
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flux melting of metal–organic frameworks†
Louis
Longley
a,
Sean M.
Collins
 a,
Shichun
Li
ab,
Glen J.
Smales
cd,
Ilknur
Erucar
e,
Ang
Qiao
f,
Jingwei
Hou
a,
Cara M.
Doherty,
g,
Aaron W.
Thornton
g,
Anita J.
Hill
g,
Xiao
Yu
h,
Nicholas J.
Terrill
d,
Andrew J.
Smith
d,
Seth M.
Cohen
h,
Paul A.
Midgley
a,
David A.
Keen
i,
Shane G.
Telfer
j and
Thomas D.
Bennett
*a
a,
Shichun
Li
ab,
Glen J.
Smales
cd,
Ilknur
Erucar
e,
Ang
Qiao
f,
Jingwei
Hou
a,
Cara M.
Doherty,
g,
Aaron W.
Thornton
g,
Anita J.
Hill
g,
Xiao
Yu
h,
Nicholas J.
Terrill
d,
Andrew J.
Smith
d,
Seth M.
Cohen
h,
Paul A.
Midgley
a,
David A.
Keen
i,
Shane G.
Telfer
j and
Thomas D.
Bennett
*a
aDepartment of Materials Science and Metallurgy, University of Cambridge, Charles Babbage Road, Cambridge, CB3 0FS, UK. E-mail: tdb35@cam.ac.uk
bInstitute of Chemical Materials, China Academy of Engineering Physics, Mianyang 621900, China
cDepartment of Chemistry, University College London, Gordon Street, London, WC1H 0AJ, UK
dDiamond Light Source Ltd, Diamond House, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK
eDepartment of Natural and Mathematical Sciences, Faculty of Engineering, Ozyegin University, Istanbul, Turkey
fState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, China
gFuture Industries, Commonwealth Scientific and Industrial Research Organisation, Clayton South, Victoria 3168, Australia
hDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92023-0358, USA
iISIS Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxon OX11 0QX, UK
jMacDiarmid Institute for Advanced Materials and Nanotechnology, Institute of Fundamental Sciences, Massey University, Palmerston North 4442, New Zealand
First published on 12th February 2019
Abstract
Recent demonstrations of melting in the metal–organic framework (MOF) family have created interest in the interfacial domain between inorganic glasses and amorphous organic polymers. The chemical and physical behaviour of porous hybrid liquids and glasses is of particular interest, though opportunities are limited by the inaccessible melting temperatures of many MOFs. Here, we show that the processing technique of flux melting, ‘borrowed’ from the inorganic domain, may be applied in order to melt ZIF-8, a material which does not possess an accessible liquid state in the pure form. Effectively, we employ the high-temperature liquid state of one MOF as a solvent for a secondary, non-melting MOF component. Differential scanning calorimetry, small- and wide-angle X-ray scattering, electron microscopy and X-ray total scattering techniques are used to show the flux melting of the crystalline component within the liquid. Gas adsorption and positron annihilation lifetime spectroscopy measurements show that this results in enhanced, accessible porosity to a range of guest molecules in the resultant flux melted MOF glass.
Introduction
Porous three dimensional materials formed by the self-assembly of inorganic nodes connected by organic ligands or, as they are commonly known, metal–organic frameworks (MOFs),1 remain of extreme interest to the scientific community. The continuation of new materials discovery, combined with an improved understanding of the relationship between structure, property and application, drives intense research into their use in carbon capture, clean water production, catalysis, drug delivery, and light harvesting.2,3 Their thermomechanical properties have also generated a surge of recent studies, revolving around flexibility,4 negative gas adsorption,5 and defect-dependent properties.6 Rapid developments have also been made in asserting control over macroscale MOF architectures,7 such as membranes,8 monoliths,9 and thin films.10 Although many MOFs can be processed into pellet forms, their mechanical instabilities are not conducive to processing of their nano-crystalline structures into bulk structures which are free from grain boundaries.11 Set against this backdrop, there remains a necessity for MOFs in macroscale architectures that retain porous properties, but circumvent the drawbacks associated with processing and handling microcrystalline powders.Zeolitic imidazolate frameworks (ZIFs) are a subset of MOFs in which tetrahedral metal centers are connected by imidazolate (Im, C3H3N2−) based ligands.12,13 Amongst these, the prototypical framework ZIF-8, Zn(mIm)2 (mIm, 2-methylimidazolate, C4H5N2−), is investigated extensively.14,15 For example, the structure, which contains pores of diameter 11.6 Å, has been shown to exhibit selectivity for the removal of Li+ ions from water, courtesy of the 3.4 Å limiting pore window size.16 The formation of mixed-matrix membranes (MMMs) through dispersal of ZIF-8 within an organic polymer has also been attempted,17 though products may suffer from poor adhesion between the two components. Cross-linking of the ZIF to the organic matrix, through either amine surface functionalization of the ZIF, or from high-temperature heat treatment of the MMM, have been investigated as potential solutions to the problem of chemical compatibility between the two components.18,19
Recently, several members of the ZIF family have been observed to melt upon heating to temperatures above 673 K.20 Cooling the liquid ZIF below these temperatures potentially allows the ZIF to be shaped and handled akin to a conventional silicate glass. However, the temperature window over which these materials remain intact in their liquid state is bounded by the temperature of thermal decomposition (Td),21 which is up to ca. 100 K higher than the melting temperature (Tm). The [Zn(Im)2] glasses produced upon cooling from the liquid state possess continuous random networks, mimicking that of amorphous SiO2. The dominant Zn–N coordination bonding in the glass state means that they form a new, 4th category of melt-quenched glasses, distinct from the inorganic (non-metallic), organic and metallic glass categories known at present.22 The melting behaviour of ZIFs, alongside that of phosphate-based porous coordination polymers,23,24 therefore opens up unexplored avenues in the synthesis and processing of new MOF-based glasses.25
The melting process in metal-imidazolate and -phosphate coordination polymer/metal–organic framework families has been observed to obey Lindemann's law,20,24,26 in which the ratio of the mean thermal atomic displacement of a species, and the distance to the nearest neighbour, approaches 0.1–0.13 at the melting temperature. A microscopic structural view of ZIF melting, obtained by molecular dynamics simulation, shows that Zn–Im bond breaking is a rare event. This rare event is followed by movement of the Im ligand away from the now under coordinated Zn2+ center, before association of a different imidazolate. This melting process, which has been likened to hydrogen bond switching in water,20 has only been observed in ZIFs containing the Im species. Other materials, including ZIF-8, do not melt27 and this places severe constrictions on the chemical and network functionality of the resultant glasses. Pathways are therefore being sought to reduce the Tm of non-melting ZIF structures below Td.
In the molten salt domain, the problem of reducing Tm is approached through use of a flux. For example, Na2O (Tm ≈ 1400 K) is used to lower the melting temperature of SiO2 (Tm ≈ 2000 K),28 whilst in addition molten oxide fluxes enable production of bulk metallic glasses.29 Organic analogies also exist, in the use of ionic liquids as solvents for secondary species.30,31 Encouraged by the similarities between inorganic glasses and those formed by melting ZIFs, we hypothesized that the high-temperature liquid state of a ZIF may serve as a flux – that is, a solvent – for other ZIFs. This strategy is successfully used to form a glass, derived from a high-temperature liquid of ZIF-62 [Zn(C3H3N2)1.75(C7H5N2)0.25] (bIm = benzimidazolate, C7H5N2−) and ZIF-8. The resultant flux melted glass displays increased porosity towards H2, compared with the pure MOF-glass.
Experimental
Synthesis
The synthesis of ZIF-62 was taken from Gustafsson et al.32 Specifically, solutions in DMF of Zn(NO3)2·6H2O (0.2 M), imidazole (1.5 M) and benzimidazole (0.2 M) were prepared, and mixed together in the ratio Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Im:bIm of 1:13.5:1.5. Solutions were heated at 403 K for 96 hours and cooled to ambient temperature. The microcrystalline product was washed three times in DMF, and dried at 373 K for 4 hours. ZIF-8 was purchased from Sigma Aldrich and used as received. A reported, steam-assisted synthesis was used for ZIF-67.33
:Im:bIm of 1:13.5:1.5. Solutions were heated at 403 K for 96 hours and cooled to ambient temperature. The microcrystalline product was washed three times in DMF, and dried at 373 K for 4 hours. ZIF-8 was purchased from Sigma Aldrich and used as received. A reported, steam-assisted synthesis was used for ZIF-67.33
The preparation of mixed samples was carried out in 0.5 g quantities. For example, 0.1 and 0.4 g of ZIF-8 and ZIF-62 were placed in a 10 mL stainless steel jar, along with 2 × 7 mm stainless steel balls. The mixture was then milled for 5 minutes (for Zn based samples) or 15 minutes (for Co based samples) in a Retsch MM400 grinder mill operating at 25 Hz (Fig. S1†). The different milling times were to accommodate the different initial particle sizes of ZIF-8, and ZIF-67 (Fig. S2†), given the larger initial particle sizes of the as-synthesized ZIF-67 phase. These crystalline mixtures were subsequently heated to 453 K for 3 hours to remove the solvent.
To form the glasses, 0.25 g of the evacuated crystalline mixture was placed in a ceramic crucible in a tube furnace, which was sealed and flushed with argon for 30 minutes. ZIF-8/ZIF-62 and ZIF-67/ZIF-62 mixtures were then heated at 10 K min−1 to the temperatures indicated in the main text. This was followed by natural cooling back to room temperature, still under flowing argon.
Characterization
Combined small and wide angle X-ray scattering data were collected at the I22 beamline at the Diamond Light Source, UK (λ = 0.9998 Å, 12.401 keV). The SAXS detector was positioned at a distance of 9.23634 m from the sample as calibrated using a 100 nm period Si3N4 grating (Silson, UK), giving a usable q range of 0.0018–0.18 Å−1. The WAXS detector was positioned at a distance of 0.16474 m from the sample as calibrated using a standard CeO2 sample (NIST SRM 674b, Gaithersburg USA), giving a usable q range of 0.17–4.9 Å−1. Samples were loaded into 1.5 mm diameter borosilicate capillaries under argon inside a glovebox and sealed with Blu-tack and Para-film to prevent the ingress of air. Samples were heated using a Linkam THMS600 capillary stage (Linkam Scientific, UK) from room temperature to 873 K at 10 K min−1. Simultaneous SAXS/WAXS data were collected every 1 K. Data were reduced to 1D using the DAWN package34,35 and standard reduction pipelines.36 Values for the power law behavior of the samples were found using the power law model of SASView 4.1.1.37 Data were fitted over the range 0.003 ≤ q ≤ 0.005 Å−1. Particle size distributions were calculated using the McSAS package,38,39 a minimal assumption Monte Carlo method for extracting size distributions from small-angle scattering data. Data were fitted over a range of 0.002 ≤ q ≤ 0.18 Å−1 with a sphere model.
X-ray total scattering data were collected at room temperature using a PANalytical Ag-source Empyrean lab diffractometer (λ = 0.561 Å). Data collection was carried out using loaded 1.0 mm diameter quartz capillaries and collection times of approximately 6 h. Background, multiple scattering, container scattering, Compton scattering and absorption corrections were performed using the GudrunX program.40,41
Results and discussion
Thermal characterization of flux melting
Selection of the two components was based upon the requirement for an accessible and reasonably wide temperature region over which the liquid, and crystalline MOFs, were both stable. That is, the two components should obey the condition Tm1 < Td2, where Tm1 refers to the melting temperature of structure 1, the liquid-forming MOF, and Td2 to the decomposition temperature, or upper stability limit, of the crystalline form of component 2. A suitable combination was found (Fig. 1a) using: (i) the comparatively low Tm of ca. 710 K established for ZIF-62 [Zn(C3H3N2)1.75(C7H5N2)0.25],46 and (ii) the relatively high Td of ZIF-8 (ca. 800 K at a heating rate of 10 K min−1).21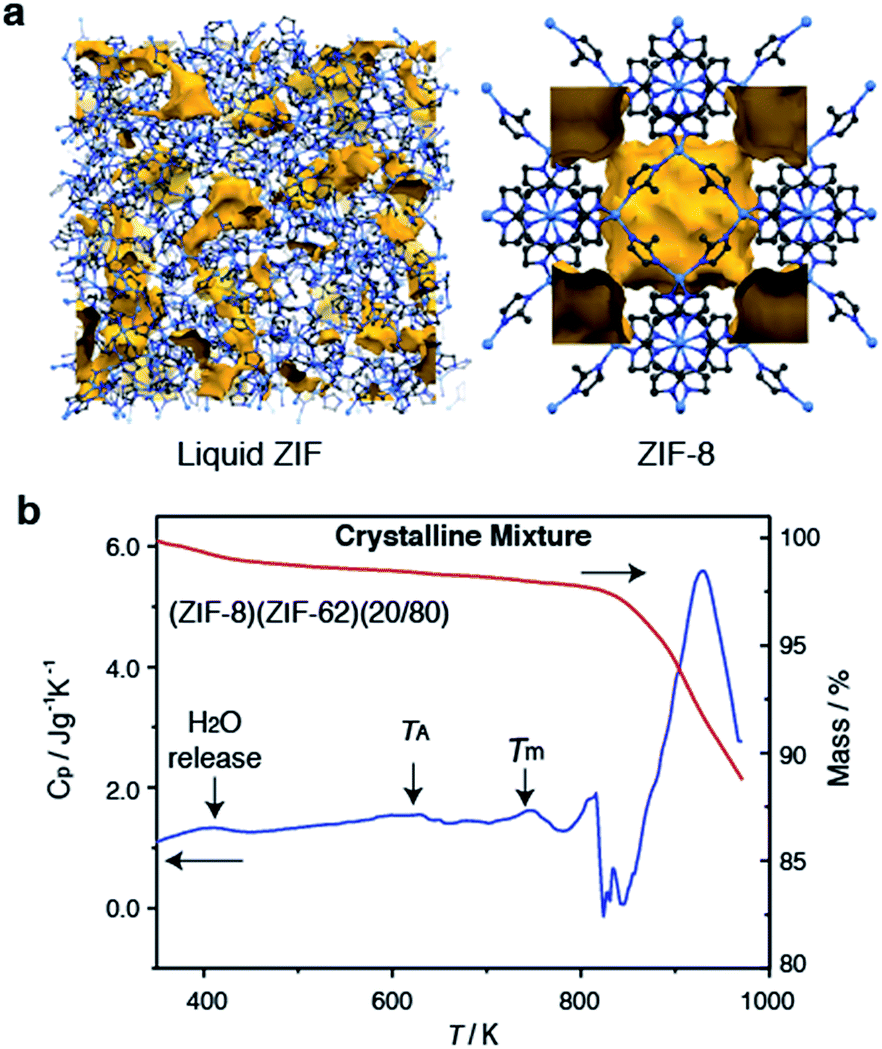 | ||
| Fig. 1 (a) Atomic configuration, ca. 50 Å3, of a high-temperature liquid ZIF obtained from a previous publication through computational, and experimental neutron and synchrotron pair distribution function modelling.20 Also included is the unit cell of ZIF-8.12 Zn – light blue, N – dark blue, C – grey, H – omitted for clarity. Void space – yellow. (b) Isobaric heat capacity (Cp) and mass as a function of temperature for (ZIF-8)(ZIF-62)(20/80), at a heating rate of 10 K min−1. | ||
ZIF-62 was synthesized according to literature procedures, and combined with a sample of commercially purchased ZIF-8. Specifically, the two microcrystalline samples were mixed together in a 20/80 (ZIF-8/ZIF-62) wt/wt ratio, and ball-milled for 5 minutes to homogenize the sample. The sample was then evacuated by heating at 453 K for 3 hours. This evacuation of solvent did not result in a change in crystal structure (Fig. S1 and S2†). The resultant mix of crystalline frameworks is hereby referred to as (ZIF-8)(ZIF-62)(20/80). Differential scanning calorimetry (DSC) experiments were performed up to 973 K in an inert argon atmosphere (Fig. 1b). A broad endotherm at ca. 600 K indicative of thermal amorphisation, followed by an endothermic melting peak at ca. 730 K was observed, broadly consistent with prior observations.46
In a separate, simultaneous differential scanning calorimetry-thermogravimetric (SDT) experiment, (ZIF-8)(ZIF-62)(20/80) was heated to 773 K at a rate of 10 K min−1, i.e. above the melting temperature of ZIF-62, and then quenched at a rate of 10 K min−1 back to room temperature. This produced a solid, self-supporting monolith, hereby referred to as ag[(ZIF-8)0.2(ZIF-62)0.8], of strikingly different external appearance to the microcrystalline mixture prior to heating (Fig. 2a inset). This terminology differentiates the flux melted glasses, from metal–organic framework blends, e.g. (ZIF-4)0.5(ZIF-62)0.5,47 in which both constituent amorphous MOF component structures appear to remain intact. Scanning electron microscopy (Fig. 2b and S2†) demonstrated that the individual particles coalesce upon their transformation into ag[(ZIF-8)0.2(ZIF-62)0.8], with no distinct, remnant particles from either ZIF-8 or ZIF-62 observable in this material. Consistent with these observations, the powder X-ray diffraction (PXRD) pattern of ag[(ZIF-8)0.2(ZIF-62)0.8] contained no Bragg scattering (Fig. 2a). A sample of pure ZIF-8 was also ball-milled for 5 minutes and heated to 773 K, then subsequently cooled to room temperature (Fig. S3†). Crystallinity was shown to remain intact.
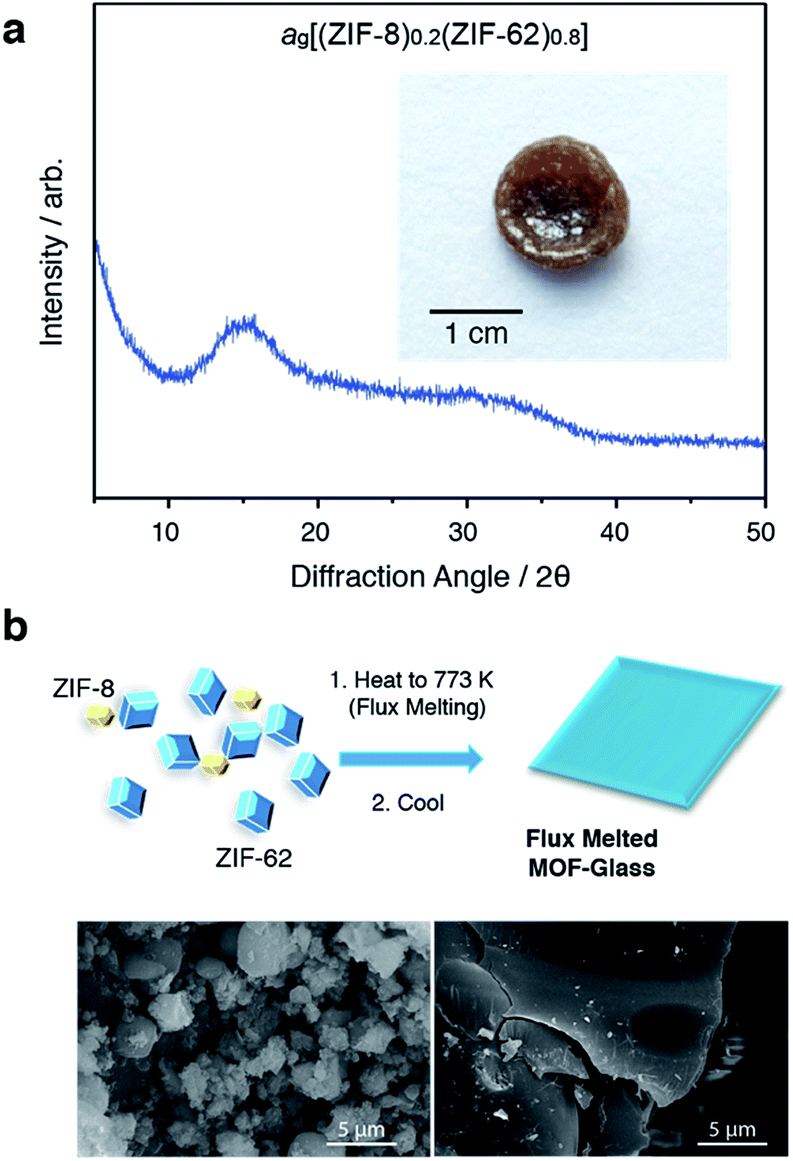 | ||
| Fig. 2 (a) Powder X-ray diffraction pattern of the glass formed after quenching from 773 K, and (inset) optical image. (b) Schematic of flux melted glass formation and SEM images of (left) (ZIF-8)(ZIF-62)(20/80) and (right) ag[(ZIF-8)0.2(ZIF-62)0.8]. | ||
The glassy nature of ag[(ZIF-8)0.2(ZIF-62)0.8] was confirmed by a second DSC heating curve (Fig. S4†), which demonstrated a glass transition of Tg = 607 K. This value is greater than that for pure ZIF-62 (Tg = 591 K).46 Thermogravimetric analysis (TGA) on the sample indicated that no mass was lost on heating to ca. 850 K (Fig. S5†). 1H nuclear magnetic resonance (NMR) spectroscopy on digested samples of ag[(ZIF-8)0.2(ZIF-62)0.8] confirmed the presence of the Im, mIm, and bIm linkers (Fig. S6†).
Structural evolution during flux melting
To further investigate the structural changes upon melting, in situ wide-angle X-ray scattering (WAXS) data were collected on a sample of (ZIF-8)(ZIF-62)(20/80) at the Diamond Light Source. As in prior studies, where the technique has proven useful in elucidating the mechanisms of MOF particle growth in solution,48 the WAXS data were combined with simultaneously collected small-angle X-ray scattering (SAXS) data. Amorphization of ZIF-62, indicated by the disappearance of the (211) peak in the temperature resolved WAXS profile, takes place at ca. 600 K (Fig. 3a), consistent with previous observations and the DSC trace (Fig. 1).46 The remaining Bragg diffraction from ZIF-8, for example the (110) peak, then disappears by ca. 650 K.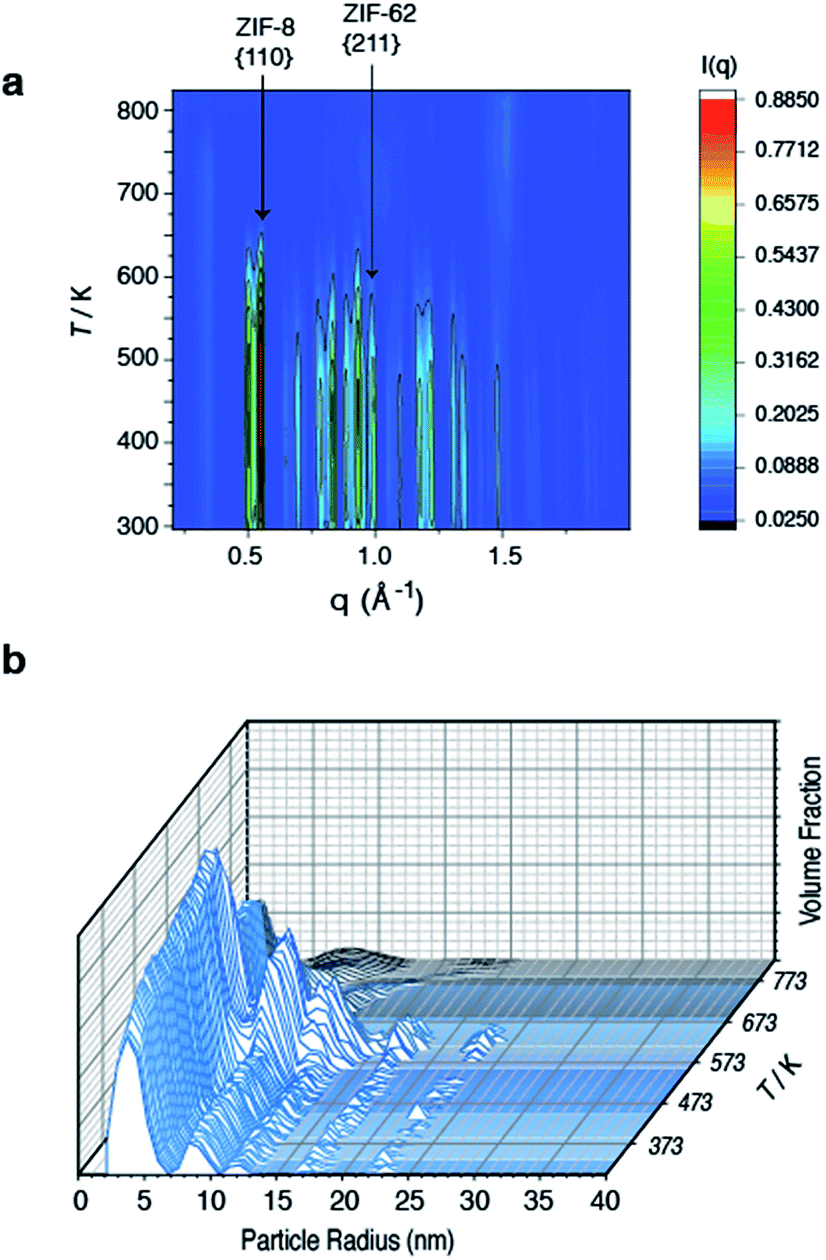 | ||
| Fig. 3 Temperature resolved diffraction of (a) WAXS profile of (ZIF-8)(ZIF-62)(20/80) and (b) volume fraction distributions of (ZIF-8)(ZIF-62)(20/80). | ||
Porod fitting of the variable temperature SAXS profile (Fig. S7 and S8†) at room temperature, reveals that the decay in SAXS signal follows power law behavior of the form q−α, where α = 3.65. This remains constant to ca. 573 K, before dropping to α = 3.55, and then starting to rapidly increase at ca. 650 K. At ca. 750 K, the signal reaches a maximum of α = 4.00 and starts to decrease rapidly. This data matches the DSC data presented (Fig. S4†), where a marked increase in heat flow to the sample starts at ca. 650 K, which then reaches a maximum at ca. 750 K.
In a previous experiment, Porod fitting of variable temperature SAXS data taken on a pure sample of ZIF-62 demonstrated α to reach a maximum of 3.9, at ca. 693 K, i.e. in the liquid state.47 The maximum value of α = 4.00 at 750 K thus indicates loss of the internal pore structure of ZIF-8 at this temperature. Calculation of the volume weighted fraction of particle sizes below the observable limit of 310 nm diameter indicates the gradual onset of particle coalescence at ca. 553 K (Fig. 3b). Scattering from the original particles above 5 nm in diameter ceases at temperatures approaching 673 K, though the population of 5 nm diameter particles continues and retains some independence.
Taken together, these data indicate that ZIF-62 amorphizes at ca. 600 K, before beginning to melt at ca. 650 K, i.e. the same temperature region in which the Bragg peaks from ZIF-8 disappear. Thus, the implication here is that the formation of the liquid phase of ZIF-62 is causal to the flux melting of ZIF-8. The apparent offset between Tm,WAXS and Tm,SAXS is therefore ascribed to amorphization before melting, which results in the disappearance of Bragg peaks (Fig. 3a). The downturn in the value of α (Fig. S8†) is almost identical in temperature to the maximum of the Tm peak in the DSC (Fig. S4†).
Flux melted glass characterization
Laboratory Ag-source total scattering experiments were carried out on crystalline ZIF-8, (ZIF-8)(ZIF-62)(20/80), and ag[(ZIF-8)0.2(ZIF-62)0.8] (Fig. 4a, b and S9†). The structure factor S(q) for (ZIF-8)(ZIF-62)(20/80) contained Bragg scattering, as expected for this crystalline mixture. On the other hand, consistent with its glassy nature, ag[(ZIF-8)0.2(ZIF-62)0.8] did not exhibit Bragg diffraction. This observation also indicated that no intact ZIF-8 crystallites remained after the melting of ZIF-62. The pair distribution functions, D(r)s, of both (ZIF-8)(ZIF-62)(20/80) and ag[(ZIF-8)0.2(ZIF-62)0.8], contain peaks at distances in the range 1.3–6 Å that are characteristic of ZIFs. The Zn–Zn correlation at ca. 6 Å in the PDFs of both (ZIF-8)(ZIF-62)(20/80) and ag[(ZIF-8)0.2(ZIF-62)0.8] corresponds well with a simple average of the Zn–Zn distances determined from the CIF files of ZIF-8 (6.007 Å) and ZIF-62 (5.913 Å),12,49 confirming that the short range order is maintained. The PDF of ag[(ZIF-8)0.2(ZIF-62)0.8] shows distinct peaks in the 6.5–8 Å region, evidencing some correlations in this region. These, through comparison with data collected previously, are ascribed to the ZIF-62 glass and not to remnant ZIF-8 crystallinity (Fig. 4b).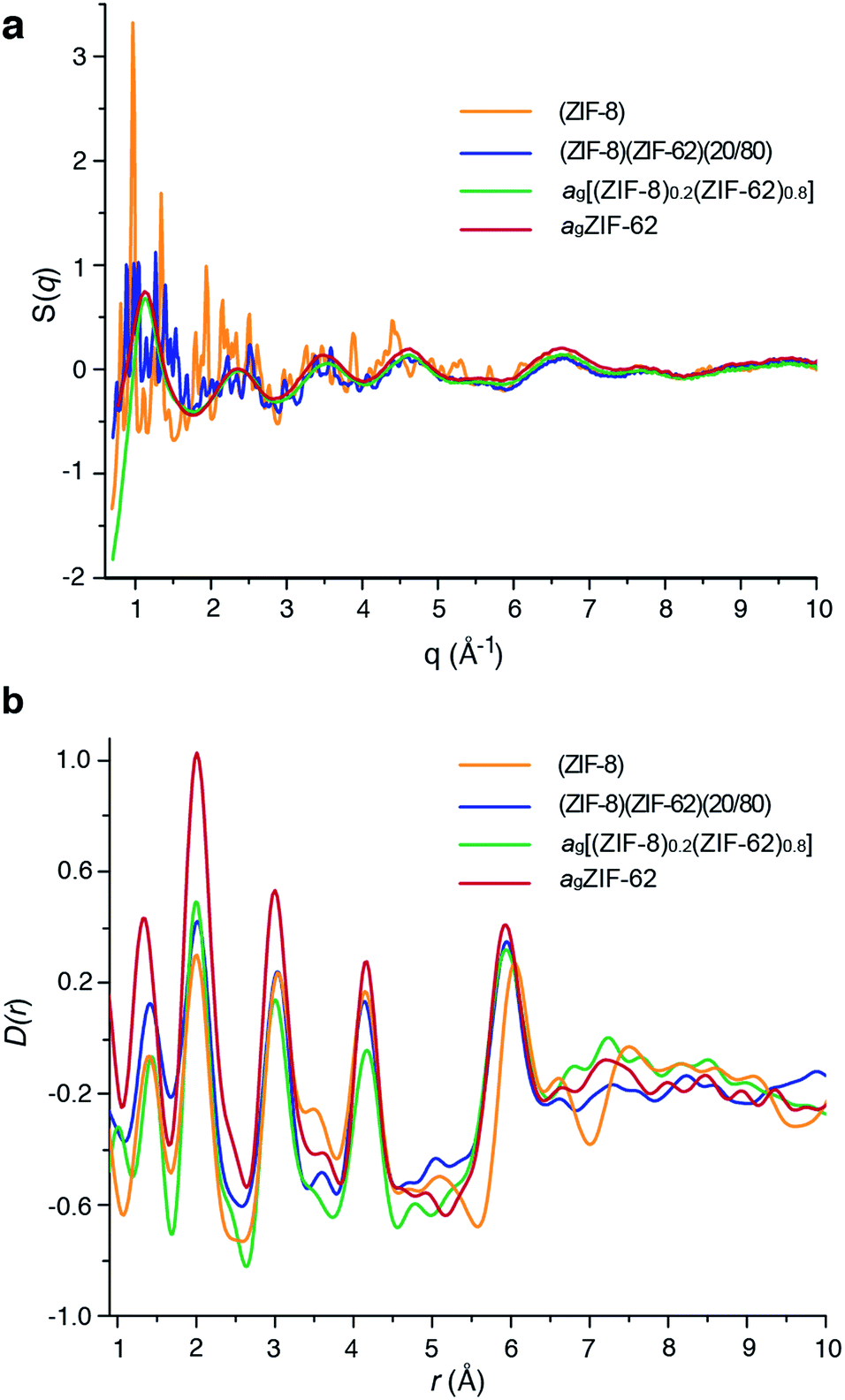 | ||
| Fig. 4 (a) Structure factors S(q) of (ZIF-8)(ZIF-62)(20/80), ag[(ZIF-8)0.2(ZIF-62)0.8] and ZIF-8, alongside that of agZIF-62 from a previous publication.46 (b) Corresponding X-ray pair distribution functions D(r). | ||
To provide chemical contrast between the two glass components, and enable the use of electron microscopy as a tool for analysing the fate of the ZIF-8 particles post-quenching, a second series of samples was prepared using ZIF-67. This framework is the isostructural cobalt(II) analogue of ZIF-8, i.e. [Co(mIm)2]. The Td of a pure sample of ZIF-67 was observed at ca. 780 K, which is below that of ZIF-8 but above the Tm of ZIF-62 (Fig. S10 and S11†).49 A sample of (ZIF-67)(ZIF-62)(20/80) was accordingly prepared by first synthesizing ZIF-67,33 and following the methodology used for the zinc based mixture. Specifically, 0.1 g of ZIF-67 was ball-milled for 15 minutes with 0.4 g ZIF-62. A sample of ag[(ZIF-67)0.2(ZIF-62)0.8] was then prepared by heating this mixture to 770 K in a tube furnace. Annular dark field (ADF) STEM, exhibiting thickness and atomic number contrast, and X-ray energy dispersive spectroscopy (EDS) were then used to provide chemical element maps in both the crystalline mixture and flux melted glass samples. In (ZIF-67)(ZIF-62)(20/80) (Fig. 5a), Zn, C, and N are observed in one set of particles, while Co, C and N are seen in a different, segregated set of particles.
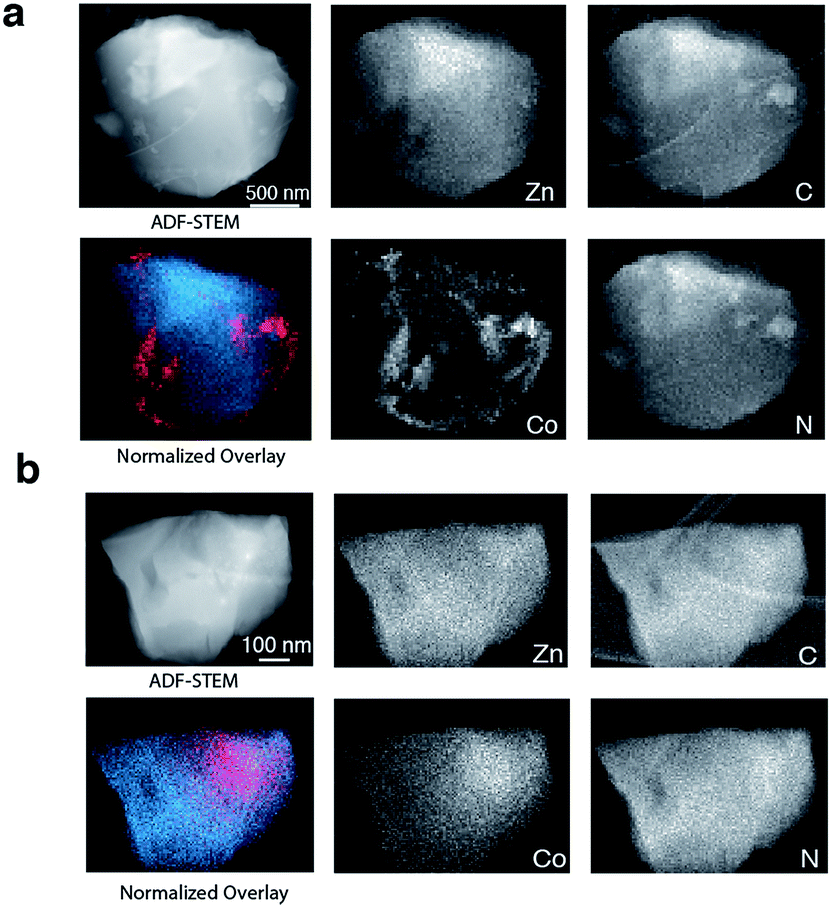 | ||
| Fig. 5 (a) ADF STEM image, EDS elemental maps for C, N, Zn and Co signals, and Zn (blue) and Co (red) component map overlay of (ZIF-67)(ZIF-62)(20/80), (b) ADF STEM image of a shard of ag[(ZIF-67)0.2(ZIF-62)0.8] and corresponding C, N, Zn and Co EDS elemental maps, and an overlay map of Zn (blue) and Co (red) components. | ||
Investigation of a shard of the flux melted glass, ag[(ZIF-67)0.2(ZIF-62)0.8], indicated a much more homogeneous distribution of Zn and Co (Fig. 5b). The interfaces between the two components in ag[(ZIF-67)0.2(ZIF-62)0.8] were also found to be much more diffuse than in (ZIF-67)(ZIF-62)(20/80), or in samples of (ZIF-67)(ZIF-62)(20/80) heated to ca. 100 K and ca. 150 K below Tm (Fig. S12–S14†). This was found to be the case across multiple particles, with elemental mapping showing similar sharp interfaces in the crystalline mixture and diffuse interfaces in the glass (Fig S15 and S16†). These maps show a two-dimensional representation of a three-dimensional interface, and therefore unambiguous analysis of individual interfaces is limited by signals arising from variation in the thickness of the particle or chemical domains within it and by uncertainty in the orientation of the interface relative to the electron beam. Here, particularly in the Co maps (Fig. 5, S15 and S16†), the preponderance of smooth interfaces observed in ag[(ZIF-67)0.2(ZIF-62)0.8] contrasts vividly with the prevalence of abrupt interfaces observed in (ZIF-67)(ZIF-62)(20/80).
The gradual variation of Zn and Co in ag[(ZIF-67)0.2(ZIF-62)0.8] suggests that zinc(II) and cobalt(II) are able to diffuse across significant distances in the flux-mediated melt.
Flux melting and porosity
The permanent porosity of a pure sample of agZIF-62 was recently characterized (Table 1),50 with H2 (at 77 K) and CO2 (at 273 K) uptakes of 9 mL STP per g and 20 mL STP per g recorded at a pressure of 1 bar. These are lower than for the crystalline ZIF-62 framework (130 mL STP per g and 39 mL STP per g respectively). The uptake of H2 and CO2 by agZIF-62 is also lower than for agZIF-76-mbIm, which displays corresponding H2 and CO2 uptakes of ca. 45 mL STP per g and 35 mL STP per g.51 Experiments performed here also demonstrated that ZIF-62 and agZIF-62 display adsorption behaviour toward CH4 at 273 K (Fig. S17†).| Gas (kinetic diameter/Å), temperature/K | H2 (2.9), 77 | CO2 (3.3), 273 | O2 (3.46), 273 | N2 (3.64), 77 | CH4 (3.76), 273 |
|---|---|---|---|---|---|
| a Denotes data taken from ref. 50. | |||||
| ZIF-62 | 130a | 39a | 0a | 0a | 27 |
| Simulated | 130.5 | 33.9 | 5.2 | 174.2 | 18.6 |
| agZIF-62 | 9.3a | 20.1a | 1.5a | 0a | 3.9 |
| Simulated | 40.5 | 7.8 | 0.7 | 45.9 | 1.4 |
| (ZIF-8)(ZIF-62)(20/80) | 104.5 | 30.7 | 4.4 | 104.3 | 15.2 |
| Simulated | 119.9 | 29.5 | 4.4 | 207.2 | 16.1 |
| ag[(ZIF-8)0.2(ZIF-62)0.8] | 27.8 | 18.7 | 1.6 | 1.1 | 4.7 |
| Simulated | 48.5 | 9.1 | 1.3 | 105.9 | 2.7 |
Our observations indicate that the gas adsorption properties of (ZIF-8)(ZIF-62)(20/80) approximate a weighted average of its two components. The N2 adsorption isotherm of (ZIF-8)(ZIF-62)(20/80) at 77 K displays type I nitrogen behaviour, from which an accessible surface area of 350 m2 g−1 was calculated using the BET model. As the adsorbate pressure approached 1 bar, the N2 uptake at 77 K plateaus around 90 mL STP per g (Fig. 6a). This is consistent with the reported experimental value for N2 uptake in ZIF-8 of ca. 400 mL STP per g,12 and our own measurements on the material (Fig. S18†). It is in broad agreement with the 20% quantity of ZIF-8 in (ZIF-8)(ZIF-62)(20/80).
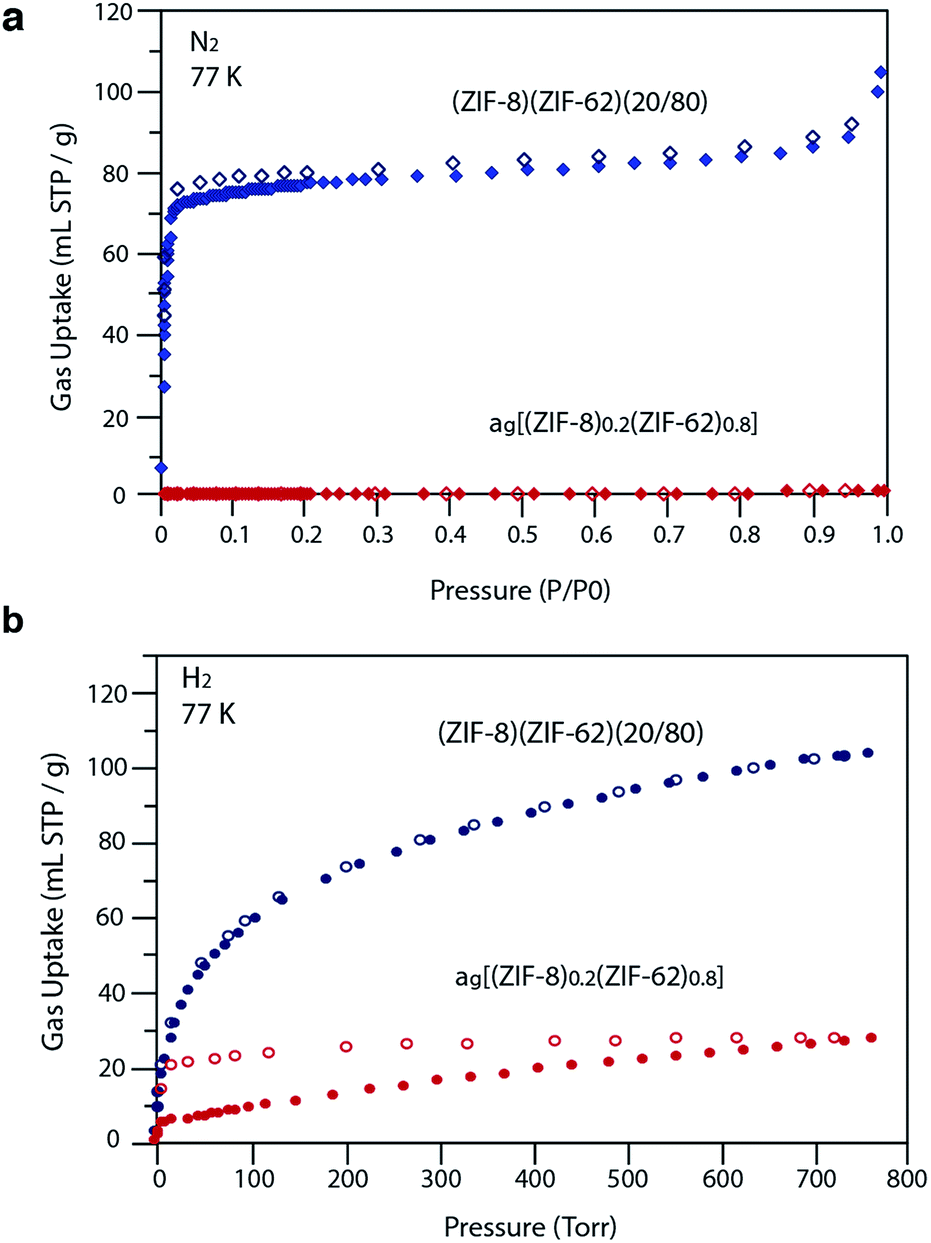 | ||
| Fig. 6 (a) N2 at 77 K and (b) H2 at 77 K gas isotherms for the zinc-based crystalline mixtures and glasses. Closed symbols represent adsorption isotherms and open symbols represent desorption isotherms. | ||
For H2 at 77 K, (ZIF-8)(ZIF-62)(20/80) takes up 105 mL STP per g which is slightly lower than both pure ZIF-62 (130 mL STP per g) and ZIF-8 (145 mL STP per g)12 (Fig. 6b, Table 1). This indicates that the ball milling process used to produce (ZIF-8)(ZIF-62)(20/80) may close off a number of small pores, which are accessible to H2 but not larger molecules such as N2. (ZIF-8)(ZIF-62)(20/80) reversibly adsorbs CO2 at 273 K (Fig. 7a). An uptake capacity of 31 mL STP per g was observed at a pressure of 1 bar, which equates to 5.7 wt%. This uptake is only slightly lower than that of ZIF-62 (39 mL STP per g),50 and ZIF-8 (53 mL STP per g).52
 | ||
| Fig. 7 (a) CO2 isotherm at 273 K and (b) O2 and CH4 at 273 K isotherms for zinc based samples. Closed symbols represent adsorption isotherms and open symbols represent desorption isotherms. | ||
A considerable degree of accessible porosity was found for ag[(ZIF-8)0.2(ZIF-62)0.8], i.e. upon flux melting and quenching of (ZIF-8)(ZIF-62)(20/80). This glass took up 19 mL STP per g of CO2 at 1 bar (Fig. 7a), which equates to 3.6 wt%. The desorption isotherm mapped back exactly on the adsorption isotherm, indicating the absence of significant barriers to gas diffusion. Further adsorption experiments using O2 and CH4 were also performed, which indicate that ag[(ZIF-8)0.2(ZIF-62)0.8] is permanently accessible to these guest molecules (Fig. 7b). The observed porosity of (ZIF-8)(ZIF-62)(20/80) towards N2 at 77 K is virtually eliminated by the vitrification process, however (Fig. 6a). The pore network of the ZIF-8 component, which is accessible in (ZIF-8)(ZIF-62)(20/80), appears to collapse in ag[(ZIF-8)0.2(ZIF-62)0.8] and places kinetic barriers to diffusion at low temperatures. This is also evident for adsorbate molecules as small as H2 (Fig. 6b). Here, the small amount of H2 uptake into ag[(ZIF-8)0.2(ZIF-62)0.8] confirms that it is accessible to incoming guest molecules. However, considerable hysteresis is evident in the desorption branch of this isotherm. This produced an isotherm that does not reach equilibrium between the adsorbed and free gas under any practical measurement regime.
Analysis of the CO2 isotherms at 273 K yielded surface areas of 325 m2 g−1 and 202 m2 g−1 for (ZIF-8)(ZIF-62)(20/80) and ag[(ZIF-8)0.2(ZIF-62)0.8], respectively. Pore size distributions were also calculated from these isotherms (Fig. S19 and S20†), which indicates the major pores in both materials have diameters of around 5 Å, while a smaller number of cavities have diameters centered on 9 Å. The pore volumes accessible to CO2 were 0.095 cm3 g−1 and 0.068 cm3 g−1, respectively.
Gas adsorption isotherms were also measured on (ZIF-67)(ZIF-62)(20/80) and ag[(ZIF-67)0.2(ZIF-62)0.8] (Fig. S21, Table S1†). As anticipated, the crystalline material adsorbs N2 at 77 K but not after vitrification. H2 is taken up at 77 K, but with hysteresis. On the other hand, CO2 adsorption at 273 K is largely preserved when the crystalline material is transformed to the glass. Surface areas of 218 m2 g−1 for (ZIF-67)(ZIF-62)(20/80) and 194 m2 g−1 for ag[(ZIF-67)0.2(ZIF-62)0.8] were estimated from these isotherms together with accessible pore volumes of 0.067 cm3 g−1 and 0.062 cm3 g−1, respectively. Estimated pore size distributions (Fig. S22 and S23†) paralleled their zinc counterparts. The rate of uptake of CO2 in these materials was measured. A comparison of these kinetics plots (Fig. S24†) reveals that the diffusion of CO2 in the glass is slower than in its crystalline precursor, which is consistent with its more constricted and tortuous pore network.
To provide further information on the pores present in the samples before and after vitrification, positron annihilation lifetime spectroscopy (PALS) experiments were carried out (Fig. S25 and S26, Table S2†). Measurements on crystalline ZIF-8 indicated one main cavity size of 9.5 Å diameter, which is close to the reported 11.6 Å value after accounting for van der Waals radii. A minor cavity of 23 Å was also observed in the study. (ZIF-8)(ZIF-62)(20/80) was also found to possess a major cavity with 5.8 Å diameter, alongside a secondary cavity with a diameter of 10 Å. This is consistent with the presence of a major cavity in pure ZIF-8 at 9.5 Å, and one in ZIF-4, which is closely related to ZIF-62, at 6.2 Å.44 PALS measurements on ag[(ZIF-8)0.2(ZIF-62)0.8] also show a bimodal distribution, with a similar cavity size distribution to a pure sample of agZIF-62, found previously (Fig. S25, Table S2†).25 The key difference between the two samples is that the smaller pore limiting aperture is larger for the ag[(ZIF-8)0.2(ZIF-62)0.8] glass at 3.1 Å compared to 2.5 Å for ag(ZIF-62). In all cases, these results are broadly consistent with the pore size distributions obtained from CO2 sorption analysis.
Grand canonical Monte Carlo (GCMC) simulations of gas adsorption by (ZIF-8)(ZIF-62)(20/80), for which structural models of each of the crystalline phases were derived from single-crystal X-ray diffraction, led to broad agreement between calculated and experimental data (Table 1). For example, CO2 uptake at 273 K in (ZIF-8)(ZIF-62)(20/80) was predicted as 30 mL STP per g, which is very close to the experimental CO2 measurement (31 mL STP per g). Similarly, the simulated CH4 uptake at 273 K of 16 mL STP per g agrees well with the experimental value of 15 mL STP per g. Simulations overestimated N2 adsorption at low temperature (77 K) in (ZIF-8)(ZIF-62)(20/80), where diffusion limitations prevented the ingress of the adsorbate in experimental isotherms. The source of error was proven by simulating N2 uptake in (ZIF-8)(ZIF-62)(20/80) at 195 K (25 mL STP per g), which agreed well with an observed experimental value at 195 K of 23 mL STP per g (ESI Table S5†).
The modelling of amorphous structures is extremely challenging due to the complexity of constructing accurate models. To provide a qualitative estimate of the gas sorption behaviour of ag[(ZIF-8)0.2(ZIF-62)0.8], we followed existing literature20,53 and used a molecular dynamics (MD) method to develop a model for agZIF-62. Initial configurations of ZIF-62 were melted in the NPT ensemble at 1 bar by heating to 1500 K at a rate of 100 K ps−1 from 300 K before quenching to 300 K at a controlled rate. Calculations of the gas adsorption behaviour of this model were then combined with those using a crystalline model of ZIF-8 (ESI†). Simulated O2 uptake (1 mL STP per g) at 273 K was in agreement with negligible experimental uptake of 2 mL STP per g for ag[(ZIF-8)0.2(ZIF-62)0.8], whilst the low predictions for CH4 adsorption at 273 K, agreed broadly with experimental data. Simulations overestimated N2 adsorption at low temperature (77 K) in ag[(ZIF-8)0.2(ZIF-62)0.8], though the simulated N2 uptake in ag[(ZIF-8)0.2(ZIF-62)0.8]at 273 K of 1 mL STP per g agrees well with the experimental value of 1 mL STP per g (ESI Table S5†). The over prediction of H2 uptake at 77 K (49 mL STP per g) compared to the experimental value (28 mL STP per g) is consistent with our assertion that the ZIF-8 structure does not remain intact within the flux melted glass (Table 1). Full details of molecular simulations are given in the ESI,† though two different configurations (imidazolate and benzimidazolate linkers with partial occupancies of 62.5% and 37.5%, respectively) of ZIF-62 were considered in molecular simulations due to the disorder in the framework. The average of the predictions agreed well with the experimental data (Table 1). Secondly, our computational approach represents the first instance where accurate predictions for the gas adsorption performances of ZIF–ZIF crystalline mixture absorbents and ZIF–ZIF glassy flux melts have been made.
Conclusions
These results show that the concept of flux melting, that is, the use of a molten salt as a solvent, may be applicable to MOF chemistry. The flux melted glass reported here is different from the intriguing mixed matrix membrane created by Kertik et al., by thermally amorphizing a ZIF-8 loaded imide polymer.19 This in situ amorphization, by heating the MMM at 623 K for up to 24 hours, was observed to cross-link ZIF-8 particles with the imide. This resulted in retention of the porous interior of the ZIF-8 component, though in an amorphous material. Here, the highly porous ZIF-8 interior does not appear to be retained, suggesting a different process to the cross-linking in the thermally treated MMM.From a fundamental view, the successful realisation of flux melting, which uses the liquid state of ZIF-62 to facilitate the melting of ZIF-8, presents a method by which the Tm of a non-melting framework can be accessed. Use of elemental contrast in the electron microscopy experiments shows that melting of the cobalt analogue of ZIF-8 occurs quickly upon formation of the liquid ZIF-62, and results in regions of the glass which contain higher concentrations of the cobalt-containing component than others. The flux melted glass contains short range ordering reminiscent of the crystalline ZIF-62 parent phase, and a continuous random network akin to that of amorphous SiO2, though with accessible porosity. The increased porosity relative to the pure ZIF-62 glass is ascribed to the ZIF-8 component disrupting the close packing of the ZIF-62 matrix in the liquid phase, rather than any retention of the nanopores belonging to crystalline ZIF-8. The demonstration of porosity towards H2 and CO2, in the flux melted samples is notable, and suggests possibilities in, for example, free-standing membrane manufacture.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TDB would like to thank the Royal Society for a University Research Fellowship (UF150021), and EPSRC (grant EP/R015481/1). LL acknowledges an EPSRC studentship. JH would like to acknowledge funding from the EPSRC (EP/R015481/1). We acknowledge the provision of synchrotron access to beamline I22 (NT18236-1) at the Diamond Light Source, Rutherford Appleton Laboratory UK. SL acknowledges the China Scholarship Council (CSC). This work benefited from the use of the SasView application, originally developed under NSF Award DMR-0520547. SasView also contains code developed with funding from the EU Horizon 2020 programme under the SINE2020 project Grant No. 654000. S. M. Collins acknowledges the Henslow Research Fellowship and Girton College, Cambridge. S. M. Collins and PAM acknowledge funding from the European Research Council under the European Union's Seventh Framework Program (No. FP7/2007-2013)/ERC Grant Agreement No. 291522-3DIMAGE, and from the EPSRC (EP/R008779/1). We are grateful to Christopher Ashling for assistance with PDF measurements. SMC and XY were supported by the NSF, Division of Chemistry, under award number CHE-1661655. CMD acknowledges support from the Australian Research Council Discovery Early Career Researcher Award (DE140101359) and the Veski Inspiring Women Fellowship.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974–986 CrossRef CAS PubMed.
- J. W. Yoon, H. Chang, S. J. Lee, Y. K. Hwang, D. Y. Hong, S. K. Lee, J. S. Lee, S. Jang, T. U. Yoon, K. Kwac, Y. Jung, R. S. Pillai, F. Faucher, A. Vimont, M. Daturi, G. Ferey, C. Serre, G. Maurin, Y. S. Bae and J. S. Chang, Nat. Mater., 2017, 16, 526–531 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Q. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. W. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- A. Schneermann, V. Bon, I. Schwedler, I. Senkoska, S. Kaskel and R. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- S. Krause, V. Bon, I. Senkoska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F. X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef PubMed.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- M. S. Denny, J. C. Moreton, L. Benz and S. M. Cohen, Nat. Rev. Mater., 2016, 1, 16078 CrossRef CAS.
- B. Bueken, N. Van Velthoven, T. Willhammar, T. Stassin, I. Stassen, D. A. Keen, G. V. Baron, J. F. M. Denayer, R. Ameloot, S. Bals, D. De Vos and T. D. Bennett, Chem. Sci., 2017, 8, 3939–3948 RSC.
- O. Shekhah, J. Liu, R. A. Fischer and C. Woll, Chem. Soc. Rev., 2011, 40, 1081–1106 RSC.
- J. C. Tan and A. K. Cheetham, Chem. Soc. Rev., 2011, 40, 1059–1080 RSC.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- Y. Q. Tian, Y. M. Zhao, Z. X. Chen, G. N. Zhang, L. H. Weng and D. Y. Zhao, Chem.–Eur. J., 2007, 13, 4146–4154 CrossRef CAS PubMed.
- I. Stassen, M. Styles, G. Grenci, H. Van Gorp, W. Vanderlinden, S. De Feyter, P. Falcaro, D. De Vos, P. Vereecken and R. Ameloot, Nat. Mater., 2016, 15, 304–310 CrossRef CAS PubMed.
- T. Wu, X. Feng, S. K. Elsaidi, P. K. Thallapally and M. A. Carreon, Ind. Eng. Chem. Res., 2017, 56, 1682–1686 CrossRef CAS.
- H. C. Zhang, J. Hou, Y. X. Hu, P. Y. Wang, R. W. Ou, L. Jiang, J. Z. Liu, B. D. Freeman, A. J. Hill and H. T. Wang, Sci. Adv., 2018, 4, eaaq0066 CrossRef PubMed.
- B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421–2454 RSC.
- S. W. Yu, S. C. Li, S. L. Huang, Z. H. Zeng, S. Cui and Y. Liu, J. Membr. Sci., 2017, 540, 155–164 CrossRef CAS.
- A. Kertik, L. H. Wee, M. Pfannmoller, S. Bals, J. A. Martens and I. F. J. Vankelecom, Energy Environ. Sci., 2017, 10, 2342–2351 RSC.
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F. X. Coudert, Nat. Mater., 2017, 16, 1149–1154 CrossRef CAS PubMed.
- J. B. James and Y. S. Lin, J. Phys. Chem. C, 2016, 120, 14015–14026 CrossRef CAS.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- S. S. Nagarkar, S. Horike, T. Itakura, B. Le Ouay, A. Demessence, M. Tsujimoto and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 4976–4981 CrossRef CAS PubMed.
- D. Umeyama, S. Horike, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2015, 137, 864–870 CrossRef CAS PubMed.
- A. Qiao, T. D. Bennett, H. T. Tao, A. Krajnc, G. Mali, C. M. Doherty, A. W. Thornton, J. C. Mauro, G. N. Greaves and Y. Z. Yue, Sci. Adv., 2018, 4, eaao6827 CrossRef PubMed.
- F. A. Lindemann, Phys. Z., 1910, 11, 609 CAS.
- T. D. Bennett, J. C. Tan, Y. Z. Yue, E. Baxter, C. D. Ducati, N. Terril, H. Y. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 CrossRef CAS PubMed.
- J. E. Shelby, Introduction to Glass Science and Technology, Royal Society of Chemistry, 2nd edn, 2005 Search PubMed.
- H. W. Kui, A. L. Greer and D. Turnbull, Appl. Phys. Lett., 1984, 45, 615–616 CrossRef CAS.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- M. Gustafsson and X. D. Zou, J. Porous Mater., 2013, 20, 55–63 CrossRef CAS.
- Q. Shi, Z. F. Chen, Z. W. Song, J. P. Li and J. X. Dong, Angew. Chem., Int. Ed., 2011, 50, 672–675 CrossRef CAS PubMed.
- M. Basham, J. Filik, M. T. Wharmby, P. C. Chang, B. El Kassaby, M. Gerring, J. Aishima, K. Levik, B. C. Pulford, I. Sikharulidze, D. Sneddon, M. Webber, S. S. Dhesi, F. Maccherozzi, O. Svensson, S. Brockhauser, G. Naray and A. W. Ashton, J. Synchrotron Radiat., 2015, 22, 853–858 CrossRef PubMed.
- J. Filik, A. W. Ashton, P. C. Y. Chang, P. A. Chater, S. J. Day, M. Drakopoulos, M. W. Gerring, M. L. Hart, O. V. Magdysyuk, S. Michalik, A. Smith, C. C. Tang, N. J. Terrill, M. T. Wharmby and H. Wilhelm, J. Appl. Crystallogr., 2017, 50, 959–966 CrossRef CAS PubMed.
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thünemann, J. Appl. Crystallogr., 2017, 50, 1800–1811 CrossRef CAS PubMed.
- M. e. a. Doucet, SASView 4.1.1, 2017 Search PubMed.
- B. R. Pauw, J. S. Pedersen, S. Tardif, M. Takata and B. B. Iversen, J. Appl. Crystallogr., 2013, 46, 365–371 CrossRef CAS PubMed.
- I. Bressler, B. R. Pauw and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 962–969 CrossRef CAS PubMed.
- A. K. Soper, Tech. Rep. RAL-TR-2011-013, 2011 Search PubMed.
- A. K. Soper and E. R. Barney, J. Appl. Crystallogr., 2011, 44, 714–726 CrossRef CAS.
- F. De la Pēna, T. Ostasevicius, V. T. Fauske, P. Burdet, P. Jokubauskas and M. Sarahan, hyperspy/hyperspy: HyperSpy 1.3, 2016 Search PubMed.
- S. J. Tao, J. Chem. Phys., 1972, 56, 5499 CrossRef CAS.
- A. W. Thornton, K. E. Jelfs, K. Konstas, C. Doherty, A. J. Hill, A. K. Cheetham and T. D. Bennett, Chem. Commun., 2016, 52, 3750–3753 RSC.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS PubMed.
- T. D. Bennett, Y. Z. Yue, P. Li, A. Qiao, H. Tao, G. N. Greaves, T. Richards, G. I. Lampronti, S. A. T. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, J. Am. Chem. Soc., 2016, 138, 3484–3492 CrossRef CAS PubMed.
- L. Longley, S. M. Collins, C. Zhou, G. J. Smales, S. E. Norman, N. J. Brownbill, C. W. Ashling, P. Chater, R. Tovey, C. B. Schönlieb, T. F. Headen, N. J. Terrill, Y. Z. Yue, A. J. Smith, F. Blanc, D. A. Keen, P. A. Midgley and T. D. Bennett, Nat. Commun., 2018, 9, 2135 CrossRef PubMed.
- M. G. Goesten, E. Stavitski, J. Juan-Alcaniz, A. Martinez-Joaristi, A. V. Petukhov, F. Kapteijn and J. Gascon, Catal. Today, 2013, 205, 120–127 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- R. N. Widmer, G. I. Lampronti, S. Anzellini, R. Gaillac, S. Farsang, C. Zhou, A. Belenguer, H. Palmer, A. K. Kleppe, M. T. Wharmby, S. A. T. R. Redfern, F. X. Coudert, S. G. Macleod and T. D. Bennett, ChemRxiv, 2018 DOI:10.26434/chemrxiv.6541190.v6541193.
- C. Zhou, L. Longley, A. Krajnc, G. J. Smales, A. Qiao, I. Eruçar, C. M. Doherty, A. W. Thornton, A. J. Hill, C. W. Ashling, O. T. Qasvini, S. J. Lee, P. Chater, N. J. Terrill, A. J. Smith, Y. Z. Yue, G. Mali, D. A. Keen, S. G. Telfer and T. D. Bennett, Nat. Commun., 2018, 9, 5042 CrossRef PubMed.
- S. S. Mondal, M. Hovestadt, S. Dey, C. Paula, S. Glomb, A. Kelling, U. Schilde, C. Janiak, M. Hartmann and H. J. Holdt, CrystEngComm, 2017, 19, 5882–5891 RSC.
- C. D. Williams, K. P. Travis, N. A. Burton and J. H. Harding, Microporous Mesoporous Mater., 2016, 228, 215–223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04044c |
| This journal is © The Royal Society of Chemistry 2019 |