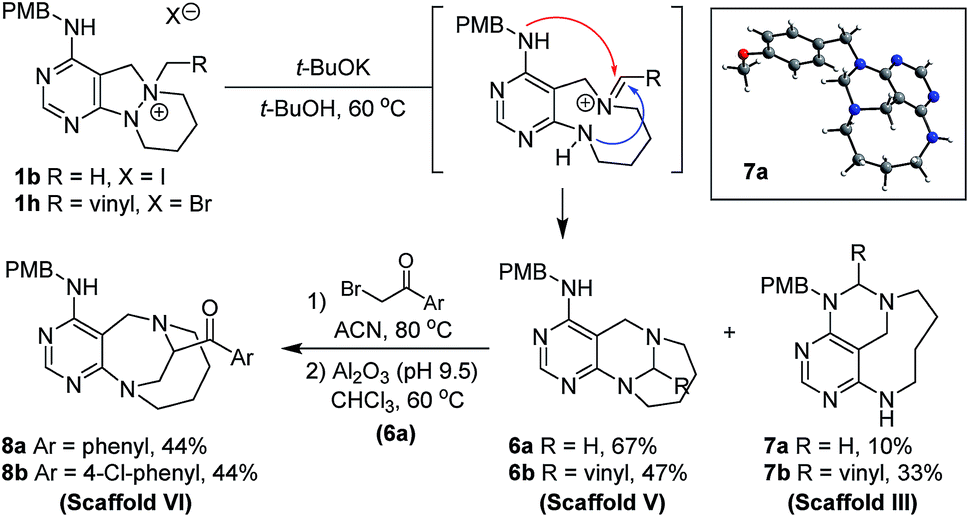
To generate bridged medium-sized ring scaffold V, we devised an intramolecular reaction that involved the migration of an N–N–C bond to an N–C–N bond. Upon treatment with base (t-BuOK) in the absence of a hydride source, 1b was converted into diazabicyclo[4.3.1]decane-fused pyrimidine 6a26 as a major product along with triazabicyclo-[6.3.1]dodecane-fused pyrimidine 7a as a minor product (Scheme 4). Under the reaction conditions, there are two nucleophiles that could attack the iminium carbon in an intramolecular fashion, so that the formation of both scaffold V (6a and 6b) and scaffold III (7a and 7b) would be possible. The structural identification of 7a was performed by X-ray crystallography. It is interesting that 6a and 7a were obtained in relatively high selectivity (67% and 10% yields, respectively) when the R group was a proton (1b), while the selectivity was reduced in the case of a vinyl R group (6b and 7b were obtained in 47% and 33% yields, respectively). In addition, further transformation of the resulting bridged structure 6a was investigated to afford ethano-bridged diazabicyclo[4.3.2]undecane-fused pyrimidines through bridge elongation. The new homologated bridged azacycles 8a and 8b (scaffold VI) were synthesized through the selective N-quaternization of 6a with α-bromoacetophenone derivatives and the subsequent migration of the α-carbon under mild basic conditions.27
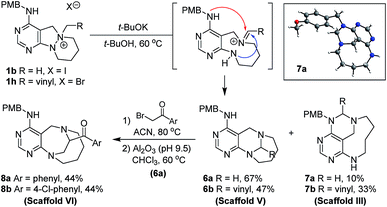 |
| | Scheme 4 Exploration of N–C–N bridging pathway (iii) for scaffold V and subsequent bridge elongation for scaffold VI. | |
Pathway (iv).
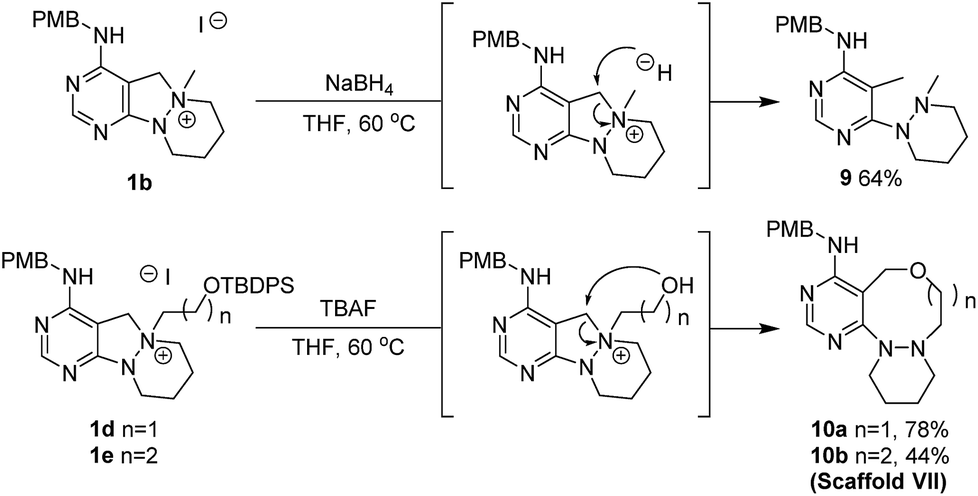
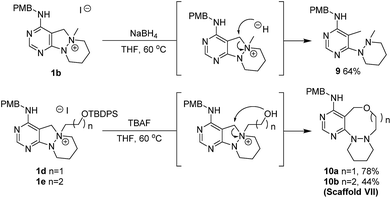
Different from the former reaction pathways, the treatment of 1b with NaBH4 in aprotic media without base yielded C–N bond-cleavage product 9 through direct hydride attack at the benzylic carbon (Scheme 5). Using this orthogonal reactivity of the key intermediate 1b, we devised the synthesis of scaffold VII through internal nucleophile-triggered dissociation of the C–N bond, and designed 1d and 1e containing a silyloxy group as a potential nucleophile. Upon treatment with tetrabutylammonium fluoride (TBAF), key intermediates 1d and 1e were respectively converted into ring- expanded 8- or 9-membered oxadiazacycles 10a and 10b as scaffold VII through removal of the silyl group followed by intramolecular nucleophilic attack of the alkoxide moiety.
 |
| | Scheme 5 Exploration of C–N bond cleavage pathway (iv) for scaffold VII. | |
Chemoinformatic analysis of the newly synthesized scaffolds
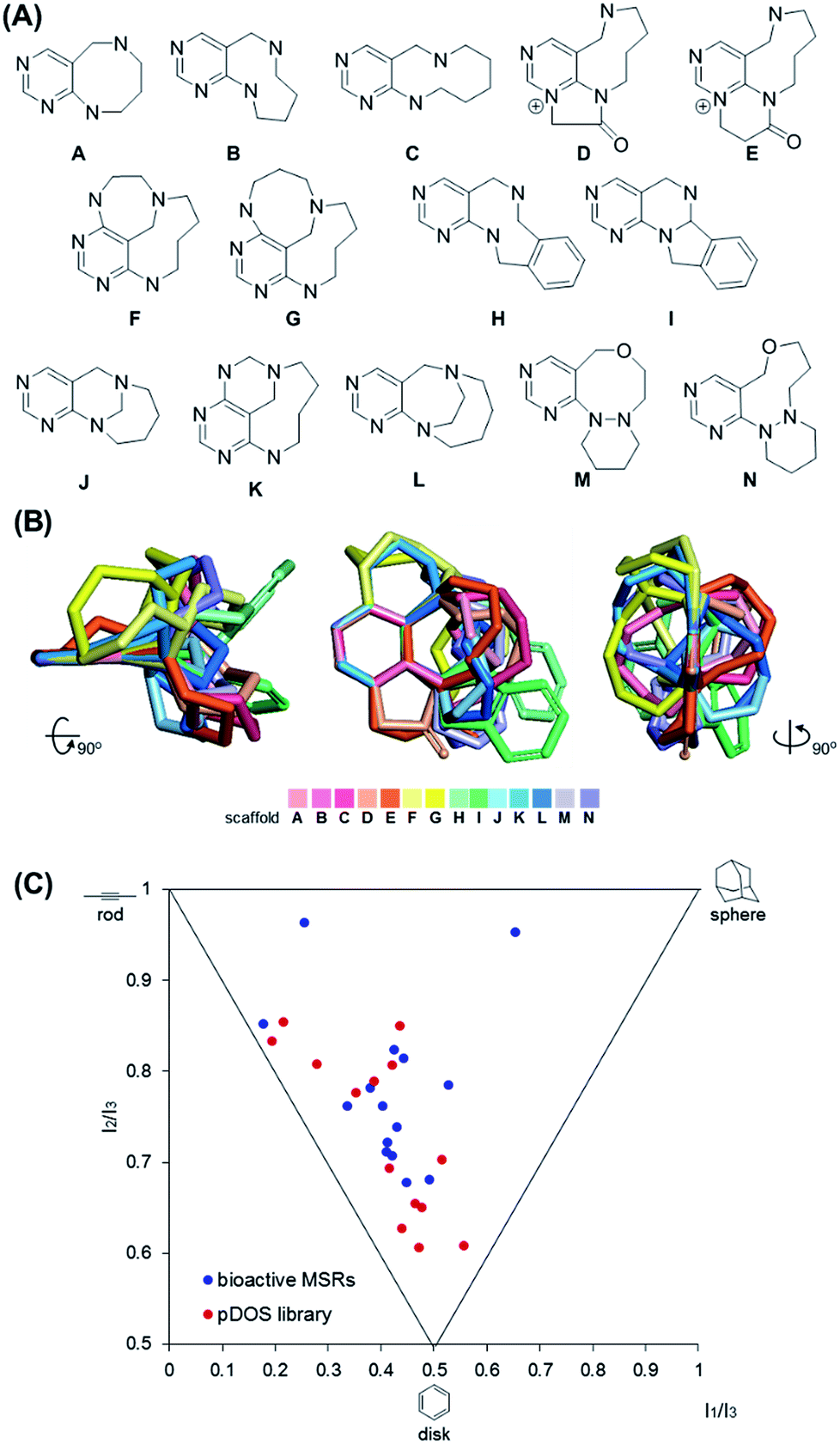
It is worth mentioning that we achieved skeletal diversity among the various medium-sized azacycles through chemoselective bond cleavage or rearrangement via the N-quaternizing strategy, while the conventional reductive N–N bond cleavage of cyclic hydrazines using transition metal-catalyzed hydrogenation allows simple cleaved azacycles. With this pDOS pathway, we synthesized 14 discrete core skeletons that contain not only synthetically challenging but also biologically relevant pyrimidine-embedded medium-sized and/or bridged azacycles (Fig. 2A). The constructed chemical library includes unique molecular frameworks such as 8- to 10- membered ring-fused diazacycles (A–E and H), five different bridged structures (F, G, and J–L), and 8- to 9-membered ring-containing oxadiazacycles (M and N). To visualize the skeletal diversity of the resulting scaffolds, we determined the energy-minimized conformer of each scaffold using quantum mechanical calculations (see ESI† for detailed procedures). As shown in Fig. 2B, we clearly visualized the skeletal diversity of the 14 scaffolds (A–N) by overlaying their calculated 3D structures, aligning their pyrimidine substructure. Furthermore, we performed a principal moment of inertia (PMI) analysis to compare the shape diversity of our pyrimidine-containing medium-sized polyheterocycles with 15 benzannulated medium-sized rings in bioactive natural products and synthetic molecules.28 As shown in Fig. 2C, a good level of molecular shape distribution was achieved in the polyheterocycles derived from our pDOS library, similar to that of the bioactive medium-sized heterocycles. We also confirmed the good structural diversity of the resulting 14 scaffolds via Tanimoto similarity matrix analysis29,30 (see Fig. S3†).
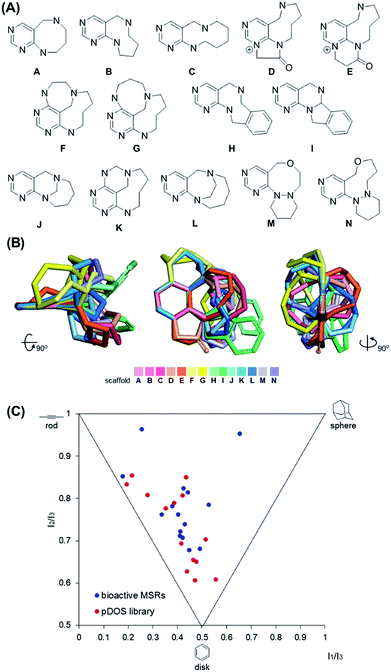 |
| | Fig. 2 Chemoinformatic analysis of discrete pyrimidine-embedded medium-sized azacycles. (A) Core skeletons of 14 discrete scaffolds (scaffold I: A–C, H; scaffold II: D, E; scaffold III: F, G, K; scaffold IV: I; scaffold V: J; scaffold VI: L; scaffold VII: M, N). (B) Overlay of energy-minimized conformers of 14 core skeletons aligned by the pyrimidine substructure. (C) Principal moment of inertia (PMI) plot. The 3D molecular shapes of pDOS library (red dots) were compared with those of 15 benzannulated medium-sized rings (MSRs) in bioactive natural products and synthetic molecules (blue dots). | |
Conclusions
We developed a new divergent synthetic pathway via an N-quaternizing strategy for efficient access to diverse pyrimidine-embedded medium-sized azacycles. Easily accessible N-quaternized key intermediate 1 could be efficiently transformed to 14 discrete scaffolds through chemoselective bond cleavage or migration. The skeletal diversity of each scaffold was demonstrated by structural alignment in 3D space and the PMI analysis of energy-minimized conformers. This unique chemical library of medium-sized azacycles might serve as a valuable resource for the discovery of novel small-molecule modulators and potential therapeutic agents, especially for modulating protein–protein interactions. The biological evaluation of the resulting compound collection will be reported in due course.
Experimental procedures
General synthetic procedure for scaffold I (2a–2f)
To a solution of 1a–1f (0.16 mmol) in ethanol (3.2 mL, 0.05 M), NaOEt (0.24 mmol) and NaBH4 (1.6 mmol) were added at room temperature. The resulting mixture was stirred at 60 °C. After completion of the reaction (checked by TLC), the reaction mixture was concentrated under reduced pressure, quenched with deionized water and saturated aq. NaHCO3, and the organic material was extracted three times with ethyl acetate (EA). The combined organic extracts were dried over anhydrous Na2SO4(s) and filtered. After the solvent was evaporated under reduced pressure, the residue was purified by silica gel flash column chromatography to obtain desired compounds 2a–2f in moderate to high yields. In the cases of 2d and 2e, 3.2 mmol of NaBH4 were used.
General synthetic procedure for scaffold II (3a–3b)
To a solution of 2b (73.0 mg, 0.23 mmol) in acetonitrile (ACN, 4.6 mL, 0.05 M), either 2-bromoacetyl bromide (for 3a, 21.4 μL, 0.25 mmol) or 3-chloropropionyl chloride (for 3b, 24.6 μL, 0.26 mmol) were added at room temperature. The resulting mixture was stirred at 80 °C (for 3a) or room temperature to 120 °C (for 3b). After completion of the reaction (checked by TLC), the reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by silica gel flash column chromatography to obtain desired compounds 3a and 3b in 80% and 67% yields, respectively.
General synthetic procedure for scaffold III (4a–4b)
Precursors for 4a and 4b were obtained through a three-step synthesis including benzylation, desilylation, and mesylation from 2d and 2e, respectively (see ESI† for detailed procedures). The prepared precursors (0.051 mmol) and NaH (60% dispersion in mineral oil, 4.1 mg, 0.102 mmol) were dissolved in dry DMF (1.0 mL, 0.05 M) under an argon atmosphere with stirring at room temperature. After completion of the reaction (checked by TLC), the resulting mixture was quenched with deionized water and saturated aq. NaHCO3, and the organic material was extracted three times with EA and three times with DCM. The combined organic extracts were dried over anhydrous Na2SO4(s) and filtered. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel flash column chromatography to obtain desired compounds 4a–4b in moderate to good yields.
General synthetic procedure for scaffold IV (5a–5b)
To a solution of 1f or 1g (0.15 mmol) in ethanol (3.0 mL, 0.05 M), NaOEt (51.0 mg, 0.75 mmol) was added at room temperature. The resulting mixture was stirred at 60 °C. After the completion of reaction checked by TLC, the reaction mixture was concentrated under reduced pressure, quenched with deionized water and saturated aq. NaCl, and the organic material was extracted four times with EA. The combined organic extracts were dried over anhydrous Na2SO4(s) and filtered. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel flash column chromatography to obtain desired compounds 5a and 5b in moderate yields.
General synthetic procedure for scaffold V (6a–6b) and scaffold III (7a–7b)
To a solution of 1b or 1h (0.23 mmol) in tert-butanol (t-BuOH, 4.6 mL, 0.05 M), potassium tert-butoxide (t-BuOK, 127.2 mg, 1.13 mmol) was added at room temperature. The resulting mixture was stirred at 60 °C. After the standard work-up procedure, the resulting residue was purified by silica gel flash column chromatography to obtain desired compounds 6a–6b as major products and 7a–7b as minor products.
General synthetic procedure for scaffold VI (8a–8b)
To a solution of 6a (0.29 mmol) in acetonitrile (5.8 mL, 0.05 M), 2-bromoacetophenone (for 8a, 63.2 mg, 0.32 mmol) or 2-bromo-4′-chloroacetophenone (for 8b, 74.2 mg, 0.32 mmol) was added at room temperature. The resulting mixture was stirred at 80 °C. After complete consumption of the starting material (checked by TLC), the reaction mixture was concentrated under reduced pressure. The residue was dissolved in CHCl3 (11.6 mL, 0.025 M) and basic alumina (Brockmann activity I, pH 9.5 ± 0.5, 400 wt%) was added at room temperature. The resulting mixture was stirred at 60 °C. When TLC indicated completion of the reaction, the reaction mixture was filtered through Celite® while washing with DCM, EA, and MeOH. The filtrate was concentrated under reduced pressure, followed by silica gel flash column chromatography to obtain desired compounds 8a–8b in moderate yields.
General synthetic procedure for scaffold VII (10a–10b)
To a solution of 1d or 1e (0.22 mmol) in tetrahydrofuran (THF, 2.2 mL, 0.1 M), TBAF (1.0 M in THF, 0.33 mL, 0.33 mmol) was added at room temperature. The resulting mixture was stirred at 60 °C. After the standard work-up procedure, the resulting residue was purified by silica gel flash column chromatography to obtain desired compounds 10a and 10b in moderate to good yields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Creative Research Initiative Grant (NRF-2014R1A3A2030423) and the Bio & Medical Technology Development Program (2012M3A9C4048780) through the National Research Foundation of Korea (NRF) funded by the Korean Government (Ministry of Science and ICT). We acknowledge Ms Jaeyoung Koo for her support in compound crystallization. Y. C. and H. K. are grateful for the BK21 Fellowship Program.
Notes and references
- S. Dandapani and L. A. Marcaurelle, Nat. Chem. Biol., 2010, 6, 861–863 CrossRef CAS PubMed.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS PubMed.
- C. J. O'Connor, L. Laraia and D. R. Spring, Chem. Soc. Rev., 2011, 40, 4332–4345 RSC.
- S. L. Schreiber, Nature, 2009, 457, 153–154 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed.
- C. J. O'Connor, H. S. G. Beckmann and D. R. Spring, Chem. Soc. Rev., 2012, 41, 4444–4456 RSC.
-
(a) H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef CAS PubMed;
(b) R. J. Rafferty, R. W. Hicklin, K. A. Maloof and P. J. Hergenrother, Angew. Chem., Int. Ed., 2014, 53, 220–224 CrossRef CAS PubMed;
(c) J. Zhang, J. Wu, B. Hong, W. Ai, X. Wang, H. Li and X. Lei, Nat. Commun., 2014, 5, 4614 CrossRef CAS PubMed;
(d) M. Garcia-Castro, L. Kremer, C. D. Reinkemeier, C. Unkelbach, C. Strohmann, S. Ziegler, C. Ostermann and K. Kumar, Nat. Commun., 2015, 6, 6516 CrossRef CAS PubMed;
(e) F. Nie, D. L. Kunciw, D. Wilcke, J. E. Stokes, W. R. J. D. Galloway, S. Bartlett, H. F. Sore and D. R. Spring, Angew. Chem., Int. Ed., 2016, 55, 11139–11143 CrossRef CAS PubMed;
(f) Y.-C. Lee, S. Patil, C. Golz, C. Strohmann, S. Ziegler, K. Kumar and H. Waldmann, Nat. Commun., 2017, 8, 14043 CrossRef PubMed.
- S. Oh and S. B. Park, Chem. Commun., 2011, 47, 12754–12761 RSC.
- J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629–14638 CrossRef CAS PubMed.
- S. Lee, Y. Nam, J. Y. Koo, D. Lim, J. Park, J. Ock, J. Kim, K. Suk and S. B. Park, Nat. Chem. Biol., 2014, 10, 1055–1060 CrossRef CAS PubMed.
- T.-J. Cho, J. Kim, S.-K. Kwon, K. Oh, J.-a. Lee, D.-S. Lee, J. Cho and S. B. Park, Chem. Sci., 2012, 3, 3071–3075 RSC.
- D. Lim, W. G. Byun, J. Y. Koo, H. Park and S. B. Park, J. Am. Chem. Soc., 2016, 138, 13630–13638 CrossRef CAS PubMed.
-
(a) H. R. Lawrence, M. P. Martin, Y. Luo, R. Pireddu, H. Yang, H. Gevariya, S. Ozcan, J.-Y. Zhu, R. Kendig, M. Rodriguez, R. Elias, J. Q. Cheng, S. M. Sebti, E. Schonbrunn and N. J. Lawrence, J. Med. Chem., 2012, 55, 7392–7416 CrossRef CAS PubMed;
(b) A. E. Wakeling, S. P. Guy, J. R. Woodburn, S. E. Ashton, B. J. Curry, A. J. Barker and K. H. Gibson, Cancer Res., 2002, 62, 5749–5754 CAS;
(c) V. Yaziji, D. Rodríguez, H. Gutiérrez-de-Terán, A. Coelho, O. Caamaño, X. García-Mera, J. Brea, M. I. Loza, M. I. Cadavid and E. Sotelo, J. Med. Chem., 2011, 54, 457–471 CrossRef CAS PubMed.
- H. Kim, T. T. Tung and S. B. Park, Org. Lett., 2013, 15, 5814–5817 CrossRef CAS PubMed.
- J. Kim, J. Jung, J. Koo, W. Cho, W. S. Lee, C. Kim, W. Park and S. B. Park, Nat. Commun., 2016, 7, 13196 CrossRef CAS PubMed.
- Y. Choi, H. Kim, Y.-H. Shin and S. B. Park, Chem. Commun., 2015, 51, 13040–13043 RSC.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- Selected examples:
(a) P. S. Wharton and M. D. Baird, J. Org. Chem., 1971, 36, 2932–2937 CrossRef CAS;
(b) M. C. Honan, A. Balasuryia and T. M. Cresp, J. Org. Chem., 1985, 50, 4326–4329 CrossRef CAS;
(c) P. Dowd and S.-C. Choi, J. Am. Chem. Soc., 1987, 109, 6548–6549 CrossRef CAS;
(d) H. Suginome, T. Kondoh, C. Gogonea, V. Singh, H. Goto and E. Osawa, J. Chem. Soc., Perkin Trans. 1, 1995, 69–81 RSC;
(e) I. J. Borowitz, G. Gonis, R. Kelsey, R. Rapp and G. J. Williams, J. Org. Chem., 1966, 31, 3032–3037 CrossRef CAS PubMed;
(f) I. J. Borowitz and R. D. Rapp, J. Org. Chem., 1968, 34, 1370–1373 CrossRef.
- F. Kopp, C. F. Stratton, L. B. Akella and D. S. Tan, Nat. Chem. Biol., 2012, 8, 358–364 CrossRef CAS PubMed.
- L. Li, Z.-L. Li, F.-L. Wang, Z. Guo, Y.-F. Cheng, N. Wang, X.-W. Dong, C. Fang, J. Liu, C. Hou, B. Tan and X.-Y. Liu, Nat. Commun., 2016, 7, 13852 CrossRef PubMed.
- R. A. Bauer, T. A. Wenderski and D. S. Tan, Nat. Chem. Biol., 2013, 9, 21–29 CrossRef CAS PubMed.
- H. H. Wasserman and H. Masuyama, J. Am. Chem. Soc., 1981, 103, 461–462 CrossRef CAS.
- H. Stetter and H. Spangenberger, Chem. Ber., 1958, 91, 1982–1988 CrossRef CAS.
- P. Aeberli and W. J. Houlihan, J. Org. Chem., 1969, 34, 2715–2720 CrossRef CAS.
- A. Nakamura and S. Kamiya, Chem. Pharm. Bull., 1972, 20, 69–75 CrossRef CAS.
- P. Aeberli and W. J. Houlihan, J. Org. Chem., 1969, 34, 2720–2723 CrossRef CAS.
- C. Michon, A. Sharma, G. Bernardinelli, E. Francotte and J. Lacour, Chem. Commun., 2010, 46, 2206–2208 RSC.
- A. Hussain, S. K. Yousuf and D. Mukherjee, RSC Adv., 2014, 4, 43241–43257 RSC.
- D. J. Rogers and T. T. Tanimoto, Science, 1960, 132, 1115–1118 CrossRef CAS PubMed.
- R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195–202 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1866157 and 1866158. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04061c |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Heejun
Kim‡
,
Heejun
Kim‡