DOI:
10.1039/C8SC04026E
(Edge Article)
Chem. Sci., 2019,
10, 843-847
Mechano-responsive circularly polarized luminescence of organic solid-state chiral emitters†
Received
10th September 2018
, Accepted 26th October 2018
First published on 29th October 2018
Abstract
Two difluoro-boron β-diketonate complexes bearing chiral amido groups have been synthesized. Their mechano-responsive luminescence and chiroptical properties have been investigated in the solid state. Both compounds display a bright blue-green emission and a significant circularly polarized luminescence (CPL) signal in the crystalline state, with |glum| values as high as 2.2–2.4 × 10−2. A bathochromic shift in emission, together with a decrease of |glum| values to c.a. 3 × 10−3, is induced upon application of a shearing stress. For the DFB-Hex-amide compound, interestingly, sign inversion of the anisotropy factor glum is observed under mechanical stimulation (“mechano-CPL effect”), which can be rationalized by a switching between monomer and excimer emission.
1 Introduction
Circularly polarized luminescence (CPL) corresponds to the differential emission of right- and left-circularly polarized light by a chiral molecule or a molecular assembly.1–4 It is a powerful tool to study the chirality of excited states and attracts growing interest in the development of new optoelectronic devices,5 OLED materials,6 and probes for enantioselective sensing.7,8 As a consequence, the development of new CPL emitters is an active field of research, with the aim of designing materials with improved anisotropy factors, and also developing CPL emissive materials responsive to various chemical,9–13 thermal,14,15 mechanical,16 or light17–19 stimuli. Amongst CPL active compounds, lanthanide complexes, especially those based on europium, are known for their high CPL signals.20 Another efficient strategy to enhance the anisotropy factor is to develop self-assembled materials displaying supramolecular chirality.21–26 Besides, mechano-responsive luminescent materials show emission properties sensitive to mechanical stress (pressure, shearing…).27 In most cases, this effect results from a supramolecular phenomenon: the mechanical stimulus triggers a transition between several crystalline or amorphous phases, or between an aggregated and a monomeric state, which is responsible for the changes in luminescence emission. The diversity of known mechano-responsive luminescent materials is rapidly expanding28 and applications are envisioned in mechanical stress sensing29,30 and security tags.31,32 In this frame, materials displaying not only a change in the emission wavelength but also in the CPL signal upon mechanical stimulation would be highly desirable. Amongst the mechano-responsive luminescent materials, difluoro-boron β-diketonate (DFB) complexes are appealing for their versatile synthesis, high fluorescence quantum yields in the solid state and marked fluorescence emission changes upon force application.33–35 We report here chiral mechano-responsive and CPL-active fluorescent DFB complexes which, to the best of our knowledge, display the first example of sign inversion of the CPL signal upon mechanical stress.
2 Results and discussion
2.1 Synthesis
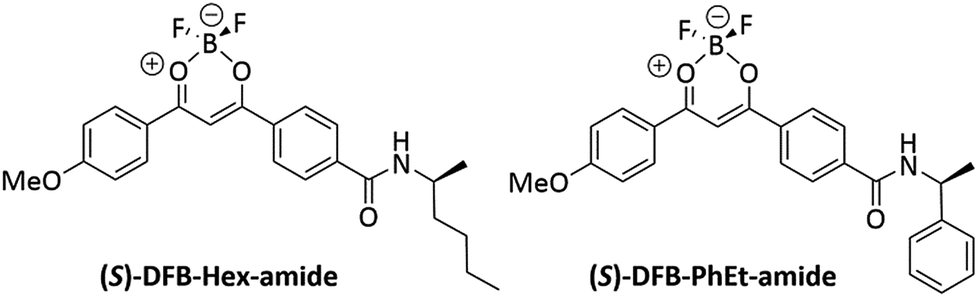
DFB complexes bearing a chiral amido group have been prepared (Fig. 1), so as to introduce a chiral substituent at the final step of the chemical synthesis (see ESI, Fig. S1†), thus affording suitable versatility of the synthetic route. We already demonstrated that a parent achiral compound with a butyl amido group, DFB-Bu-amide,36 possesses an amorphous phase that is stable over time after mechanical shearing, which is a prerequisite to allow CPL measurements in the solid state. Chiral compounds, (R/S)-DFB-Hex-amide and (R/S)-DFB-PhEt-amide, were prepared in four steps. The first step consists of a Claisen condensation between dimethyl terephthalate and 4-methoxyacetophenone,37 followed by saponification of the ester group and peptidic coupling with (R/S)-2-aminohexane and (R/S)-methylbenzylamine, respectively. These chiral amides with a relatively low steric hindrance were chosen in order to preserve the supramolecular interactions between the diketonate moieties which are essential for the mechano-responsive properties. In the last step, complexation of the diketone with BF3(OEt)2 leads to the four desired target derivatives. Full synthetic procedures are given in the ESI (Section I.1),† together with photophysical properties in solution (ESI, Section II.2).†
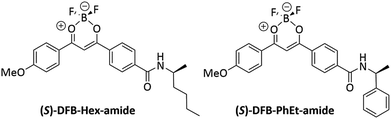 |
| | Fig. 1 Structures of (S)-DFB-Hex-amide and (S)-DFB-PhEt-amide. | |
2.2 Structure determination
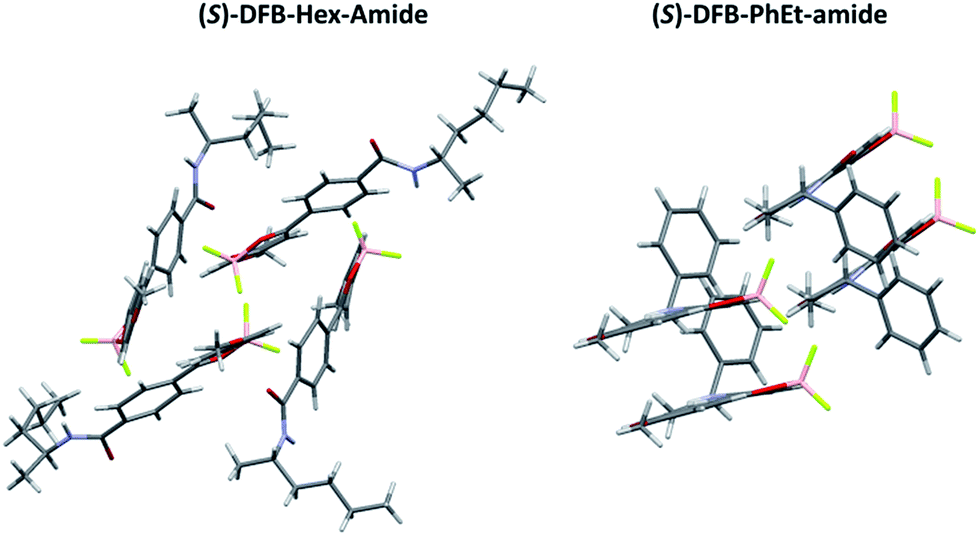
Single crystals suitable for X-ray diffraction were obtained for both (S)-DFB-Hex-amide and (S)-DFB-PhEt-amide and exhibit very different crystalline packing modes (Fig. 2). On the one hand, the (S)-DFB-Hex-amide derivative crystallizes in triclinic P1. In the crystal, the molecule is non-planar with dihedral angles of 8.16° and −22.60° between the dioxaborinine center and the methoxyphenyl and para-amido phenyl moieties, respectively. The crystalline unit cell is composed of four molecules organized in a cage-like framework, without clear π-stacking between DFB molecular units. The fluorine atoms of the BF2 fragments are oriented towards the hydrogens of neighbouring methoxy groups. On the other hand, the (S)-DFB-PhEt-amide compound crystallizes in monoclinic P21. The phenyl rings of the molecule are almost coplanar: the methoxyphenyl and the para-amido phenyl moieties are twisted by 0.37° and 4.71° with respect to the dioxaborinine center, respectively. The molecules are organized in dimers, as often observed in related DFB complexes.38,39 A partial overlap of the dioxaborinine center and the phenyl side-groups can be noticed resulting in weak π-stacking interactions. Hydrogen bonding interactions between the fluorine atoms and the hydrogen atom of the amide substituent are observed.
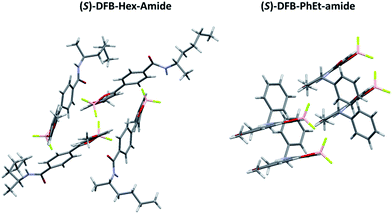 |
| | Fig. 2 Packing modes of (left) (S)-DFB-Hex-amide and (right) (S)-DFB-PhEt-amide. | |
2.3 Solid-state spectral characterization and thermal treatment
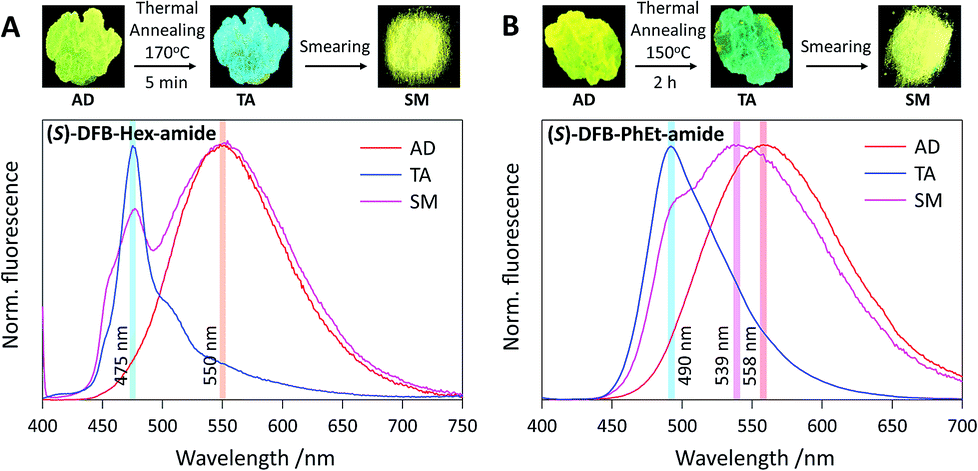
Photophysical properties were investigated on thin films deposited on a paper substrate, prepared by drop casting from a concentrated chloroform solution of DFB derivatives. Samples thus obtained, as-deposited (AD), are known to be mostly amorphous,40,41 and yellow fluorescence is observed with emission maxima at 550 nm for DFB-Hex-amide and 558 nm for DFB-PhEt-amide (Fig. 3 and ESI, Fig. S2†). To induce crystallization, a thermal annealing treatment (TA) was performed on both DFB-Hex-amide (170 °C for 5 min) and DFB-PhEt-amide (150 °C for 2 h). Upon thermal annealing, both compounds showed a significant emission hypsochromic shift. This blue shift from 550 nm to 475 nm (2850 cm−1, resulting in a blue-green emission) for the DFB-Hex-amide and from 558 nm to 490 nm (2500 cm−1, resulting in a green emission) for the DFB-PhEt-amide, together with a sharp fluorescence band can be due to stronger intermolecular interactions in the crystalline phase (see Fig. 3). Powder X-ray diffraction (PXRD) of TA states demonstrates sharp diffraction peaks indicating crystalline ordering of molecules (see Fig. S6†), however with significant discrepancies compared to the simulation based on bulk crystalline structures (see Fig. S7†). In the PXRD data, the d-spacing (defined from Bragg's law n × λ = 2 × d × sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ) of 2.1 and 1.0 nm for (R/S)-DFB-Hex-amide can be correlated with the average length along the molecular long axis and the thickness along the vertical direction. Similarly, the characteristic d-spacing of ∼1.5 nm for (R/S)-DFB-PhEt-amide corresponds to the average molecular length. The observed d-spacing of ∼0.7–0.5 nm can be assigned to molecular packing modes in different dimensions which can be attributed to higher order diffraction patterns.42
θ) of 2.1 and 1.0 nm for (R/S)-DFB-Hex-amide can be correlated with the average length along the molecular long axis and the thickness along the vertical direction. Similarly, the characteristic d-spacing of ∼1.5 nm for (R/S)-DFB-PhEt-amide corresponds to the average molecular length. The observed d-spacing of ∼0.7–0.5 nm can be assigned to molecular packing modes in different dimensions which can be attributed to higher order diffraction patterns.42
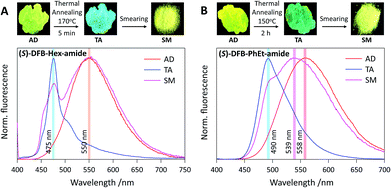 |
| | Fig. 3 (Top) Photographs of the samples after deposition (AD), after annealing (TA) and after smearing (SM) (excitation 365 nm), and (bottom) their associated fluorescence spectra of (A) (S)-DFB-Hex-amide and (B) (S)-DFB-PhEt-amide. | |
2.4 Mechanofluorochromism
The mechanofluorochromic properties of the two compounds were probed by smearing (SM) the thermally annealed samples and visualized through emission colour change (Fig. 3). The (R/S)-DFB-Hex-amide and (R/S)-DFB-PhEt-amide showed a bathochromic shift of the emission band of 75 nm (2850 cm−1) and 49 nm (1850 cm−1), respectively. Both compounds remain highly fluorescent in the solid state, before and after mechanical stimulation (ϕF in the range 0.10–0.35, see Table 1), although fluorescence quantum yields are significantly lower than those observed in solution (ϕF = 0.80 and 0.94 in THF, respectively, see Table S1†), attesting intermolecular interactions in the crystalline and amorphous states. The reversibility of the system was established by a second thermal annealing step of the SM samples (see Fig. S3 and S4†). The emission spectra of the (R/S)-DFB-Hex-amide and (R/S)-DFB-PhEt-amide amorphous phases were continuously monitored for 1 hour (see Fig. S5†) and appeared to be perfectly stable over such a period of time, which is a requisite condition for CPL measurements (vide infra). PXRD spectra of the DFB derivatives suggest that the crystalline-rich TA samples became amorphous-rich in the SM state (see Fig. S6†). It should be noted that some remaining diffraction peaks in SM samples may come from incomplete conversion from crystal to amorphous phases, with small microcrystalline domains. The degree of this mechanically induced phase-conversion may not be fully reproducible as discussed below.
Table 1 Photophysical properties in the solid state
| Compound |
λ
max(TA)/nm |
ϕ
F
|
λ
max(SM)/nm |
ϕ
F
|
|
Determined in the powder crystalline state for both enantiomers. The difference between the values for the two enantiomers is in the range of uncertainty for solid state quantum yield measurements (±0.02).
Determined on smeared paper samples.
|
|
(R/S)-DFB-Hex-amide
|
475 |
0.13/0.10 |
550 |
0.35 |
|
(R/S)-DFB-PhEt-amide
|
490 |
0.20/0.23 |
539 |
0.11 |
2.5 Circularly polarized luminescence (CPL) properties
CPL in solution.
CPL measurements of both DFB derivatives were first conducted in chloroform solution. (R) and (S) isomers of DFB-Hex-amide exhibited mirror image CPL spectra with negative and positive signals, respectively. The CPL signals for both isomers were too weak to determine the luminescence dissymmetry factor, as |glum| < 1 × 10−3 (at 455 nm), where |glum| = 2(IL − IR)/(IL + IR), for which IL and IR represent the luminescence intensities of left and right circularly polarized light, respectively. DFB-PhEt-amide exhibits mirror image CPL signals with opposite signs for (R) and (S) isomers as compared to DFB-Hex-amide (see Fig. S8†).
CPL in the solid state.
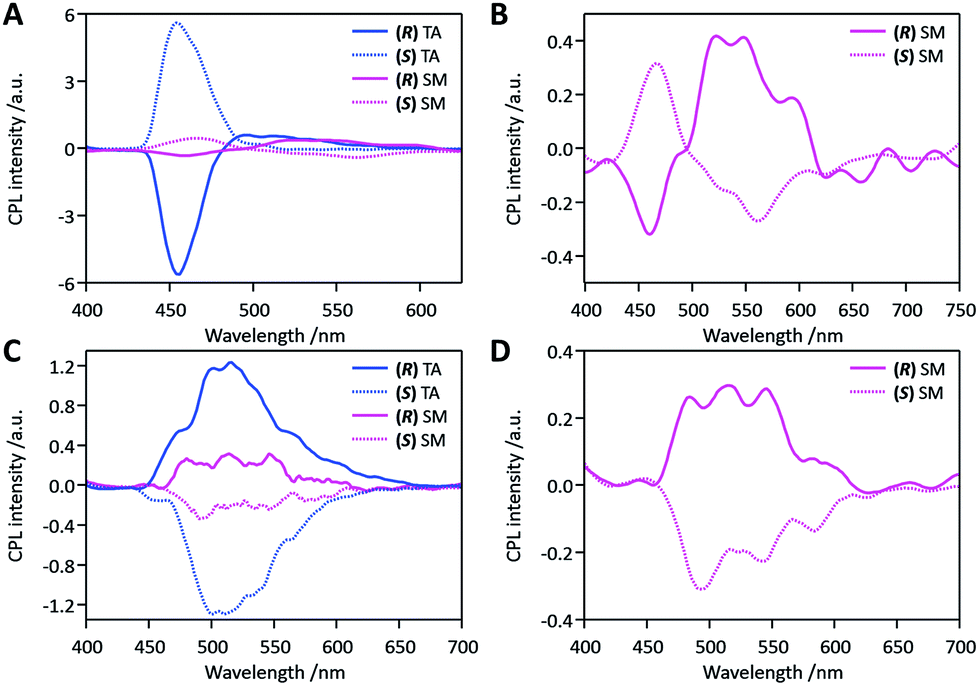
CPL of both DFB derivatives was investigated for thin films deposited on a paper substrate and their different behaviours are highlighted in Fig. 4. In the AD state, both compounds showed very weak signals (see Fig. S9†). In marked contrast, the TA samples of DFB-Hex-amide and DFB-PhEt-amide clearly offered CPL signals at 455 nm and 500 nm, respectively (Fig. 4 A and C). The CPL intensity and |glum| values were much enhanced after the annealing treatment (see Fig. S10†). The |glum| values in the TA state were 2.4 × 10−2 (at 455 nm) and 2.2 × 10−2 (at 500 nm) for (R/S)-DFB-Hex-amide and (R/S)-DFB-PhEt-amide, respectively. The mirror-image CPL profiles were clearly demonstrated for the isomers of opposite handedness of both compounds. This supports the reliability of these CPL data. Moreover, interestingly, (R)-DFB-Hex-amide showed negative CPL signals which is opposite to that of (R)-DFB-PhEt-amide, while the (S)-isomers behaved in an opposite manner. In these measurements, we employed the 2ω AC-component compensation procedure for suppressing effects of the linearly polarized emission on the CPL signal.23
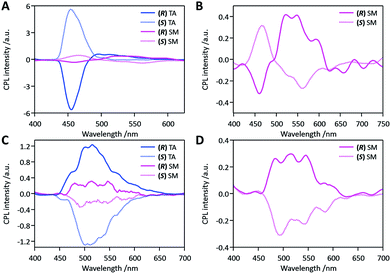 |
| | Fig. 4 CPL spectra of solid-state samples deposited on paper: thermally annealed (TA) in blue and mechanically smeared (SM) in pink for (A and B) (R/S)-DFB-Hex-amide and (C and D) (R/S)-DFB-PhEt-amide, respectively. (R) isomers are plotted as solid lines and (S) isomers as dashed lines. | |
2.6 Mechano-CPL effect
We then studied the effects of mechanical stress on the CPL properties, namely the mechano-CPL effect. CPL spectra of both compounds in the SM state are presented in Fig. 4 (pink curves). The quasi-mirror-image CPL signals for the two enantiomers were again observed in these SM samples. Minor fluctuations in the mechanical smearing may be responsible for the less symmetric CPL profiles of opposite stereo-isomers. The SM state of DFB-Hex-amide showed two distinctive and inverted CPL bands at 450 nm and at ca. 550 nm. The CPL band at 450 nm was similar to that of the TA state in shape and sign of the signal. This was thus assigned to microcrystals remaining in the SM sample, which were also monitored in its emission profile (Fig. 3A). The broad CPL band at 550 nm was assigned to the amorphous state after comparison with the emission profile. Although this CPL band is weak with a |glum| value as low as ca. 3 × 10−3 in the wavelength range of 500–550 nm, it is still notably larger than that of the solution phase. This suggests possible chiral-exciton coupling between chromophores in the amorphous SM state. The CPL band is thus recognized as a remarkable indicator of short-distance ordering of chromophores in the smeared amorphous state. It must be highlighted that the CPL signals in this region were opposite to those of the crystalline state at 450 nm in both SM and TA samples. As reasonably expected for the SM state, we observed CPL profiles not fully reproducible and less mirror-symmetric, but markedly depending on the conversion ratio of derived amorphous and persisting crystalline phases. The SM state of (R/S)-DFB-PhEt-amide displayed CPL profiles with mirror symmetry in the same wavelength range as TA samples. Although the dissymmetry again decreased with the |glum| value being ca. 3 × 10−3, the sign of CPL signals of both stereo-isomers remained the same as those of their corresponding TA samples. Thus this CPL band at 500 nm was again assigned to the crystalline parts remaining after the mechanical treatment. These changes in emission profiles and CPL profiles were reproduced in a quasi-reversible way with alternating thermal treatment and mechanical smearing processes.
The remarkable enhancement of |glum| values in the TA state compared to those in solution, AD and SM states for both compounds is ascribed to the chiral supramolecular arrangement of chromophores in the crystal lattice. Preliminary quantum chemical TD-DFT calculations for isolated molecules and molecules in crystalline lattices (see Fig. S12–S16†) showed an enhancement of the CD intensity when going from one isolated molecule to four (for DFB-Hex-amide) or two (for DFB-PhEt-amide) molecules in the crystal packing. It is thus supposed that significant exciton interactions operate between the chromophores in the TA crystalline structures.24,43–45
Besides previous studies reporting enhanced CPL in the condensed phase,15,17,46 it is the first time, to the best of our knowledge, that such a mechano-induced shift coupled with an inversion of the CPL signal is demonstrated in the solid state. Nevertheless, related characteristics were observed in solution by Asami et al.47 on N,N′-dipyrenyldiamine derivatives by changing the fluorophore concentration in toluene. Such changes in the CPL spectra were attributed to a switching between monomer and excimer emission. Yet, it is known that the bathochromic emission shift of DFB derivatives under mechanical stress can be explained by the formation of excimers or emitting dimers in the amorphous state.33,37,48 Time-resolved fluorescence measurements on as-deposited (AD), thermally annealed (TA) and smeared (SM) samples were performed for (S)-DFB-Hex-amide, showing that a short rise-time and a long decay-component are present in AD and SM samples, but absent in TA samples (Fig. S11†). This observation confirms the hypothesis of excimers in the amorphous state, responsible for the change of the CPL signal. It has also been recently studied that the aggregation of chromophores with chiral hierarchical order substantially enhances the CPL dissymmetry.4,21–26 The |glum| of about 3 × 10−3 seems typical for π-conjugated fluorescent chromophores forming chiral dimers and higher aggregates.
In contrast, examination of the crystal structure of (S)-DFB-Hex-amide reveals that the formation of dynamic excimers is unfavourable in the crystalline state, which is consistent with the small shift observed between the emission recorded in solution and in the crystalline TA state (1600 cm−1). The mechanical perturbations applied to the system would then change the intermolecular arrangement in the solid and favour the formation of excimers in the SM state. This transition to the excimer-like emission would be responsible for these shift and sign inversion of the CPL signal. The decreased but notable CPL activity suggests that the excimer in the SM state is associated in a chiral manner. In the case of (R/S)-DFB-PhEt-amide, the molecules are organized in a dimer form in the crystal structure, which is very favourable for the formation of emissive dimers (dynamic excimers or preformed dimers). Dimeric emission is compatible with the more pronounced bathochromic shift observed between the solution and the crystal TA emission (2150 cm−1). Indeed, excimer emission has been evidenced in the parent achiral compound DFB-Bu-amide which also crystallizes with molecules organized in dimers and also displays green fluorescence with λmax(em) = 490 nm.36 Such emissive dimers have also been evidenced for related compounds in their crystalline state.37,39 The mechanical shearing stress applied to the crystalline DFB-PhEt-amide sample promotes the formation of an amorphous phase. Therefore, the formation of low-energy excimers in a different configuration is allowed, due to increased conformational freedom, which would explain the large bathochromic shift observed in the fluorescence spectrum. As the nature of the emitting species is not profoundly modified, the sign of glum was retained for AD, TA and SM samples.
Conclusions
In summary, we have prepared new chiral difluoro-boron diketonate complexes and investigated their mechanochromic fluorescence as well as their chiroptical properties. Both compounds display a significant mechanofluorochromic response in the solid state. An important enhancement of their CPL signals is observed in the crystalline state compared to that in solution, to reach high |glum| values of 2.2–2.4 × 10−2. DFB-Hex-amide demonstrated a wavelength shift and a sign inversion in CPL, which can be rationalized by a change of the chiral environment of the emitters as well as a switching between monomer and excimer emission upon mechanical stimulation. The CPL may provide distinctive insights into solid state fluorescent materials with chiral molecules ordered at short distance.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support of this work by the European Research Council (ERC StG MECHANO-FLUO No 715757 to C. A.) and by the IDEX Paris-Saclay (PhD fellowship to M. L.). T. K. acknowledges the JSPS KAKENHI Grant Number JP26107006 in Scientific Research on Innovative Areas “Photosynergetics”.
Notes and references
- F. S. Richardson and J. P. Riehl, Chem. Rev., 1977, 77, 773–792 CrossRef CAS.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS.
- E. M. Sanchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef CAS PubMed.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS.
- S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed.
- S. Shuvaev, M. Starck and D. Parker, Chem.–Eur. J., 2017, 23, 9974–9989 CrossRef CAS PubMed.
- R. Sethy, J. Kumar, R. Métivier, M. Louis, K. Nakatani, N. M. T. Mecheri, A. Subhakumari, K. G. Thomas, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2017, 56, 15053–15057 CrossRef CAS PubMed.
- H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266–9269 CrossRef CAS PubMed.
- N. Saleh, B. Moore, 2nd, M. Srebro, N. Vanthuyne, L. Toupet, J. A. Williams, C. Roussel, K. K. Deol, G. Muller, J. Autschbach and J. Crassous, Chem.–Eur. J., 2015, 21, 1673–1681 CrossRef CAS PubMed.
- Y. Nagata, T. Nishikawa and M. Suginome, Chem. Commun., 2014, 50, 9951–9953 RSC.
- J. Li, C. Yang, X. Peng, Q. Qi, Y. Li, W. Y. Lai and W. Huang, Org. Biomol. Chem., 2017, 15, 8463–8470 RSC.
- H. Maeda and Y. Bando, Pure Appl. Chem., 2013, 85, 1967–1978 CAS.
- J. Kumar, T. Nakashima and T. Kawai, Langmuir, 2014, 30, 6030–6037 CrossRef CAS PubMed.
- Q. Ye, D. Zhu, H. Zhang, X. Lu and Q. Lu, J. Mater. Chem. C, 2015, 3, 6997–7003 RSC.
- K. Okano, M. Taguchi, M. Fujiki and T. Yamashita, Angew. Chem., Int. Ed., 2011, 50, 12474–12477 CrossRef CAS PubMed.
- H. Hayasaka, T. Miyashita, K. Tamura and K. Akagi, Adv. Funct. Mater., 2010, 20, 1243–1250 CrossRef CAS.
- Y. Hashimoto, T. Nakashima, D. Shimizu and T. Kawai, Chem. Commun., 2016, 52, 5171–5174 RSC.
- A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505–10509 CrossRef CAS PubMed.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899–11907 CrossRef CAS PubMed.
- H. Tsumatori, T. Nakashima and T. Kawai, Org. Lett., 2010, 12, 2362–2365 CrossRef CAS PubMed.
- T. Ikeda, T. Masuda, T. Hirao, J. Yuasa, H. Tsumatori, T. Kawai and T. Haino, Chem. Commun., 2012, 48, 6025–6027 RSC.
- J. Kumar, T. Nakashima, H. Tsumatori and T. Kawai, J. Phys. Chem. Lett., 2014, 5, 316–321 CrossRef CAS PubMed.
- J. Kumar, H. Tsumatori, J. Yuasa, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2015, 54, 5943–5947 CrossRef CAS PubMed.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS PubMed.
-
J. Xu and Z. Chi, Mechanochromic Fluorescent Materials: Phenomena, Materials and Applications, The Royal Society of Chemistry, 2014 Search PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- A. Battisti, P. Minei, A. Pucci and R. Bizzarri, Chem. Commun., 2016, 53, 248–251 RSC.
- H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef PubMed.
- X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, S. Liu and H. Zhu, Chem. Commun., 2014, 50, 12951–12954 RSC.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- F. Ito, Y. Suzuki, J. Fujimori, T. Sagawa, M. Hara, T. Seki, R. Yasukuni and M. Lamy de la Chapelle, Sci. Rep., 2016, 6, 22918 CrossRef CAS PubMed.
- L. Wilbraham, M. Louis, D. Alberga, A. Brosseau, R. Guillot, F. Ito, F. Labat, R. Métivier, C. Allain and I. Ciofini, Adv. Mater., 2018, 30, 1800817 CrossRef PubMed.
- M. Louis, A. Brosseau, R. Guillot, F. Ito, C. Allain and R. Métivier, J. Phys. Chem. C, 2017, 121, 15897–15907 CrossRef CAS.
- G. R. Krishna, R. Devarapalli, R. Prusty, T. Liu, C. L. Fraser, U. Ramamurty and C. M. Reddy, IUCrJ, 2015, 2, 611–619 CrossRef CAS PubMed.
- A. Sakai, E. Ohta, Y. Yoshimoto, M. Tanaka, Y. Matsui, K. Mizuno and H. Ikeda, Chem.–Eur. J., 2015, 21, 18128–18137 CrossRef CAS PubMed.
- N. D. Nguyen, G. Zhang, J. Lu, A. E. Sherman and C. L. Fraser, J. Mater. Chem., 2011, 21, 8409–8415 RSC.
- G. Zhang, J. Lu and C. L. Fraser, Inorg. Chem., 2010, 49, 10747–10749 CrossRef CAS PubMed.
- D. Ke, C. Zhan, A. D. Li and J. Yao, Angew. Chem., Int. Ed., 2011, 50, 3715–3719 CrossRef CAS PubMed.
- S. Sato, A. Yoshii, S. Takahashi, S. Furumi, M. Takeuchi and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13097–13101 CrossRef CAS PubMed.
- R. Tempelaar, A. Stradomska, J. Knoester and F. C. Spano, J. Phys. Chem. B, 2011, 115, 10592–10603 CrossRef CAS PubMed.
- X. Shang, I. Song, H. Ohtsu, Y. H. Lee, T. Zhao, T. Kojima, J. H. Jung, M. Kawano and J. H. Oh, Adv. Mater., 2017, 29, 1605828 CrossRef PubMed.
- J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2018, 30, 2737–2747 Search PubMed.
- S. Ito, K. Ikeda, S. Nakanishi, Y. Imai and M. Asami, Chem. Commun., 2017, 53, 6323–6326 RSC.
- W. A. Morris, T. Liu and C. L. Fraser, J. Mater. Chem. C, 2015, 3, 352–363 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic protocols and characterization, equipment information, sample preparation, spectroscopic properties in solution and in the solid state, CPL measurements, time-resolved fluorescence, DFT optimization, and X-ray diffraction analyses. CCDC 1850803 and 1817892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04026e |
| ‡ Dr M. Louis and R. Sethy contributed equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Jatish
Kumar
b,
Jatish
Kumar