
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the electronic properties of hexanuclear cobalt sulfide superatoms via ligand substitution†
Gaoxiang
Liu
 a,
Andrew
Pinkard
b,
Sandra M.
Ciborowski
a,
Vikas
Chauhan
c,
Zhaoguo
Zhu
a,
Alexander P.
Aydt
b,
Shiv N.
Khanna
*c,
Xavier
Roy
*b and
Kit H.
Bowen
*a
a,
Andrew
Pinkard
b,
Sandra M.
Ciborowski
a,
Vikas
Chauhan
c,
Zhaoguo
Zhu
a,
Alexander P.
Aydt
b,
Shiv N.
Khanna
*c,
Xavier
Roy
*b and
Kit H.
Bowen
*a
aDepartment of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA. E-mail: kbowen@jhu.edu
bDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: xr2114@columbia.edu
cDepartment of Physics, Virginia Commonwealth University, 701 W. Grace St., Richmond, Virginia 23284, USA. E-mail: snkhanna@vcu.edu
First published on 3rd December 2018
Abstract
Molecular clusters are attractive superatomic building blocks for creating materials with tailored properties due to their unique combination of atomic precision, tunability and functionality. The ligands passivating these superatomic clusters offer an exciting opportunity to control their electronic properties while preserving their closed shells and electron counts, which is not achievable in conventional atoms. Here we demonstrate this concept by measuring the anion photoelectron spectra of a series of hexanuclear cobalt sulfide superatomic clusters with different ratios of electron-donating and electron-withdrawing ligands, Co6S8(PEt3)6−x(CO)x (x = 0–3). We find that Co6S8(PEt3)6 has a low electron affinity (EA) of 1.1 eV, and that the successive replacement of PEt3 ligands with CO gradually shifts its electronic spectrum to lower energy and increases its EA to 1.8 eV. Density functional theory calculations reveal that the increase of EA results from a monotonic lowering of the cluster highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO). Our work provides unique insights into the electronic structure and tunability of superatomic building blocks.
Introduction
Electron affinity and ionization energy are fundamental properties of the elements. Together they govern the interactions and bonding of close-contacting atoms and control the collective properties of solids. Because they are intrinsic to each element, however, the electron affinity and ionization energy of a given atom cannot be altered. This presents an immense synthetic challenge for the design of tunable materials as substituting atoms often leads to entirely new structures, interactions and collective behaviors.By analogy to “atomic” building blocks, certain clusters can be used as “superatomic” building blocks for the assembly of novel materials.1–22 Within this context, the family of metal chalcogenide molecular clusters has recently received renewed attention for the creation of functional solids with tunable properties, including ferromagnetism, electrical conductivity, tunable optical gaps and thermal switching.1,7,23–32 One of the key advantages of this approach over traditional atomic solids is that the characteristics of the building blocks can be tuned pre-assembly without changing the total electron count of the superatom. Using the molecular cluster Co6Te8L6 (L = passivating ligand) as a model system, Khanna et al. recently predicted theoretically that changing L from PEt3 to CO would increase its electron affinity, in effect transforming the cluster from a superatomic alkali metal toward a superatomic halogen.33,34 The potential to alter the donor/acceptor property of these superatomic clusters sheds light on assembling superatomic materials with fine-tuned properties. Experimentally demonstrating this concept by measuring the electron affinity and/or ionization energy of metal chalcogenide clusters is challenging, however, because it requires bringing the charged clusters into the gas phase without damaging the inorganic core or dissociating the ligands.
In this work, we use anion photoelectron spectroscopy to probe the electron affinity (EA) and electronic structure of a series of cobalt sulfide clusters, whose ligand shells consist of differing combinations of PEt3 and CO. The clusters Co6S8(PEt3)6−x(CO)x are synthesized from the parent compound Co6S8(PEt3)6 by ligand substitution with CO. These clusters are then brought into the gas phase, where electrons are attached to form anions, using a unique infrared desorption/photoelectron-emission/supersonic helium expansion source. Mass spectrometry confirms the existence of carbonylated products with x up to 3 (i.e. Co6S8(PEt3)5(CO)−, Co6S8(PEt3)4(CO)2−, and Co6S8(PEt3)3(CO)3−). We find that the electron affinity and vertical detachment energy increase with the number of CO ligands, demonstrating the electronic spectral tunability of this family of superatoms. These results are further examples of the superatom concept, and an important step towards designing materials from programmable building blocks.
Methods
Synthesis
Dicobalt octacarbonyl and sulfur were purchased from Strem Chemicals. Triethylphosphine was purchased from Alfa Aesar. All other reagents and solvents were purchased from Sigma Aldrich. Dry and deoxygenated solvents were prepared by elution through a dual-column solvent system. Unless otherwise stated, all reactions and sample preparations were carried out under inert atmosphere using standard Schlenk techniques or in a N2-filled glovebox. While a multi-step synthesis of Co6S8(PEt3)6 has been previously reported,35 we have developed an alternative one-pot approach detailed below.Anion photoelectron spectroscopy
Anion photoelectron spectroscopy was conducted by crossing a mass-selected negative ion beam with a fixed energy photon beam and analyzing the energies of the resultant photodetached electrons. This technique is governed by the well-known energy-conserving relationship, hν = EBE + EKE, where hν, EBE, and EKE are the photon energy, electron binding energy (photodetachment transition energy), and the electron kinetic energy, respectively. The details of our apparatus have been described elsewhere.36 Briefly, the photoelectron spectra were collected on an apparatus consisting of an ion source, a linear time-of-flight mass spectrometer for mass analysis and selection, and a magnetic-bottle photoelectron spectrometer for electron energy analysis (resolution ∼35 meV at 1 eV EKE). The third harmonic (355 nm, 3.49 eV per photon) of a Nd:YAG was used to photodetach electrons from the cluster anion of interest. Photoelectron spectra were calibrated against the well-known atomic lines of the copper anion, Cu−.To make the parent anions Co6S8(PEt3)6−x(CO)x− in the gas phase, a specialized infrared desorption/laser photoemission (IR/PE) supersonic helium expansion source is employed.37 Briefly, a low power IR pulse (1064 nm) from a Nd:YAG laser hits a translating graphite bar thinly coated with the Co6S8(PEt3)6−x(CO)x sample. Because the graphite absorbs most of the energy, a localized thermal shock lasting a few nanoseconds propels the clusters into the gas phase. Almost simultaneously, a high power pulse of 532 nm light from a second Nd:YAG laser strikes a close-by photoemitter (Hf wire), creating a shower of electrons that attach to the evaporated neutral clusters. Also, almost simultaneously, a plume of ultrahigh purity helium gas rapidly expands from a pulsed valve located slightly upstream, cooling the nascent anions and directing them into the mass spectrometer, where they are analyzed. Because the Co6S8(PEt3)6−x(CO)x clusters are slightly sensitive to air and moisture, they are coated onto the graphite bar in a N2-filled glove box. The graphite bar is then enclosed inside an air-tight “suitcase” container, which is only opened under high vacuum after being transferred to the vacuum chamber.
Theoretical calculations
First-principles electronic structure calculations on the anion and neutral forms of Co6S8(PEt3)6−x(CO)x (x = 0–3) clusters were carried out within density functional theory formalism. The ADF set of codes were used to perform the calculations where a PBE0 hybrid functional comprised of the PBE generalized gradient functional and 25% Hartree–Fock exchange, as proposed by Ernzerhof et al., is used to incorporate exchange and correlation effects.38–40 The atomic wave functions are expressed in terms of Slater-type orbitals (STO) located at the atomic sites and the cluster wave functions are constructed from a linear combination of these atomic orbitals.41 A TZ2P basis set and a large frozen electron core was used. The zero-order regular approximation (ZORA) is used to include scalar-relativistic effect.42,43 The trial structures of the clusters were taken from the previously optimized structure of the Co6Te8(PEt3)6 where the Te sites were replaced by S atoms.33 The quasi-Newton method without any symmetry restriction is used to determine the lowest energy arrangement for each trial structure of the clusters. The calculations also covered the several possible spin states for all the clusters during optimization. The adiabatic electron affinity (AEA) is calculated as the total energy difference between the anion in its ground state geometry and the neutral cluster in its ground state geometry, while the vertical detachment energy (VDE) is given by the total energy difference between the anion in its ground state and the neutral cluster in the geometry of the anion.Results and discussions
Fig. 1a depicts the structure of Co6S8(PEt3)6−x(CO)x. Our strategy to synthesize these clusters begins by preparing the parent compound Co6S8(PEt3)6 on a multigram scale. To achieve this, we have developed a new approach detailed in the Methods section. Briefly, under a CO atmosphere, Co6S8(PEt3)6 is reacted with six equivalents of S in toluene at ∼100 °C to partially substitute CO for PEt3, which is trapped as S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PEt3. Using the mass spectrometry technique described below, we observe a mixture of clusters in which up to three CO ligands have been substituted for PEt3. It is possible to separate these different species using column chromatography,30 but for this study we can use the mixture of clusters without further purification.
PEt3. Using the mass spectrometry technique described below, we observe a mixture of clusters in which up to three CO ligands have been substituted for PEt3. It is possible to separate these different species using column chromatography,30 but for this study we can use the mixture of clusters without further purification.
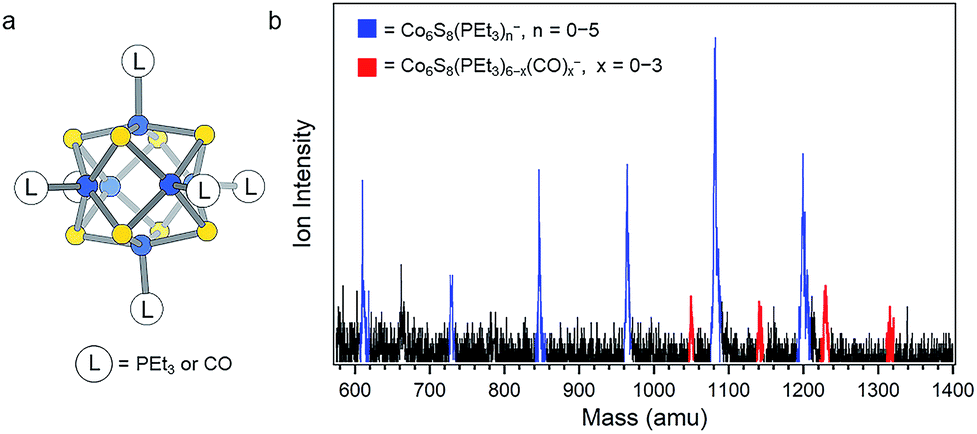 | ||
| Fig. 1 (a) Structure of Co6S8(PEt3)6−x(CO)x. (b) Anion mass spectrum of Co6S8(PEt3)6−x(CO)x− generated using IR/PE anion source. This work focuses on the red peaks in the mass spectrum. | ||
Measuring the electron affinity and electronic structure of Co6S8(PEt3)6−x(CO)x requires a negative ion photoelectron spectroscopic study on their parent anions, Co6S8(PEt3)6−x(CO)x−, in the gas phase. Common ionization methods (e.g. electrospray ionization and matrix-assisted laser desorption/ionization), however, fail to generate the parent anions Co6S8(PEt3)6−x(CO)x− in the gas phase due to the effect of the solvent/matrix on neutral Co6S8(PEt3)6−x(CO)x clusters: the obtained anions are either dissociated or tagged by the solvent/matrix molecules. To make the parent anions in the gas phase, we instead employ a unique infrared desorption/photoemission (IR/PE) source as described in the Methods section. A typical anion mass spectrum of Co6S8(PEt3)6−x(CO)x− obtained with the IR/PE source is shown in Fig. 1b. The spectrum contains two major series of anions: Co6S8(PEt3)n− (n = 0–5) and Co6S8(PEt3)6−x(CO)x− (x = 0–3). The Co6S8(PEt3)n− clusters are from the sequential dissociation of PEt3 from Co6S8(PEt3)6 during infrared desorption, while the Co6S8(PEt3)6−x(CO)x− clusters are the anions of interest in this study. Though the Co6S8(PEt3)6−x(CO)x− signals are weaker, their high photodetachment cross-sections allow us to collect their anion photoelectron spectra.
Fig. 2 presents the anion photoelectron spectra of Co6S8(PEt3)6−x(CO)x− (x = 0–3) from which the adiabatic electron affinity (AEA) and vertical detachment energy (VDE) of each cluster are determined. The value of the AEA is taken to be the onset of the lowest electron binding energy (EBE) peak in the photoelectron spectrum. The VDE is the vertical transition energy from the ground state of the anion to the neutral state at the anion geometry. It is determined as the EBE value at the intensity maximum of the peak of interest, i.e. typically the first EBE peak. Based on the anion photoelectron spectra, the AEA values of Co6S8(PEt3)6−x(CO)x are 1.1, 1.3, 1.7 and 1.8 eV for x = 0, 1, 2, 3, respectively, while the corresponding anion VDE values are 1.30, 1.51, 1.95 and 2.09 eV, respectively. By sequentially substituting half of the PEt3 ligands with CO, the AEA of this superatom increases from 1.1 eV to 1.8 eV, all while maintaining the same oxidation state for the Co6S8 core. As a reference, the reducing agent SO2 and oxidizing agent SO3 have AEA values of 1.11 eV and 1.90 eV,44,45 respectively, where the increase in AEA is the result of the change in the S atom oxidation state. Thus, the observed changes in AEA values for the ligated superatoms are significant, given that the oxidation state of the Co6S8 core does not change across the ligation series: these results underscore the remarkable characteristics of the superatoms. The observed unambiguous trend of increasing AEA and VDE values with ligand substitution is a direct evidence that the electronic properties of this superatom can be tuned effectively in this way.
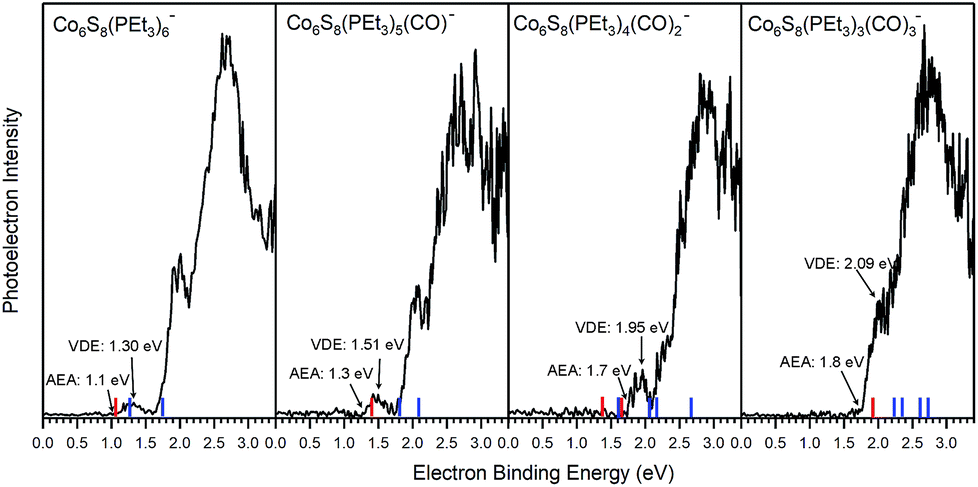 | ||
| Fig. 2 Negative ion photoelectron spectra of Co6S8(PEt3)6−x(CO)x− (x = 0–3) anions collected using 355 nm (3.49 eV) photons from a Nd:YAG laser. The arrows point to the experimental values of AEA and VDE; the red lines correspond to theoretical AEA, and the blue lines correspond to vertical transitions from the anion to the singlet and triplet neutral states. | ||
The tuning of the electronic properties can be further demonstrated through the spectral features located at higher electron binding energies than that of the first EBE peak. These higher EBE peaks arise from transitions from the anion ground state to various excited electronic states of the neutral cluster. The shape of these features relates to the electronic structure of the neutral clusters. While the photoelectron spectra of Co6S8(PEt3)6−x(CO)x− shift to higher electron binding energy as x increases, the overall spectral shapes are remarkably similar, confirming that the sequential exchange of PEt3 with CO ligand leaves the essential electronic structure of Co6S8(PEt3)6−x(CO)x superatoms largely unchanged. What is changing with sequential ligation, however, is a monotonic decrease in the energies of the highest occupied and lowest unoccupied molecular orbital (HOMO and LUMO) levels, even though the gap between them remains relatively constant. This is discussed further below.
To support these experimental observations, we modeled the ground state structures of Co6S8(PEt3)6−x(CO)x−, and calculated their AEA and VDE using density functional theory formalism (Fig. 3). In agreement with previous calculations of the Co6Te8(PEt3)6−x(CO)x system,33 we find that the anionic clusters Co6S8(PEt3)6−x(CO)x− have doublet spin ground states, while the neutral species have singlet spin states. For Co6S8(PEt3)4(CO)2−, there are two possible structures: trans-Co6S8(PEt3)4(CO)2− and cis-Co6S8(PEt3)4(CO)2−, with the latter lower in energy by 0.20 eV. The replacement of three PEt3 ligands with CO leads to fac-Co6S8(PEt3)3(CO)3− and mer-Co6S8(PEt3)3(CO)3− isomers, with the latter isomer lower in energy by 0.07 eV. The small energy difference between the Co6S8(PEt3)3(CO)3− isomers suggests their co-existence in the experimental beam. Table S1† contains more calculation results on neutral and anionic species.
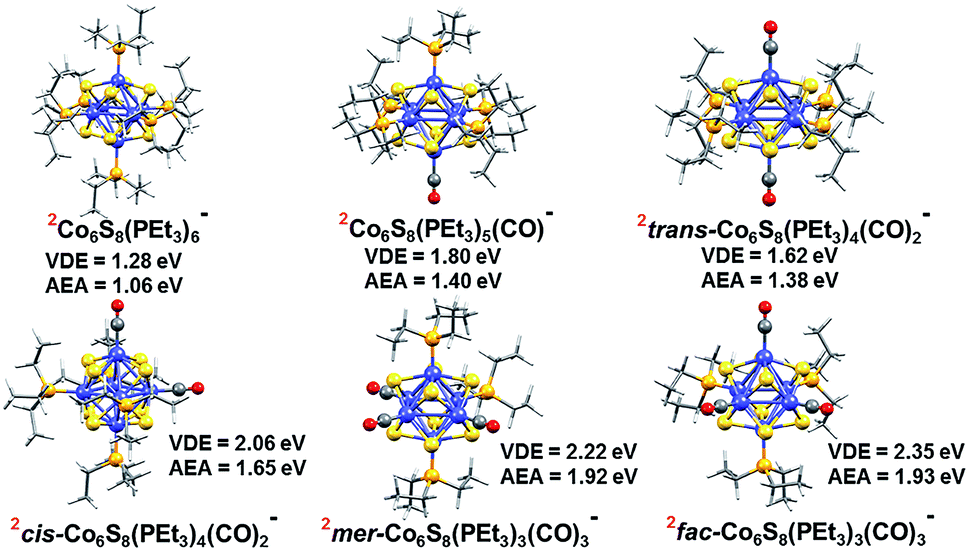 | ||
| Fig. 3 Optimized ground state structure of anionic Co6S8(PEt3)6−x(CO)x− (x = 0–3) clusters. The red superscript indicates the spin multiplicity (2S + 1) of each cluster. | ||
We modeled the electronic structure of the superatoms using the anion photoelectron spectra as fingerprints of the ground state geometry and electronic states of both the neutral and anionic clusters.46 Three points of comparison are used to establish the theoretical ground state of the system: AEA, VDE and VDE*. VDE and VDE* are the vertical transitions from the anion ground state to the neutral with singlet and triplet spin multiplicity, respectively. Experimentally, VDE and VDE* correspond to the electron binding energies of the first (lowest EBE) and second (next highest EBE) peaks in the photoelectron spectra.
Table 1 presents both theoretical and experimental values of AEA, VDE and VDE* for all the superatoms considered here. Using AEA, VDE and VDE* as references to validate the computation, we observe close agreement between experimental and calculated values. The agreement between experiments and calculations is further demonstrated by including the computed values in the anion photoelectron spectra in Fig. 2. The key result is that the AEA and both VDE values increase as PEt3 ligands are successively replaced with CO. Starting with Co6S8(PEt3)6−, the calculated AEA, VDE, and VDE* values of 1.06, 1.28, and 1.74 eV are close to the experimental values of 1.1, 1.30 eV, and 1.91 eV, respectively. Experimentally, the replacement of one PEt3 with a CO ligand results in a ∼0.2 eV increase in the AEA (1.3 eV) and VDE values (1.51 and 2.05 eV), whereas the calculated AEA (1.40 eV) and VDE values (1.80 and 2.08 eV) for Co6S8(PEt3)5CO− are only slightly higher. For cis-Co6S8(PEt3)4(CO)2− and trans-Co6S8(PEt3)4(CO)2−, the computed ground states have AEA values of 1.65 and 1.38 eV, VDE values of 2.06 and 1.62 eV, and VDE* values of 2.66 and 2.16 eV, respectively. A comparison between the experimental and theoretical results for Co6S8(PEt3)4(CO)2− confirms that this cluster is primarily the cis-isomer: the experimental values AEA = 1.7 eV, VDE = 1.95 eV, and VDE* = 2.51 eV all agree very well with the cis-isomer calculations. We note that the small shoulder at 2.19 eV in the spectrum (Fig. 2) matches the calculated VDE* of the trans-isomer, raising the possibility that a small amount of the trans-isomer is also present. For n = 3, both the mer-Co6S8(PEt3)3(CO)3− and fac-Co6S8(PEt3)3(CO)3− have similar computed AEA and VDE values that are consistent with the experimental data. Because the mer- and fac-clusters are close in energy in their neutral and anionic forms, it is very likely that they both exist in the ion beam, and that the Co6S8(PEt3)3(CO)3− photoelectron spectrum contains transitions from both isomers.
| Anionic clusters | AEAT (eV) | VDET (eV) | AEAE (eV) | VDEE (eV) | ||
|---|---|---|---|---|---|---|
| VDE | VDE* | VDE | VDE* | |||
| a The superscripts T and E indicate theoretical and experimental values, respectively. | ||||||
| Co6S8(PEt3)6− | 1.06 | 1.28 | 1.74 | 1.1 | 1.30 | 1.91 |
| Co6S8(PEt3)5(CO)− | 1.40 | 1.80 | 2.08 | 1.3 | 1.51 | 2.05 |
| cis-Co6S8(PEt3)4(CO)2− | 1.65 | 2.06 | 2.66 | 1.7 | 1.95 | 2.51 |
| trans-Co6S8(PEt3)4(CO)2− | 1.38 | 1.62 | 2.16 | |||
| mer-Co6S8(PEt3)3(CO)3− | 1.92 | 2.22 | 2.71 | 1.8 | 2.09 | 2.67 |
| fac-Co6S8(PEt3)3(CO)3− | 1.93 | 2.35 | 2.60 | |||
The replacement of PEt3 ligands by CO concurrently lowers the energies of the HOMO and LUMO, with the result that the HOMO–LUMO gap is essentially unchanged across the cluster series. Fig. 4a illustrates this trend for the neutral species while their absolute energy values are given in Table S1.† The lowering of the HOMO and LUMO can be in part understood in terms of the vastly different ligand field effect of the cluster surface passivation: PEt3 is a strong σ-donor that increases the electron density in the core while CO is a strong π-acceptor that removes electron density from the core, thus lowering the energy of the electronic spectrum.33 This behavior can be seen in the Fig. S1 and S2† which show the one-electron energy levels of Co6S8(PEt3)6−x(CO)x (x = 0–3). The change in the AEA closely tracks the change in the LUMO as the electron is attached to the LUMO of the neutral. This is illustrated in Fig. 4b, which shows the changes in the energy of the LUMO (ΔLUMO), theoretical AEA (ΔAEAT) and experimental AEA (ΔAEAE) of Co6S8(PEt3)6−x(CO)x as x increases from 0 to 3. The molecular orbital iso-surfaces of the HOMO and LUMO in Co6S8(PEt3)6−x(CO)x are shown in Fig. 4c. A Mulliken population analysis of the molecular orbitals reveals that hybridization between the p-states of S and the 3d-states of Co forms the HOMO in each cluster, while the LUMO is an antibonding state comprising the 3dz2-orbital of Co and the lone pair of the ligand (PEt3/CO). An additional phenomenon combines with the traditional ligand field effect to explain the changes in AEA and VDE: PEt3 and CO form charge transfer complexes, and the induced dipoles at the cluster surface play a critical role in changing the electronic properties. As a reference, surface dipoles can either increase or decrease the work function of metals.47,48 In this case, as PEt3 ligands are replaced by CO, the sign of the dipole changes and hence AEA increases as more CO are bound to the core.
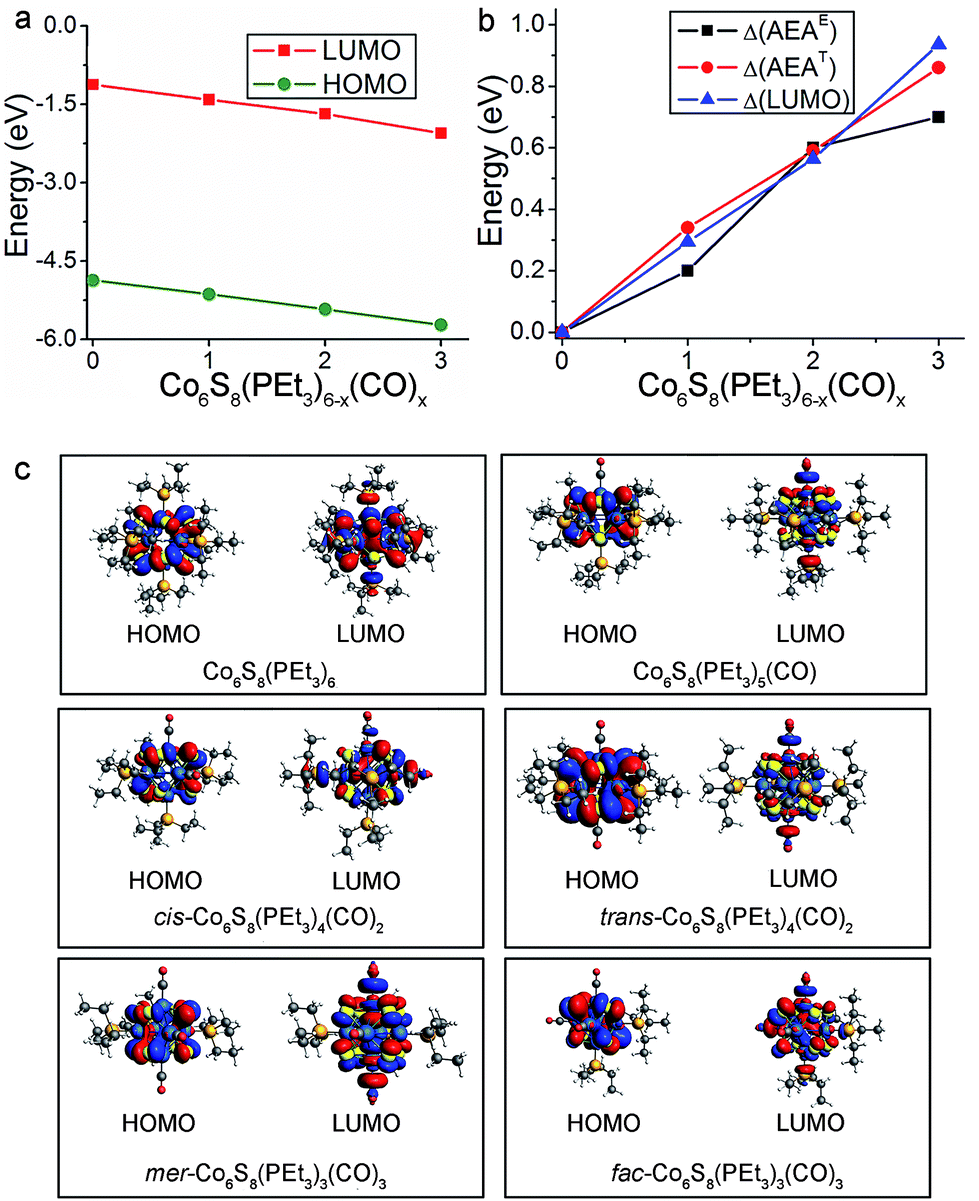 | ||
| Fig. 4 (a) The absolute energy values of the HOMO and LUMO for neutral Co6S8(PEt3)6−x(CO)x (x = 0–3). (b) Incremental differences in the experimental AEA values, the theoretical AEA values, and the LUMO values of Co6S8(PEt3)6−x(CO)x, each with respect to Co6S8(PEt3)6. (c) Molecular orbital iso-surfaces of the HOMO and LUMO in Co6S8(PEt3)6−x(CO)x (x = 0–3) clusters. | ||
Lastly, though the ionization energy of Co6S8(PEt3)6−x(CO)x is not directly measured, the successive rise in AEA and lowering of HOMO are evidence of a trend of increasing ionization energy as PEt3 is replaced by CO.
Conclusions
By combining cluster synthesis, gas-phase anion photoelectron spectroscopy, and electronic structure calculations, we have demonstrated that the electronic properties of metal chalcogenide superatomic clusters can be tuned by varying their capping ligands. While the total electron count of the closed-shell core remains unchanged, substitution of the ligands leads to a simultaneous rise or fall of the HOMO and LUMO levels, resulting in a change of the cluster donor/acceptor behavior.These results are significant because such drastic change in the cluster properties is rarely observed in other superatomic systems. Besides changes of electron affinity and ionization potential, ligand substitution of a ligated cluster almost always results in changes of other core chemical properties such as the HOMO–LUMO gap, electron configuration, and electronic structure. For example, in phosphine-passivated gold clusters approximated by the confined nearly free electron gas model, charge transfer from the phosphine ligands results in significant changes to the core electron count and HOMO–LUMO gap.49 The unpredictable outcome of ligand substitution can impose important challenges in designing programmable cluster building blocks. In this context, the metal chalcogenide cluster presented in this work is unique: ligand substitution can be used to tune its electron affinity and ionization potential while maintaining its HOMO–LUMO gap, electronic structure and effective electron count, thus enabling us to control its oxidative and reductive properties. Such tunability may prove crucial for applications as catalysts, semiconductor dopants, and superatomic building blocks for cluster-assembled materials. In one example, we recently showed that related clusters can be used as electron-donating or electron-accepting surface dopants for two-dimensional semiconductors, where the band structures can be controlled by the choice of ligands.32,50 Such dopants could also be made magnetic because of their finite spin moments.51 More broadly, our results open the door to the design of tunable functional materials from superatomic building blocks.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This material is based on work supported by the Office of Naval Research (ONR), Multidisciplinary University Research Initiative (MURI) under Grant No. N00014-15-1-2681 (K. H. B.), and by the Air Force Office of Scientific Research (AFOSR) under grant number, FA9550-15-1-0259 (K. H. B.). The synthetic work was supported by the NSF MRSEC program through the Center for Precision Assembly of Superstratic and Superatomic Solids (DMR-1420634) (X. R., A. P., A. P. A.) and the US Air Force Office of Scientific Research (AFOSR) Grant No. FA9550-18-1-0020 (X. R., A. P., A. P. A.). Theoretical studies (V. C. and S. N. K.) were supported by a grant from Air Force Office of Scientific Research (AFOSR) Grant No. FA9550-18-1-0511. Part of the theoretical computations used the resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 (V. C. and S. N. K.). A. P. was supported by the NSF Graduate Research Fellowship Program (DGE-16-44869).Notes and references
- A. Pinkard, A. M. Champsaur and X. Roy, Acc. Chem. Res., 2018, 51, 919–929 CrossRef CAS PubMed
.
- S. A. Claridge, A. W. Castleman, S. N. Khanna, C. B. Murray, A. Sen and P. S. Weiss, ACS Nano, 2009, 3, 244–255 CrossRef CAS PubMed
- E. G. Tulsky and J. R. Long, Chem. Mater., 2001, 13, 1149–1166 CrossRef CAS
- S. A. Baudron, P. Batail, C. Coulon, R. Clerac, E. Canadell, V. Laukhin, R. Melzi, P. Wzietek, D. Jerome, P. Auban-Senzier and S. Ravy, J. Am. Chem. Soc., 2005, 127, 11785–11797 CrossRef CAS
- B. Yoon, W. D. Luedtke, R. N. Barnett, J. Gao, A. Desireddy, B. E. Conn, T. Bigioni and U. Landman, Nat. Mater., 2014, 13, 807–811 CrossRef CAS PubMed
- J. F. Corrigan, O. Fuhr and D. Fenske, Adv. Mater., 2009, 21, 1867–1871 CrossRef CAS
- X. Roy, C.-H. Lee, A. C. Crowther, C. L. Schenck, T. Besara, R. A. Lalancette, T. Siegrist, P. W. Stephens, L. E. Brus, P. Kim, M. L. Steigerwald and C. Nuckolls, Science, 2013, 341, 157–160 CrossRef CAS PubMed
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS
- A. W. Castleman and S. N. Khanna, Clusters, J. Phys. Chem. C, 2009, 113, 2664–2675 CrossRef CAS
- A. C. Reber and S. N. Khanna, Acc. Chem. Res., 2017, 50, 255–263 CrossRef CAS PubMed
- C.-H. Lee, L. Liu, C. Bejger, A. Turkiewicz, T. Goko, C. J. Arguello, B. A. Frandsen, S. C. Cheung, T. Medina, T. J. S. Munsie, R. D'Ortenzio, G. M. Luke, T. Besara, R. A. Lalancette, T. Siegrist, P. W. Stephens, A. C. Crowther, L. E. Brus, Y. Matsuo, E. Nakamura, Y. J. Uemura, P. Kim, C. Nuckolls, M. L. Steigerwald and X. Roy, J. Am. Chem. Soc., 2014, 136, 16926–16931 CrossRef CAS PubMed
- X. Roy, C. L. Schenck, S. Ahn, R. A. Lalancette, L. Venkataraman, C. Nuckolls and M. L. Steigerwald, Angew. Chem., Int. Ed., 2012, 51, 12473–12476 CrossRef CAS PubMed
- M. T. Trinh, A. Pinkard, A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, L. M. Campos, X. Roy and X.-Y. Zhu, Sci. Adv., 2017, 3, e1700241 CrossRef PubMed
- V. Chauhan, S. Sahoo and S. N. Khanna, J. Am. Chem. Soc., 2016, 138, 1916–1921 CrossRef CAS PubMed
- S. N. Khanna and A. C. Reber, Nat. Chem., 2017, 9, 1151–1152 CrossRef CAS PubMed
- D. A. Tomalia and S. N. Khanna, Chem. Rev., 2016, 116, 2705–2774 CrossRef CAS PubMed
- Z. Zheng, J. R. Long and R. H. Holm, J. Am. Chem. Soc., 1997, 119, 2163–2171 CrossRef CAS
- M. Cargnello, A. C. Johnston-Peck, B. T. Diroll, E. Wong, B. Datta, D. Damodhar, V. V. T. Doan-Nguyen, A. A. Herzing, C. R. Kagan and C. B. Murray, Nature, 2015, 524, 450–453 CrossRef CAS PubMed
- M. N. O'Brien, M. R. Jones, B. Lee and C. A. Mirkin, Nat. Mater., 2015, 14, 833–839 CrossRef PubMed
- M. A. Boles and D. V. Talapin, J. Am. Chem. Soc., 2015, 137, 4494 CrossRef CAS PubMed
- C. L. Poyser, T. Czerniuk, A. Akimov, B. T. Diroll, E. A. Gaudling, A. S. Salasyuk, A. J. Kent, D. R. Yakovlev, M. Bayer and C. B. Murray, ACS Nano, 2016, 10, 1163–1169 CrossRef CAS PubMed
- W.-L. Ong, S. M. Rupich, D. V. Talapin, A. J. H. McGaughey and J. A. Malen, Nat. Mater., 2013, 12, 410–415 CrossRef CAS PubMed
- A. Voevodin, L. M. Campos and X. Roy, J. Am. Chem. Soc., 2018, 140, 5607–5611 CrossRef CAS PubMed
- A. M. Champsaur, J. Yu, X. Roy, D. W. Paley, M. L. Steigerwald, C. Nuckolls and C. M. Bejger, ACS Cent. Sci., 2017, 3, 1050–1055 CrossRef CAS PubMed
- A. M. Champsaur, C. Mézière, M. Allain, D. W. Paley, M. L. Steigerwald, C. Nuckolls and P. Batail, J. Am. Chem. Soc., 2017, 139, 11718–11721 CrossRef CAS PubMed
- E. S. O'Brien, M. T. Trinh, R. L. Kann, J. Chen, G. A. Elbaz, A. Masurkar, T. L. Atallah, M. V. Paley, N. Patel, D. W. Paley, I. Kymissis, A. C. Crowther, A. J. Millis, D. R. Reichman, X.-Y. Zhu and X. Roy, Nat. Chem., 2017, 9, 1170–1174 CrossRef PubMed
- G. Lovat, B. Choi, D. W. Paley, M. L. Steigerwald, L. Venkataraman and X. Roy, Nat. Nanotechnol., 2017, 12, 1050–1054 CrossRef CAS PubMed
- W.-L. Ong, E. S. O'Brien, P. S. M. Dougherty, D. W. Paley, C. Fred Higgs Iii, A. J. H. McGaughey, J. A. Malen and X. Roy, Nat. Mater., 2017, 16, 83–88 CrossRef CAS PubMed
- B. Choi, J. Yu, D. W. Paley, M. Tuan Trinh, M. V. Paley, J. M. Karch, A. C. Crowther, C. H. Lee, R. A. Lalancette, X. Zhu, K. Kim, M. L. Steigerwald, C. Nuckolls and X. Roy, Nano Lett., 2016, 16, 1445–1449 CrossRef CAS PubMed
- A. M. Champsaur, A. Velian, D. W. Paley, B. Choi, X. Roy, M. L. Steigerwald and C. Nuckolls, Nano Lett., 2016, 16, 5273–5277 CrossRef CAS PubMed
- A. Turkiewicz, D. W. Paley, T. Besara, G. Elbaz, A. Pinkard, T. Siegrist and X. Roy, J. Am. Chem. Soc., 2014, 136, 15873–15876 CrossRef CAS PubMed
- J. Yu, C. H. Lee, D. Bouilly, M. Han, P. Kim, M. L. Steigerwald, X. Roy and C. Nuckolls, Nano Lett., 2016, 16, 3385–3389 CrossRef CAS PubMed
- V. Chauhan, A. C. Reber and S. N. Khanna, J. Am. Chem. Soc., 2017, 139, 1871–1877 CrossRef CAS PubMed
- V. Chauhan and S. N. Khanna, J. Phys. Chem. A, 2018, 122(28), 6014–6020 CrossRef CAS PubMed
- F. Cecconi, C. A. Ghilardi, S. Midollini and A. Orlandini, Inorg. Chim. Acta, 1983, 76, 183–184 CrossRef
- X. Zhang, G. Liu, G. Gantefoer, K. H. Bowen and A. N. Alexandrova, J. Phys. Chem. Lett., 2014, 5, 1596–1601 CrossRef CAS PubMed
- A. Grubisic, H. Wang, X. Li, Y. J. Ko, S. Kocak, M. R. Pederson, K. H. Bowen and B. W. Eichhorn, PNAS, 2011, 108, 14757–14762 CrossRef CAS PubMed
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS
- C. Adamo, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS
- E. van Lenthe, A. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943 CrossRef CAS
- M. R. Nimlos and G. B. Ellison, J. Phys. Chem., 1986, 90, 2574–2580 CrossRef CAS
- E. W. Rothe, S. Y. Tang and G. P. Reck, J. Chem. Phys., 1975, 62, 3829 CrossRef CAS
- P. J. Robinson, G. Liu, S. Ciborowski, C. Martinez-Martinez, J. R. Chamorro, X. Zhang, T. M. McQueen, K. H. Bowen and A. N. Alexandrova, Chem. Mater., 2017, 29, 9892–9896 CrossRef CAS
- P. C. Rusu and G. Brocks, J. Phys. Chem. B, 2006, 110, 22628–22634 CrossRef CAS PubMed
- V. Chauhan, A. C. Reber and S. N. Khanna, Nat. Commun., 2018, 9, 2357 CrossRef PubMed
- D. Mollenhauer and N. Gaston, Phys. Chem. Chem. Phys., 2016, 18, 29686 RSC
- A. C. Reber and S. N. Khanna, npj Comput. Mater., 2018, 4, 33 CrossRef
- V. Chauhan, A. C. Reber and S. N. Khanna, Phys. Chem. Chem. Phys., 2017, 19, 31940–31948 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Calculated energies, one-electron energy levels, and Cartesian coordinates of neutral and anionic Co6S8(PEt3)6−x(CO)x clusters. See DOI: 10.1039/c8sc03862g |
| This journal is © The Royal Society of Chemistry 2019 |