
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemiluminescence-initiated and in situ-enhanced photoisomerization for tissue-depth-independent photo-controlled drug release†
Yufu
Tang
a,
Xiaomei
Lu
b,
Chao
Yin
a,
Hui
Zhao
a,
Wenbo
Hu
 a,
Xiaoming
Hu
a,
Yuanyuan
Li
a,
Zhen
Yang
a,
Feng
Lu
a,
Quli
Fan
*a and
Wei
Huang
abc
a,
Xiaoming
Hu
a,
Yuanyuan
Li
a,
Zhen
Yang
a,
Feng
Lu
a,
Quli
Fan
*a and
Wei
Huang
abc
aKey Laboratory for Organic Electronics and Information Displays, Jiangsu Key Laboratory for Biosensors, Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing University of Posts & Telecommunications (NUPT), Nanjing 210023, China. E-mail: iamqlfan@njupt.edu.cn
bKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (Nanjing Tech), Nanjing 211816, China
cShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), Xi'an 710072, China
First published on 10th November 2018
Abstract
Tissue-penetration-depth-independent self-luminescence is highly expected to perform photoisomerization-related bioapplications in vivo to overcome the limitation of shallow tissue-penetration from external photoexcitation. However, it remains extremely challenging because of lacking a target-specific high-intensity self-luminescence to precisely and effectively drive the photoisomerization. Here, we first report a target-specific tissue-depth-independent photoisomerization in vivo by developing a target-specific initiated and in situ-enhanced chemiluminescence (one of self-luminescence) strategy that overcomes the limitation of lacking target-specific high-intensity self-luminescence. Considering that photoisomerization shows boundless glamour in drug-controlled release for disease-specific chemotherapy, we demonstrated applicability of our strategy to apply it in tumor-specific self-luminescence-controlled drug chemotherapy. Specifically, a chemiluminescence substrate and chemiluminescence fluorophore (antitumor drug, CPT) were co-encapsulated in host–guest carriers composed of cyclodextrin and the photoisomerization molecule azobenzene. Tumor-specific H2O2-induced chemiluminescence preliminarily isomerizes azobenzene, triggering the partial dissociation of host–guest carriers and CPT release. Particularly, the initially released CPT again functions as a chemiluminescence enhancer to achieve in situ enhanced chemiluminescence, assuring target-specific enhanced isomerization and CPT release. With high tumor-inhibition-rate (73%) and no obvious therapy-side-effect in vivo indicates the good efficiency and target-specificity of our chemiluminescence-driven photoisomerization. Although we only demonstrated one example of a photoisomerization-related bioapplication, namely photoisomerization-controlled drug chemotherapy, our work provides guidelines to design various target-specific tissue-depth-independent photoisomerization for bioapplications.
Introduction
Photoisomerization that can spatiotemporally change structure between isomers upon photoexcitation is one of the most promising strategies for precisely controlling biological structures/function in a living body.1 This has been widely studied in biomedicine, including photo-controlled drug release,2–4 optogenetics,5 and optopharmacology.6 Unfortunately, all these applications in vivo are severely limited to shallow tissue because most photoisomerization materials can only be excited by UV/Vis light which has poor tissue penetration.7,8 Recently, the use of near-infrared light with longer wavelength (650–1000 nm) through upconversion, such as two-photon,4,5 upconverting nanoparticles,2 and triplet–triplet annihilation,9 is proposed to improve tissue penetration, but it only increases to millimetre depths. Self-luminescence, including chemiluminescence (CL),10–23 bioluminescence,24–27 and Cerenkov radiation,28 are now deemed to have more potential as tissue-penetration-depth-independent light and has been successfully applied in the highly sensitive bioimaging field.8 However, self-luminescence-driven photoisomerization in vivo remains extremely challenging because such a low-intensity light source insufficiently implements a good photoisomerization effect.8 Thus, developing a self-luminescence source that inherits an ability of external light to specifically select a target site and also provide sufficient intensity is highly desirable for implementing target-specific tissue-depth-independent photoisomerization for in vivo application.As one of frequently used self-luminescence techniques, CL usually results from a CL substrate rapidly and selectively reacting with hydrogen peroxide (H2O2) to produce chemical reaction energy for exciting nearby CL fluorophores.10–23 Since a high H2O2 level is a feature of many target tissues, for example, the higher H2O2 level (ranging from 0.1 to 1 mM) in tumors29–31 than in normal tissues (∼10−4 mM),15,32 it can endow CL with the ability of target-specific initiation. While CL intensity shows rapid recession along with the consumption of H2O2, and consequently very few high-intensity-light-implemented bioapplications (e.g., photodynamic/photothermally therapy and photochemical bond cleavage) using CL as a light source have been developed except for two recent attempts in tumor photodynamic therapy.14,18 One employed direct intratumor injection of a CL source and H2O2 and the other used oral administration of H2O2 enhancer to realize enhanced CL.14,18 However, these CL enhancement examples lacked a strategy to precisely amplify the signal at the target site, which is inadequate for implementing target-specific photoisomerization in vivo.
Herein, we report a precise tissue-depth-independent photoisomerization in vivo by developing a target-specifically initiated and in situ-enhanced CL strategy that solves restrictions of low self-luminescence intensity and lacking a method to precisely amplify the signal at the target site. Furthermore, considering that photoisomerization-controlled drug release is one of crucial embranchment of photoisomerization-related bioapplications, we first demonstrated the applicability of our strategy to construct target-specific tissue-depth-independent photo-controlled drug release systems (CLDRSs) for tumor chemotherapy. Specifically, the CL substrate peroxyoxalate (CPPO) and CL fluorophore (antitumor drug camptothecin, CPT) were used as two building units to construct the CL source. Host–guest nanoparticles (PEAZO-PCD NPs) self-assembled from a photoisomerization molecule, azobenzene (EAZO)-pendant polymer (PEAZO) and a cyclodextrin-pendent polymer (PCD), served as the carrier to encapsulate CPPO and CPT (Scheme 1). In a tumor, the EAZO groups can preliminarily be isomerized by the existing-H2O2-induced CL from the drug CPT, thus triggering partial dissociation of the host–guest carrier and release of the drug CPT. Subsequently, the initially released CPT again functions as a H2O2 enhancer to induce a higher H2O2 level in a tumor. Such a positive-feedback-mechanism-improved the H2O2 level and, in turn, promoted enhanced CL (in situ-enhanced CL) of subsequently accumulative CLDRSs and incompletely dissociative CLDRSs in tumors to realize enhanced EAZO photoisomerization and CPT release. In contrast, a low-level-H2O2-induced weak CL in normal tissues which barely initiated the isomerization, not to mention an enhanced isomerization effect. Such target-specific initiated and self-enhanced CL by a positive feedback mechanism endowed CL with the ability to precisely accomplish EAZO isomerization and drug CPT release. High tumor-inhibition-rate (73%) and no obvious therapy-side-effect in vivo indicated good efficiency and target-specificity of our chemiluminescence-driven photoisomerization. Considering that many diseases have the feature of high level H2O2 and that CPT can easily be replaced by other UV/Vis fluorophores or H2O2 enhancers (e.g., vitamin C), thus, although we only demonstrated one example of target-specific self-luminescence-driven photoisomerization in a bioapplication, namely photoisomerization for drug-controlled release, our work provides guidelines to design various other photoisomerization-related biomedical applications with target-specificity and a depth-independent manner in vivo.
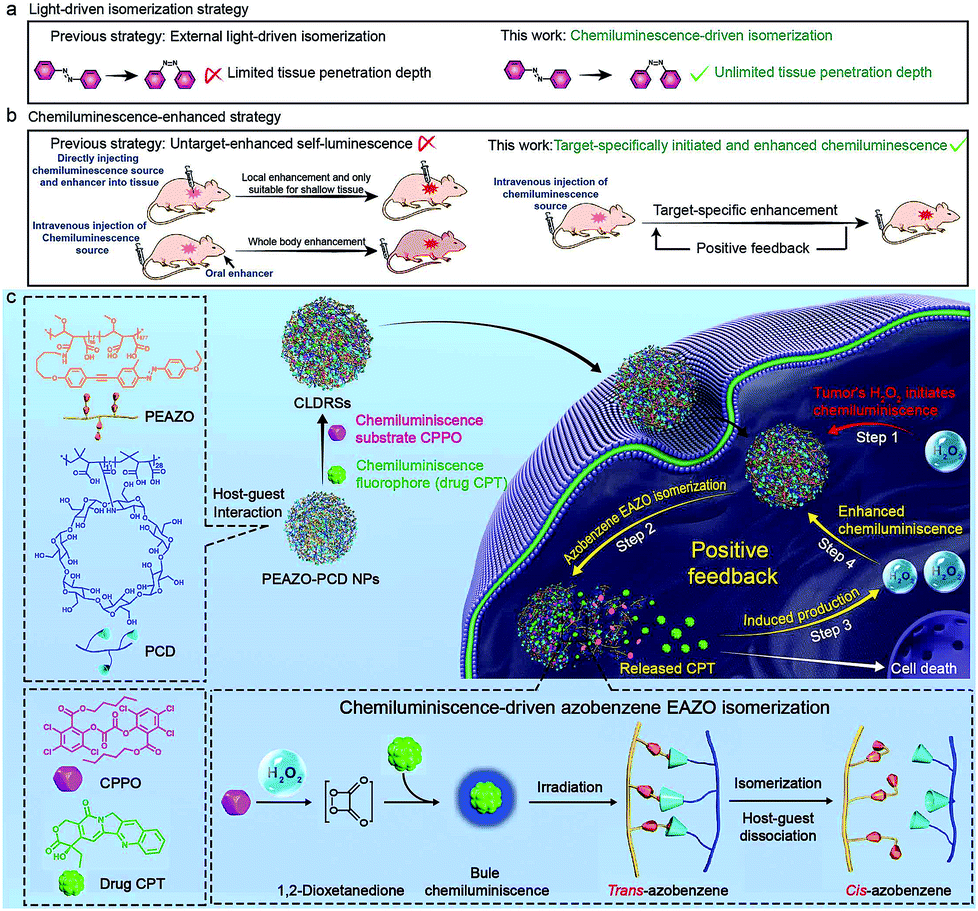 | ||
| Scheme 1 (a and b) Schematic illustration of the differences between previous works and our works. (c) Schematic illustration of the concept of target-specific tissue-depth-independent photoisomerization and one example of this concept bioapplication in the photo-controlled drug release field for tumor chemotherapy. | ||
Results and discussion
EAZO, (4-ethoxy-2′-methyl-4′-(6-hydroxyhexyloxyphenylethynyl)-azobenzene), can be easily isomerized within a few seconds under low-intensity sunlight or ultralow-power external UV-Vis light (even as low as 1 mW cm−2,9,33 far below the currently reported34 minimum light power of ∼10 mW cm−2 for photodynamic therapy). Since CL has been successfully used instead of external light or sunlight for photodynamic therapy14,16–18 and plant photosynthesis,35 we envisioned that this EAZO might also be isomerized by CL irradiation. PCD and PEAZO (see Scheme 1c) were synthesized using an amidation reaction by conjugation of 3A-amino-3A-deoxy-(2AS,3AS)-β-cyclodextrin hydrate (β-CD-NH2) and azobenzene EAZO groups into poly(isobutylene-alt-maleic anhydride) (Mn = 6 kDa) and poly(methyl vinyl ether-alt-maleic anhydride) (Mn = 80 kDa), respectively. The average numbers of EAZO and β-CD-NH2 molecules within each PEAZO and PCD chain were about 11 and 36, respectively (see the ESI†). The stability constant (Kc) of β-CD-NH2 cyclodextrin with the azobenzene EAZO precursor was measured to be 4 × 104 M−1 (Fig. S1†), close to that of cyclodextrin with azobenzene (trans, about 105 M−1) in previous literature reports.36,37 Given that it is much higher than the Kc of cyclodextrin with CPT (266 M−1)3,36,38 and CPPO (1890 M−1) (Fig. S2†), cyclodextrin preferentially forms inclusion complexes with EAZO in our system, which is consistent with previous reports.3,36 Then, water-soluble host–guest nanoparticles (PEAZO-PCD NPs, molar ratio of EAZO/β-CD-NH2 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) were successfully constructed from PEAZO and PCD using a previously reported method.39,40 Furthermore, CLDRS (CPT + CPPO@PEAZO-PCD NPs) were prepared by encapsulating hydrophobic CPT and CPPO into the PEAZO-PCD NPs. The UV-Vis spectrum of the CLDRS exhibited the characteristic absorption peaks of CPT and CPPO (Fig. 1a), indicating their successful loading. The loading capacity of CPPO and CPT (if ever present) were 151 and 186 μg mg−1, respectively (details in the ESI†). Two other nanoparticles (NPs) simply containing CPT (CPT@PEAZO-PCD NPs) and CPPO (CPPO@PEAZO-PCD NPs) were also prepared using the same method as the controls.
:2) were successfully constructed from PEAZO and PCD using a previously reported method.39,40 Furthermore, CLDRS (CPT + CPPO@PEAZO-PCD NPs) were prepared by encapsulating hydrophobic CPT and CPPO into the PEAZO-PCD NPs. The UV-Vis spectrum of the CLDRS exhibited the characteristic absorption peaks of CPT and CPPO (Fig. 1a), indicating their successful loading. The loading capacity of CPPO and CPT (if ever present) were 151 and 186 μg mg−1, respectively (details in the ESI†). Two other nanoparticles (NPs) simply containing CPT (CPT@PEAZO-PCD NPs) and CPPO (CPPO@PEAZO-PCD NPs) were also prepared using the same method as the controls.
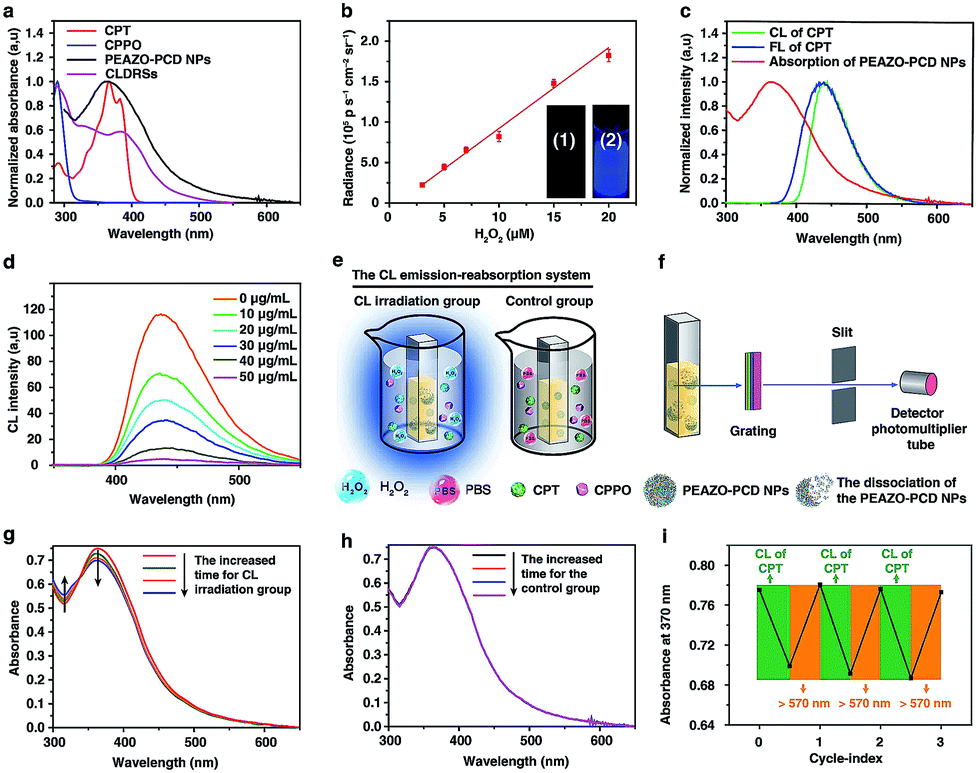 | ||
| Fig. 1 Photoisomerization of the EAZO moieties induced by CL. (a) Normalized absorption spectra of CPT and CPPO in a 1:9 DMF–PBS solution, PEAZO-PCD NPs and CLDRSs in PBS. (b) CL intensities of CPT (5 μg mL−1, 1 mL), CPPO (5 μg mL−1, 1 mL) and PCD (10 μg mL−1, 1 mL) as a function of H2O2 concentration in a 1:9 DMF–PBS solution. The inset is a photograph of the before (1) and after (2) generated CL in the dark, taken at 10 s after addition of H2O2 (0.05 M) with error bars and standard deviation (s.d.) (n = 3 replicates). (c) Normalized absorption of PEAZO-PCD NPs in PBS (pH = 7.4) and fluorescence (FL) and CL spectra of CPT in a 1:9 DMF–PBS solution. (d) The CL emission intensity of CPT which vanishes when the PEAZO-PCD NPs increases. (e) Schematic layout of the emission–reabsorption system. (f) Schematic layout of the change in the absorption spectra of EAZO moieties measurement system with a spectrophotometer containing the photomultiplier tube. Change in the absorption spectra of the PBS solution of PEAZO-PCD NPs (g) with and (h) without the CL irradiation for different times. (i) Multiple rounds of cis–trans isomerization of EAZO with alternating CL of CPT and external yellow light (>570 nm) irradiation in PBS solution. | ||
To investigate properties of CL of CPT in CLDRSs, CPT combined with CPPO and PCD (a CPT + CPPO + PCD mixture) was used in the presence of H2O2 in a 1:9 DMF–PBS solution (pH = 7.4). As expected, a blue CL of CPT was observed (Fig. 1b, inset). The CL intensity was proportional to the H2O2 concentration (Fig. 1b) and ∼900-fold higher as compared to other reactive oxygen species (Fig. S3†), demonstrating that the peroxyoxalate CL system had H2O2-specific luminescence ability. Also, it showed a long half-life of ∼2 hours and its intensity only decreased by three-fifths even after 3 hours (Fig. S4†). Such long half-life and high intensity may be because cyclodextrin slows down the reaction between CPPO and H2O2 and improves the luminous efficiency of CPT,41 which is helpful for providing light energy to trigger EAZO photoisomerization. It also should be beneficial for in vivo applications.13,18
As shown in Fig. 1c, the fluorescence (FL) spectrum of CPT closely resembled its CL spectrum and matched well with the broad and strong absorption band of EAZO moieties in PEAZO-PCD NPs at 300–600 nm, which provides a great possibility for the emission–reabsorption process between CL emission and azobenzene EAZO moieties absorption. The dramatically decreased CL intensity of CPT at 435 nm after adding PEAZO-PCD NPs confirmed that CL of CPT can be efficiently absorbed by EAZO moieties through the emission–reabsorption process (Fig. 1d). Since this EAZO underwent trans–cis photoisomerization upon exposure to light at <570 nm and cis-EAZO returned to trans-forms with light at >570 nm irradiation,42,43 we envisioned that the CL of CPT was suitable for EAZO isomerization. To confirm whether the CL could realize trans–cis isomerization of EAZO, we attempted to provided classical evidence of the change in a trans–cis absorption spectrum of EAZO upon CL irradiation.2,9 However, because of the severe overlap between the absorption spectra of CPT and PEAZO-PCD NPs (Fig. 1a), we could not directly evaluate the change in trans–cis absorption spectrum change of EAZO moieties in our CLDRS. Thus, the emission–reabsorption process using an experimental setup (Fig. 1e) as previously reported9 was applied for this purpose. The CL source (CPT + CPPO + PCD mixture and 2 mM H2O2) were filled in a 1:9 DMF–PBS solution (pH = 7.4) in a beaker (Fig. 1e, left), and the generated CL was used to irradiate a PBS solution of the PEAZO-PCD NPs in an optically transparent cell. The control group (Fig. 1e, right) used the exact same procedure as the CL irradiation group (Fig. 1e, left) except using the same volume of PBS instead of H2O2. The changes in trans–cis absorption spectra of EAZO moieties were then measured with an experimental setup which was analogous to that in Fig. 1f. Upon exposure to the CL of CPT with increasing time, as previously reported,9,43 the characteristic absorbance of the trans-EAZO group at approximately 370 nm obviously decreased, while the peak of the cis-EAZO group at 320 nm was gradually elevated (Fig. 1g). By contrast, no absorption change was observed in the control group (Fig. 1h), indicating that the CL of CPT can cause photoisomerization of the EAZO moieties in the PEAZO-PCD NPs. Additionally, multiple rounds of trans–cis isomerization of EAZO moieties in PEAZO-PCD NPs with alternating CL of CPT and external yellow light (>570 nm) irradiation in PBS solution showed that the CL-irradiation-isomerized cis-EAZO can be recovered to the trans-EAZO (Fig. 1i), further confirming the successful CL-triggered photoisomerization of the EAZO group. Thanks to the closer distance between CPT and EAZO in CLDRSs (CPT + CPPO@PEAZO-PCD NPs), we believe a much better CL emission–reabsorption process will occur, which can acquire more effective photoisomerization of EAZO moieties in CLDRSs.
Fig. 2a shows a classical CL process.12,44 CPPO rapidly and selectively decomposes in the presence of H2O2 to form the high-energy 1,2-dioxetanedione intermediate known to excite the nearby fluorescent drug CPT to emit blue CL. The CL subsequently induces EAZO isomerization to cause the dissociation of EAZO from β-CD-NH2 cyclodextrin, further triggering the dissociation of CLDRSs and CPT release. To verify the feasibility of CL-driven azobenzene EAZO photoisomerization for CPT release, we first studied the time-dependent stability of the NPs (CPT@PEAZO-PCD NPs, CPPO@PEAZO-PCD NPs and CLDRSs) through DLS analysis and a dialysis experiment (Fig. S5–S8†). These NPs were stable after storage in the dark for 24 hours in PBS and FBS because the supramolecular polymer PEAZO-PCD has a higher stability constant compared with the monovalent complex.45 Then, we added H2O2 into CLDRSs to induce EAZO isomerization for a drug release study. The CLDRSs (CPT + CPPO@PEAZO-PCD NPs) collapsed into irregular aggregates and their hydrodynamic size changed from predominantly 70 nm to a random distribution from H2O2-induced CL irradiation (Fig. 2b). Comparatively, PEAZO-PCD NPs, CPT@PEAZO-PCD NPs, and CPPO@PEAZO-PCD NPs showed little changed sizes because they cannot generate an effective CL in the presence of H2O2 to isomerize EAZO (Fig. 2b). These results demonstrated that the CL effectively drove EAZO isomerization in CLDRSs and thus successfully dissociated the CLDRS. Drug release ability was further studied by dialysis of those NPs in PBS (pH = 7.4) under H2O2-induced CL irradiation. We first added H2O2 into the CPT@PEAZO-PCD NPs. Drug release amounts showed no obvious differences whether there was H2O2 or not (Fig. 2c), indicating that H2O2 itself did not result in drug CPT release without the CL substrate CPPO. For our CLDRSs (CPT + CPPO@PEAZO-PCD NPs), without adding H2O2, only a relatively small amount of CPT (approximately 10 wt%) was released within 24 h (Fig. 2d). By contrast, significant amounts of CPT were released from CLDRSs, reaching 35 wt% release in 24 h in the presence of H2O2 (0.5 mM)-induced CL irritation and even 72 wt% release in the presence of H2O2 (1 mM)-induced CL irritation (Fig. 2d). The 17 wt% release can still be obtained when irradiated by H2O2 (0.05 mM)-induced CL. However, the drug release was ignorable using H2O2 (10−4 mM)-induced CL irradiation. These results clearly showed that the drug release was caused by CL irradiation and the released amounts are highly correlated with H2O2-induced CL intensity. Considering the H2O2 concentration in a tumor (concentrations ranging from 0.1 mM to 1 mM (ref. 29)) was higher than in normal tissues (∼10−4 μM),32 it may trigger in the tumor environment-specific dissociation of the CLDRS. Interestingly, two hours later following the first H2O2 (0.5 mM)-induced CL irradiation, an additional 15 wt% CPT was released upon a secondary 0.5 mM H2O2 (Fig. 2e). Even employing 0.05 mM H2O2 can produce an additional 6 wt% CPT release (Fig. 2e). All these results (Fig. 1b, 2d and e) indicated that maintaining a high concentration of H2O2 can effectively achieve high CL-intensity to further enhance azobenzene EAZO photoisomerization, leading to increased drug release.
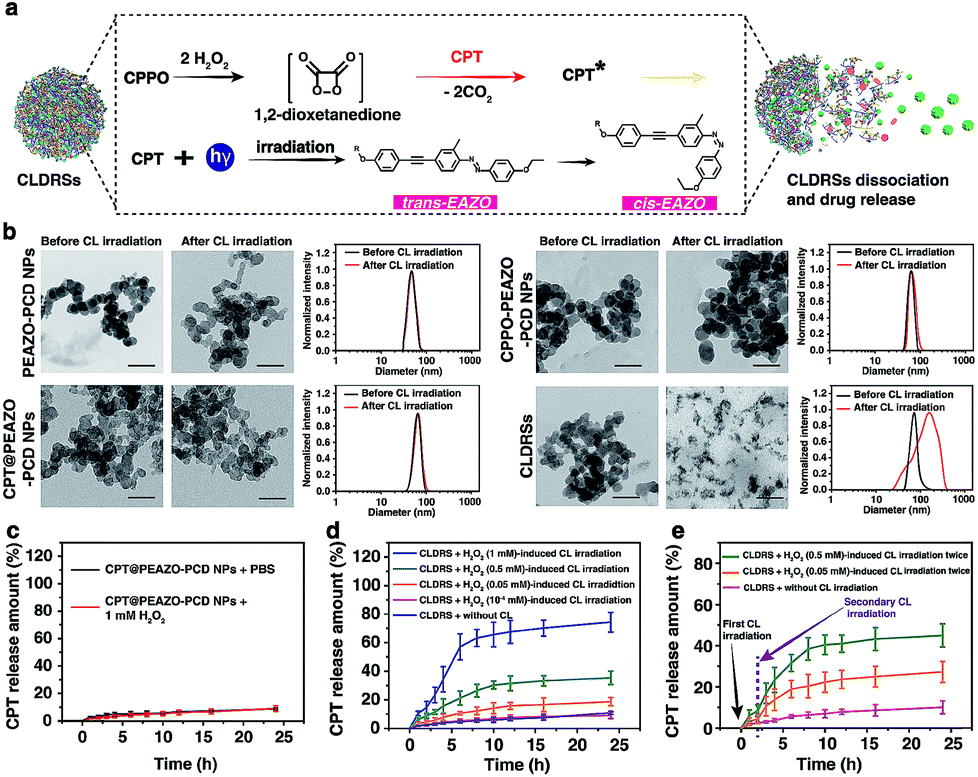 | ||
| Fig. 2 CL-driven photoisomerization for in vitro CPT-controlled release from CLDRSs. (a) Illustration of CL-triggered drug release mechanism of CLDRSs. (b) Changes in TEM and DLS of PEAZO-PCD NPs, CPT@PEAZO-PCD NPs, CPPO@PEAZO-PCD NPs, and CLDRSs after CL of CPT irradiation (CL induced by 2 mM H2O2). Scale bar, 100 nm. (c) Drug release curves of CPT@PEAZO-PCD NPs under H2O2 (1 mM) treatment. (d) Drug release curves of CLDRSs under different intensity of CL of CPT irradiation. (e) Drug release curves of CLDRSs under CL of CPT irradiation. Secondary CL irradiation indicates addition of 0.05 mM H2O2 or 0.5 mM H2O2 to enhance CL at the 16th hour. Error bars indicate the s.d. (n = 3). | ||
CPT is an antitumor drug with the ability to induce a H2O2-rich microenvironment in cells.46,47 To verify that the CPT released in our CLDRS could induce H2O2 production in tumor cells, we first performed time-dependent measurements of the H2O2 level in 4T1 breast tumor cells and NIH/3T3 fibroblast cells (the control) in vitro with a H2O2 Assay Kit using 2′,7′-dichlorofluorescin diacetate (DCFH-DA, producing green fluorescence after oxidization by H2O2). 4T1 tumor cells and 3T3 normal cells were cultured with DCFH-DA for 30 min, and then confocal fluorescence images were recorded over time after adding free CPT, CPT@PEAZO-PCD NPs, CPPO@PEAZO-PCD NPs, and the CLDRSs (Fig. 3a). For 4T1 tumor cells, the relative fluorescence intensity (F/F0, where F0 indicates the fluorescence intensity at 0 min and F is the fluorescence intensity at different time points) for the free CPT group dramatically increased from 1.00 ± 0.11 at 0 min to 9.85 ± 1.13 at 240 min, testifying that CPT in 4T1 cells can cause significant H2O2 production (Fig. 3b). The PEAZO-PCD NP group showed no influence on the H2O2 level, while the CPPO@PEAZO-PCD NP group exhibited a rapid decrease because of H2O2 consumption by CPPO. Thus, we can evaluate the release feasibility of drug CPT in our CLDRS through the F/F0 increase. The negligible F/F0 increase in the CPT@PEAZO-PCD NP group indicated CPT was rarely released into the tumor cells without CL substrate CPPO. Intriguingly, the F/F0 for the CLDRS (CPT + CPPO@PEAZO-PCD NP) group showed an obvious increase from 1.00 ± 0.11 at 0 min to 3.96 ± 0.74 at 240 min (Fig. 3b), demonstrating the effective release of CPT in 4T1 cells by their H2O2-initiated CL irradiation. Noticeably, this enhanced F/F0 value (∼3.96 fold) was lower than that of the free CPT group (∼11 fold, Fig. 3b) mainly due to the simultaneous consumption of H2O2 by CPPO in this system. Our results clearly manifested that the CPT release from this CLDRS can be triggered under CL initiation by the pristine H2O2 level in tumor cells and the released CPT can provide an elevated H2O2 level for furthering CL enhancement and the resultant self-amplified release. To investigate tumor-specificity of the CLDRS, 3T3 normal cells were used for comparison. We observed no obvious F/F0 increase in normal cells for CPT@PEAZO-PCD NP, CPPO@PEAZO-PCD NP, and CPT + CPPO@PEAZO-PCD NP groups. These results illustrated that the relatively low H2O2 level in normal cells can hardly produce sufficient CL to isomerize the azobenzene group EAZO and trigger the dissociation of CLDRSs, consistent with CLDRS behavior under similar H2O2 concentrations in vitro (Fig. 2d). It is noted that free CPT can also induce H2O2 production in normal 3T3 cells (Fig. 2a). Fluorescence of the H2O2 probe DCFH-DA in free-CPT-treated normal 3T3 cells was obviously lower than that of free-CPT-treated tumor cells at 240 min probably because the initial concentration of H2O2 was obviously lower than that in the tumor cells. Thus, our CLDRS can promote H2O2 production only in tumor cells and is inoperative in normal cells, which is beneficial to achieve enhanced CL with excellent tumor specificity.
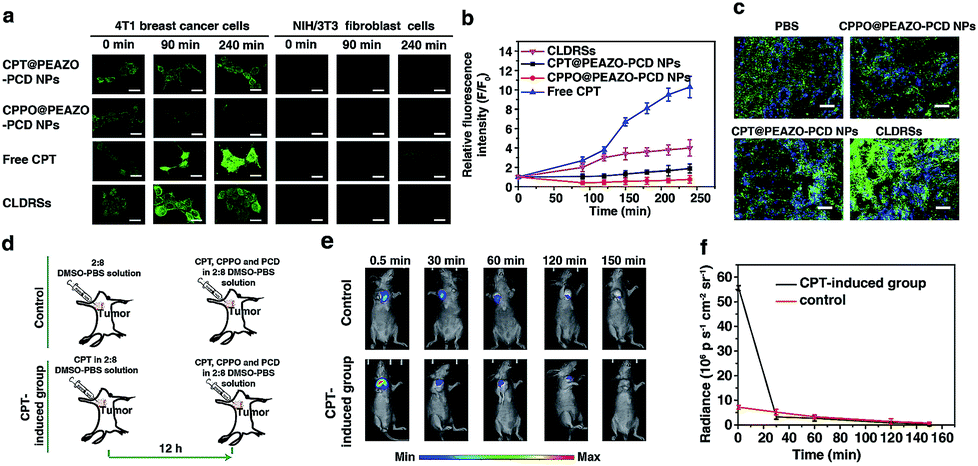 | ||
| Fig. 3 CPT-induced H2O2 production in tumor cells and tissues to achieve self-enhanced CL. (a) Time-dependent measurements of the H2O2 level using H2O2 probe DCFH-DA (10 μM) in 4T1 tumor cells and 3T3 normal cells with different treatments. Scale bar, 30 μm. (b) Quantitatively relative fluorescence intensity of (a) for 4T1 tumor cells. (c) Ex vivo H2O2 generation intravenous injection of various interventions in tumor measured using DCFH-DA (green) and Hoechst 33342 (blue) staining in tumor sections. Scale bar, 10 μm. (d) Schematic illustration of a mouse bearing a 4T1 tumor treated with CPT for CL imaging. (e) CL images of CPT induction group and control. (f) Quantification of CL intensity for the in vivo images in (e). Error bars indicate the s.d. (n = 3). | ||
To further prove the CPT-promoted H2O2 production in vivo, DCFH-DA staining in tumor sections was performed to study the H2O2 level induced by the released drug CPT. Various NPs were tail-vein injected into mice bearing 4T1 xenograft tumors using PBS as a control. After 12 h, equal amounts of a DCHF-DA working solution were intratumorally injected and then 10 μm cryo-sections of the tumor tissues were prepared and incubated with Hoechst 33342 for nuclei staining. Similar to findings in the cell experiment, the fluorescence intensity of the H2O2 probe DCFH-DA in the CLDRS group was significantly stronger than that in the control group, indicating that CPT was successfully released from our CLDRS and effectively induced H2O2 production in tumors (Fig. 3c).
Furthermore, to investigate whether drug CPT-induced H2O2 production can achieve enhanced CL, we explored the CL intensity and duration in the presence of CPT in tumors. CPT (5 μg mL−1) in a 1:9 DMSO–PBS solution as a H2O2 induction group and the 1:9 DMSO–PBS solution as control were intratumorally injected into mice bearing 4T1 tumors. After 12 h, equal amounts of a CPT + CPPO + PCD solution were intratumorally injected and both groups were visualized under CL images (Fig. 3d). The CL intensity of the CPT-inducted group was about 10 times more than the control group at 0.5 min (Fig. 3e and f) and obviously stronger than the control group within 30 min, demonstrating that CPT efficiently induced the significantly enhanced CL. Within 30 min, a higher H2O2 level of the CPT-inducted group consumed more quantitative CPPO than the control group. However, with the same dosage of CL substrate CPPO at the initial status, the CL intensity of the CPT-inducted group was lower than the control after 30 min. Thus, the observed production of high H2O2 concentrations in tumor cells and tissues by CPT successfully induced its CL enhancement, allowing us to achieve amplified azobenzene EAZO isomerization and construct an amplified drug release system for efficient drug release in tumors.
Next, the MTT assay was used to determine in vitro cytotoxicity of the CLDRSs. The drug carrier PEAZO-PCD NPs at concentrations up to 500 μg mL−1 showed no obvious cytotoxicity in normal NIH 3T3 cells and tumor 4T1, U-87 MG, as well as B16F10 cells (Fig. S9†). Just as Fig. 4a illustrated the mechanism of positive-feedback in cells, the CLDRSs indeed exhibited a marked cytotoxicity to tumor 4T1, U-87 MG, and B16F10 cells (Fig. 4b). The half-maximal inhibitory concentration (IC50) of CLDRSs was about 35 μg mL−1 for tumor 4T1, U-87 MG, and B16F10 cells (Fig. 4b), indicating its good treatment effect. By contrast, the CPT@PEAZO-PCD NPs did not result in a significant decrease in cell viability for 4T1, U-87 MG, or B16F10 cells, indicating a negligible release of CPT in the absence of the CL substrate CPPO (Fig. 4c). Additionally, CPPO@PEAZO-PCD NPs also showed no obvious cytotoxicity for 4T1, U-87 MG, and B16F10 cells (Fig. 4c). These results indicated that EAZO can be isomerized only in CLDRSs for effective drug release. Interestingly, the same experiment performed in normal 3T3 cells with CLDRSs showed no significant decrease in cell viability because low-level-H2O2-induced weak CL in normal cells was not enough to drive EAZO isomerization for effective drug release (Fig. 4b and c). Additionally, a H2O2-suppressed experiment was performed. Cells were incubated with CLDRSs. After 2 hours, the cells were treated with glutathione (GSH), an antioxidant and nucleophilic scavenger of H2O2 (Fig. 4d),13 the cell viability returned to a high value for tumor 4T1, U-87 MG, and B16F10 cells (Fig. 4e), indicating that the scavenging of H2O2 in cells by GSH can weaken the effect of CL-driven azobenzene EAZO isomerization and inhibit drug release from CLDRSs. All these results demonstrated that the CLDRSs possess the potential to achieve tumor-specific EAZO isomerization for tumor-precise therapy in vivo, maximizing therapy effects with reduced side effects. The chemotherapy efficacy of CLDRSs compared with chemotherapy with external light irradiation was also evaluated in vitro (Fig. 4f). We used CPT@PAZO-PCD NPs containing the same concentration of CPT with CLDRSs to culture tumor 4T1, U-87, and B16F10 cells. After irradiation at 436 nm with different light doses from 0.0 to 0.10 J cm−2, chemotherapy of CLDRSs with CL irradiation strategy yielded similar efficacy compared to that with ∼0.08 J cm−2 external light irradiation in tumor 4T1, U-87, and B16F10 cells (Fig. 4b, f and g). Additionally, a previous study indicated that PDT with a bioluminescence irradiation strategy yielded similar efficacy compared to that with 0.6–0.8 J cm−2 external light irradiation.24 Our lower light dose indicated that the CL-driven isomerization strategy may be extended to bioluminescence-driven isomerization.
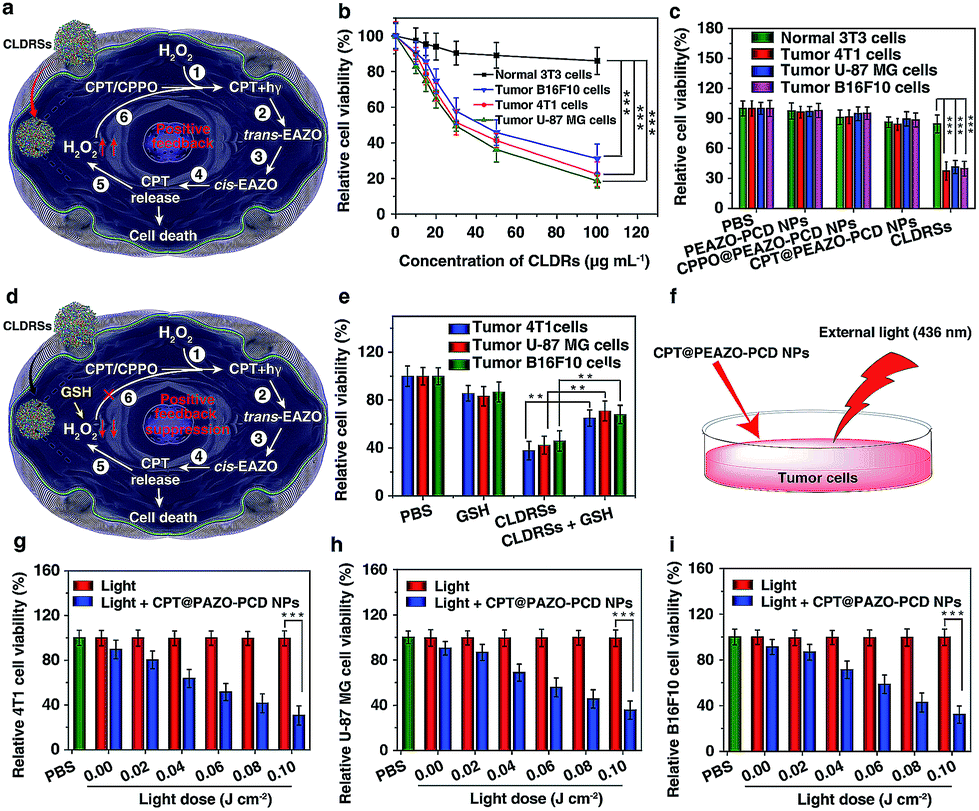 | ||
| Fig. 4 CL-driven photoisomerization for CPT release in in vitro antitumor effect. (a) Schematic illustration of positive-feedback-promoted H2O2 production mechanism and amplified isomerization mechanism of EAZO in cells. (b) Cytotoxicity of normal 3T3 cells as well as tumor 4T1, U-87 MG, and B16F10 cells after 24 h incubation with CLDRSs with different concentrations. ***P < 0.001. (c) Cell viability of normal 3T3 cells as well as tumor 4T1, U-87 MG, and B16F10 cells with various treatments (CLDRSs, 50 μg mL−1; equal 9.3 μg mL−1 of CPT). ***P < 0.001. (d) Schematic illustration of H2O2-suppressed mechanism in cells by adding GSH. Cells were incubated with CLDRSs. After 2 hours, the cells were washed twice with PBS and GSH (10 μM) added. (e) Cytotoxicity of CLDRSs (50 μg mL−1; equal 9.3 μg mL−1 of CPT) treatment with and without GSH. **P < 0.01. (f) Schematic illustration of the chemotherapy efficacy of CLDRSs compared with chemotherapy with external light irradiation. Cell viability of CPT@PEAZO-PCD NPs (equal 9.3 μg mL−1 of CPT) with different light dose (436 nm) in tumor (g) 4T1, (h) U-87 MG, and (i) B16F10 cells. ***P < 0.001. Error bars indicate the s.d. (n = 3). | ||
For an in vivo application, we first evaluated the blood retention time of CLDRSs. Due to its lack of imaging signals, a near infrared fluorescence dye, IR825, was encapsulated to form IR825-loaded CLDRS (IR-825 + CPT + CPPO@PEAZO-PCD NPs) for this study (Fig. S10†). Because of the similar size, morphology, and stability of IR825-loaded CLDRS with CLDRSs (CPT + CPPO@PEAZO-PCD NPs) by DLS and TEM observations (Fig. 2b and S11†), IR825-loaded CLDRSs were suitable to modulate our CLDRS for in vivo investigations. The blood concentration of IR825-loaded CLDRSs declined to half that level at 8 h after injection (Fig. S12†), showing its long blood retention time. Upon tail-vein injection, the tumor fluorescence signals increased gradually over time and reached a peak value at 5 h after injection, suggesting that IR825-loaded CLDRSs were able to accumulate in tumors (Fig. S13 and S14†). Furthermore, to examine the amount of IR825-loaded CLDRSs in a tumor and other organs, the tumors and major organs of the 4T1 tumor-bearing mice after intravenous injection at 5 h were then taken for ex vivo imaging (Fig. S15†). Integrated fluorescence intensity of both the tumor and liver were obviously much stronger than the heart, spleen, lung, and kidney (Fig. S16†). The tumor and liver fluorescence ratio reached up to ∼0.65, demonstrating the high efficiency of tumor accumulation of the CLDRSs via an enhanced permeability and retention effect. The high tumor accumulation of the CLDRSs has great potential for improving therapeutic effects.
To evaluate the therapeutic efficacy in vivo, 4T1 tumor-bearing mice were injected via tail vein 1 time every 2 days with 200 μL of PEAZO-PCD NPs, CPPO@PEAZO-PCD NPs, CPT@PEAZO-PCD NPs, and CLDRSs at a CPT-equivalent dose of 25 mg kg−1 with PBS as a control (Fig. 5a). The tumor volumes and body weights of the tumor-bearing mice were measured every 2 days from 0 to 14 days post injection. After 14 days of treatment, body weights of tumor-bearing mice increased gradually with time (Fig. 5b). Tumor volumes in the mice treated with CLDRSs were much smaller than those in the mice treated with other groups (Fig. 5c and d). Compared with the PBS group, the tumor inhibitory rate (calculated from the tumor volume) in the CLDRS group (73%) is much higher than those in the PEAZO-PCD NP (4%), CPPO@PEAZO-PCD NP (8%), and CPT@PEAZO-PCD NP (21%) groups (Fig. 5e). These results demonstrated that CL effectively drove azobenzene group EAZO photoisomerization to implement drug-controlled release for excellent therapeutic efficacy against tumors.
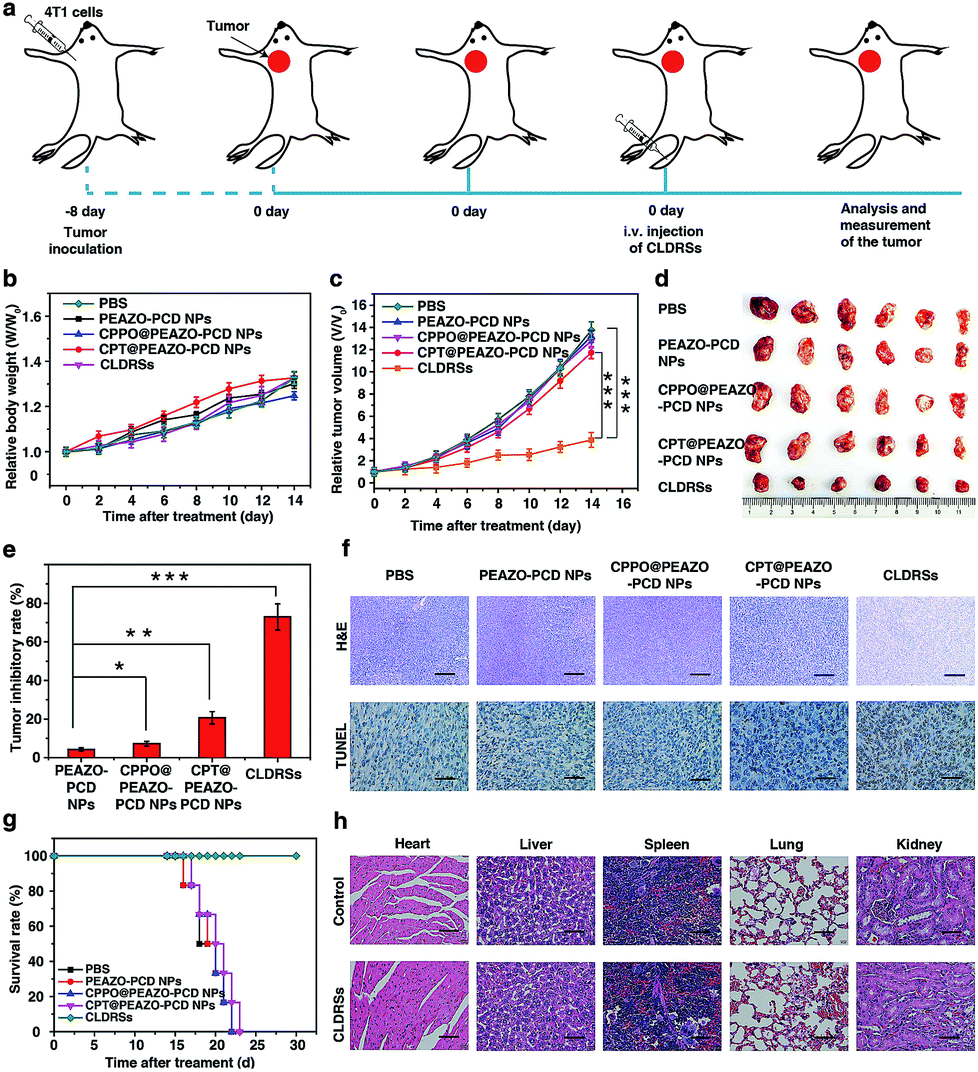 | ||
| Fig. 5 (a) Schematic illustration of CLDRSs therapy to inhibit tumors. (b) Relative body weight and (c) relative tumor volume growth curves. (d) Representative images of tumor tissues after various treatments, respectively. (e) Tumor-inhibitory-rate after various treatments. Error bars indicate standard deviations of 6 separate measurements. Statistical significance: *P < 0.05; **P < 0.01; ***P < 0.001. (f) Tumors were sectioned and stained with H&E and TUNEL. Scale bar, 100 μm. (g) Survival rates of tumor-bearing mice with different treatments within 30 days. (h) H&E stained images of different organs' tissue sections at 30 days after CLDRSs treatment and the healthy mice as control. Scale bar, 30 μm. | ||
To further evaluate the antitumor efficacy of treatments with various NPs, immunohistochemical analysis was performed. In the CLDRS-treated groups, tumor cells displayed nuclear shrinkage and fragmentation and, most importantly, a large necrotic area (Fig. 5f). Conversely, obvious malignant necrosis was not observed in the other groups. Similar results were obtained by TdT-mediated dUTP Nick-End Labelling (TUNEL) analysis (Fig. 5f). Therefore, both H&E and TUNEL analysis further confirmed the excellent antitumor efficacy of the CLDRS in vivo.
Potential in vivo side effects of the CLDRSs were further evaluated. After 14 days of treatment, body weights of tumor-bearing mice increased gradually with time, illustrating almost negligible side effects of CLDRSs for tumor therapy (Fig. 5b). After 30 days of treatment, the survival rate of mice treated with the CLDRSs remained at 100%, which was significantly improved compared with the 0% survival rate in other groups (Fig. 5g). Next, long-term damage to major organs was also assessed. Due to the self-repairing damage capability of organs, at 30 days after treatment, the major organ slices stained with H&E exhibited no noticeable signs of organ damage (Fig. 5h). These results confirmed the high selectivity for tumors and minimal off-target damage from the CLDRSs used for in vivo therapy. Such no obvious side-effects also proves that enhanced CL effectively achieved effective azobenzene photoisomerization at precise target sites.
Conclusions
In summary, we have demonstrated the concept of target-specific self-luminescence-driven photoisomerization in vivo by developing a target-specific initiated and in situ-enhanced CL strategy that solves restrictions of lacking a target-specific high-intensity self-luminescence. Furthermore, we first demonstrated the applicability of our strategy to construct target-specific self-luminescence-controlled drug release systems for tumor chemotherapy. Our target-specific enhanced mechanism not only conveniently enhanced the CL to provide sufficient energy, but also tactfully ensured target-specific enhancement. As we know, target-specific photoisomerization with external photoexcitation completely relies on a selective illumination region with artificial consciousness. It is difficult to ensure an accurate position in practical applications because most target tissues (e.g., tumor) have small areas and irregular shapes. In comparison, our target-specific initiated and enhanced CL had abilities of self-selected target tissue regions through a high H2O2 level in diseased tissues.Although we only demonstrated one example of a photoisomerization-related bioapplication, namely CL-controlled drug chemotherapy, our work of CL-driven isomerization provides a common guideline to design various target-specific depth-independent photoisomerization in bioapplications. In this CL-driven isomerization system, its CL wavelength was determined by the fluorescence of the fluorophore itself and the light wavelength of trans–cis photoisomerization of EAZO was within a fairly large wavelength range (<570 nm).42,43 Thus, we can easily select applicable fluorophores as needed to replace fluorophore CPT. Additionally, for a CL enhancer, some non-drug enhancers (e.g., D-amino acid oxidase48 or vitamin C and its derivatives49) also can be linked with a disease-microenvironment-specific response group to induce H2O2 production only in target tissue to achieve target-specific enhanced CL for other non-treatment applications. Considering that many diseases, such as inflammation, neurodegenerative diseases, and chronic obstructive pulmonary diseases,13 are closely associated with a high H2O2 level, our CL-driven isomerization strategy may therefore be applied to these diseases.
Moreover, we can also use other self-luminescence to isomerize this azobenzene due to their similar light energy levels, thus providing new advantages for photoisomerization-related studies. For instance, we can use genetic engineering to accomplish target-specific bioluminescence for this azobenzene isomerization in vivo. Furthermore, our proposed target-specific initiated an in situ-enhanced self-luminescence method by a positive feedback mechanism which provided a robust and versatile approach to implement target-specific enhanced self-luminescence, which will greatly broaden the application of self-luminescence. In future work, our self-luminescence-driven isomerization strategy may be generalized to explore photo-switchable biomolecules for a variety of in vivo applications, such as those used in artificial intelligence, optogenetics, and even the photo-controlling every aspect of a cell's inner workings, in a depth-independent manner with high target specificity, thus prospectively overcoming the Achilles' heel of photoisomerization.
Ethical statement
All animal experiments of this study were performed at the Experimental Animal Center of Simcere Pharmaceutical Group in full compliance with the guidelines approved by Jiangsu Administration of Experimental Animals. Approximately 3 × 106 4T1 cells in 200 μL of PBS were inoculated in 5 week-old 4T1 female Balb/c nude mice by subcutaneous injection into their right limb armpits.All tumor bearing nude mice were purchased from Jiangsu KeyGEN BioTECH Corp., Ltd. and used according to the guidelines of the Laboratory Animal Center of Jiangsu KeyGEN BioTECH Corp., Ltd.
Author contributions
Y. T. and Q. F. conceived, supervised, and designed the study; Y. T. and X. L. performed experiments. Other people analyzed data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21674048 and 21574064), the Jiangsu Province “333 high-level Personnel Training project”, the Primary Research & Development Plan of Jiangsu Province (No. BE2016770), and the Natural Science Foundation of Jiangsu Province of China (No. BK20160884).Notes and references
- W. A. Velema, J. P. van der Berg, M. J. Hansen, W. Szymanski, A. J. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924 CrossRef CAS PubMed.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., Int. Ed., 2013, 52, 4375 CrossRef CAS PubMed.
- J. Croissant, A. Chaix, O. Mongin, M. Wang, S. Clement, L. Raehm, J. O. Durand, V. Hugues, M. Blanchard-Desce, M. Maynadier, A. Gallud, M. Gary-Bobo, M. Garcia, J. Lu, F. Tamanoi, D. P. Ferris, D. Tarn and J. I. Zink, Small, 2014, 10, 1752 CrossRef CAS PubMed.
- J. Croissant, M. Maynadier, A. Gallud, H. Peindy N'dongo, J. L. Nyalosaso, G. Derrien, C. Charnay, J. O. Durand, L. Raehm, F. Serein-Spirau, N. Cheminet, T. Jarrosson, O. Mongin, M. Blanchard-Desce, M. Gary-Bobo, M. Garcia, J. Lu, F. Tamanoi, D. Tarn, T. M. Guardado-Alvarez and J. I. Zink, Angew. Chem., Int. Ed., 2013, 52, 13813 CrossRef CAS PubMed.
- M. Izquierdo-Serra, M. Gascon-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, E. Ferrer, R. Alibes, F. Busque, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178 CrossRef CAS PubMed.
- W. Szymanski, J. M. Beierle, H. A. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114 CrossRef CAS PubMed.
- M. Karimi, P. Sahandi Zangabad, S. Baghaee-Ravari, M. Ghazadeh, H. Mirshekari and M. R. Hamblin, J. Am. Chem. Soc., 2017, 139, 4584 CrossRef CAS PubMed.
- Z. Jiang, M. Xu, F. Li and Y. Yu, J. Am. Chem. Soc., 2013, 135, 16446 CrossRef CAS PubMed.
- O. Green, S. Gnaim, R. Blau, A. Eldar-Boock, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2017, 139, 13243 CrossRef CAS PubMed.
- D. Lee, S. Khaja, J. C. Velasquez-Castano, M. Dasari, C. Sun, J. Petros, W. R. Taylor and N. Murthy, Nat. Mater., 2007, 6, 765–769 CrossRef CAS PubMed.
- A. J. Shuhendler, K. Pu, L. Cui, J. P. Uetrecht and J. Rao, Nat. Biotechnol., 2014, 32, 373 CrossRef CAS PubMed.
- X. Zhen, C. Zhang, C. Xie, Q. Miao, K. L. Lim and K. Pu, ACS Nano, 2016, 10, 6400 CrossRef CAS PubMed.
- H. Yuan, H. Chong, B. Wang, C. Zhu, L. Liu, Q. Yang, F. Lv and S. Wang, J. Am. Chem. Soc., 2012, 134, 13184 CrossRef CAS PubMed.
- Y.-D. Lee, C.-K. Lim, A. Singh, J. Koh, J. Kim, I. C. Kwon and S. Kim, ACS Nano, 2012, 6, 6759 CrossRef CAS PubMed.
- Y. Zhang, L. Pang, C. Ma, Q. Tu, R. Zhang, E. Saeed, A. E. Mahmoud and J. Wang, Anal. Chem., 2014, 86, 3092–3099 CrossRef CAS PubMed.
- H. Yuan, H. Bai, L. Liu, F. Lv and S. Wang, Chem. Commun., 2015, 51, 722 RSC.
- D. Mao, W. B. Wu, S. L. Ji, C. Chen, F. Hu, D. L. Kong, D. Ding and B. Liu, Chem, 2017, 3, 991 CAS.
- E. S. Lee, V. G. Deepagan, D. G. You, J. Jeon, G. R. Yi, J. Y. Lee, D. S. Lee, Y. D. Suh and J. H. Park, Chem. Commun., 2016, 52, 4132 RSC.
- L. Zhang, N. He and C. Lu, Anal. Chem., 2015, 87, 1351 CrossRef CAS PubMed.
- C.-K. Lim, Y.-D. Lee, J. Na, J. M. Oh, S. Her, K. Kim, K. Choi, S. Kim and I. C. Kwon, Adv. Funct. Mater., 2010, 20, 2644 CrossRef CAS.
- M. Iranifam, TrAC, Trends Anal. Chem., 2014, 59, 156 CrossRef CAS.
- N. Hananya, A. Eldar Boock, C. R. Bauer, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2016, 138, 13438 CrossRef CAS PubMed.
- C. Y. Hsu, C. W. Chen, H. P. Yu, Y. F. Lin and P. S. Lai, Biomaterials, 2013, 34, 1204 CrossRef CAS PubMed.
- T. Kuchimaru, T. Suka, K. Hirota, T. Kadonosono and S. Kizaka-Kondoh, Sci. Rep., 2016, 6, 34311 CrossRef CAS PubMed.
- L. Xiong, A. J. Shuhendler and J. Rao, Nat. Commun., 2012, 3, 1193 CrossRef PubMed.
- Y. Yang, W. Hou, S. Liu, K. Sun, M. Li and C. Wu, Biomacromolecules, 2018, 19, 201 CrossRef CAS PubMed.
- N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370 CrossRef CAS PubMed.
- J. Kim, H. R. Cho, H. Jeon, D. Kim, C. Song, N. Lee, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2017, 139, 10992 CrossRef CAS PubMed.
- B. Halliwell, M. V. Clement and L. H. Long, FEBS Lett., 2000, 486, 10 CrossRef CAS PubMed.
- T. P. Szatrowski and C. F. Nathan, Cancer Res., 1991, 51, 794 CAS.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722 CrossRef CAS PubMed.
- K. Okano, A. Shishido and T. Ikeda, Macromolecules, 2006, 39, 145 CrossRef CAS.
- L. Huang, Z. Li, Y. Zhao, Y. Zhang, S. Wu, J. Zhao and G. Han, J. Am. Chem. Soc., 2016, 138, 14586 CrossRef CAS PubMed.
- H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Commun., 2013, 49, 10685 RSC.
- T. Sun, Q. Wang, Y. Bi, X. Chen, L. Liu, C. Ruan, Z. Zhao and C. Jiang, J. Mater. Chem. B, 2017, 5, 2644 RSC.
- B. L. May, J. Gerber, P. Clements, M. A. Buntine, D. R. Brittain, S. F. Lincoln and C. J. Easton, Org. Biomol. Chem., 2005, 3, 1481 RSC.
- J. Kang, V. Kumar, D. Yang, P. R. Chowdhury and R. J. Hohl, Eur. J. Pharm. Sci., 2002, 15, 163 CrossRef CAS PubMed.
- N. Li, Y. Chen, Y. M. Zhang, Y. Yang, Y. Su, J. T. Chen and Y. Liu, Sci. Rep., 2014, 4, 4164 CrossRef PubMed.
- J. H. Lee, K. J. Chen, S. H. Noh, M. A. Garcia, H. Wang, W. Y. Lin, H. Jeong, B. J. Kong, D. B. Stout, J. Cheon and H. R. Tseng, Angew. Chem., Int. Ed., 2013, 52, 4384 CrossRef CAS PubMed.
- E. J. Woolf and M. L. Grayeski, J. Lumin., 1987, 39, 19–27 CrossRef CAS.
- F. Cheng, Y. Zhang, R. Yin and Y. Yu, J. Mater. Chem., 2010, 20, 4888 RSC.
- R. Yin, W. Xu, M. Kondo, C.-C. Yen, J.-i. Mamiya, T. Ikeda and Y. Yu, J. Mater. Chem., 2009, 19, 3141 RSC.
- R. Bos, S. A. Tonkin, G. R. Hanson, C. M. Hindson, K. F. Lim and N. W. Barnett, J. Am. Chem. Soc., 2009, 131, 2770 CrossRef CAS PubMed.
- A. L. Nielsen, K. Steffensen and K. L. Larsen, Colloids Surf., B, 2009, 73, 267 CrossRef CAS PubMed.
- T. Ozben, J. Pharm. Sci., 2007, 96, 2181 CrossRef CAS PubMed.
- S. Simizu, M. Takada, K. Umezawa and M. Imoto, J. Biol. Chem., 1998, 273, 26900 CrossRef CAS PubMed.
- J. Fang, D. Deng, H. Nakamura, T. Akuta, H. Qin, A. K. Iyer, K. Greish and H. Maeda, Int. J. Cancer, 2008, 122, 1135 CrossRef CAS PubMed.
- I. D. Podmore, H. R. Griffiths, K. E. Herbert, N. Mistry, P. Mistry and J. Lunec, Nature, 1998, 392, 559 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04012e |
| This journal is © The Royal Society of Chemistry 2019 |