
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterisation of the biosynthetic pathway to agnestins A and B reveals the reductive route to chrysophanol in fungi†
Agnieszka J.
Szwalbe
a,
Katherine
Williams
a,
Zhongshu
Song
a,
Kate
de Mattos-Shipley
a,
Jason L.
Vincent
b,
Andrew M.
Bailey
 c,
Christine L.
Willis
a,
Russell J.
Cox
ade and
Thomas J.
Simpson
*a
c,
Christine L.
Willis
a,
Russell J.
Cox
ade and
Thomas J.
Simpson
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: tom.simpson@bristol.ac.uk
bSyngenta, Jealott's Hill International Research Centre, Bracknell, RG42 6EY, UK
cSchool of Biological Sciences, 24 Tyndall Avenue, Bristol, BS8 1TQ, UK
dInstitute for Organic Chemistry, Leibniz University of Hannover, 30167, Germany
eBMWZ, Leibniz University of Hannover, 30167, Germany
First published on 26th November 2018
Abstract
Two new dihydroxy-xanthone metabolites, agnestins A and B, were isolated from Paecilomyces variotii along with a number of related benzophenones and xanthones including monodictyphenone. The structures were elucidated by NMR analyses and X-ray crystallography. The agnestin (agn) biosynthetic gene cluster was identified and targeted gene disruptions of the PKS, Baeyer–Villiger monooxygenase, and other oxido-reductase genes revealed new details of fungal xanthone biosynthesis. In particular, identification of a reductase responsible for in vivo anthraquinone to anthrol conversion confirms a previously postulated essential step in aromatic deoxygenation of anthraquinones, e.g. emodin to chrysophanol.
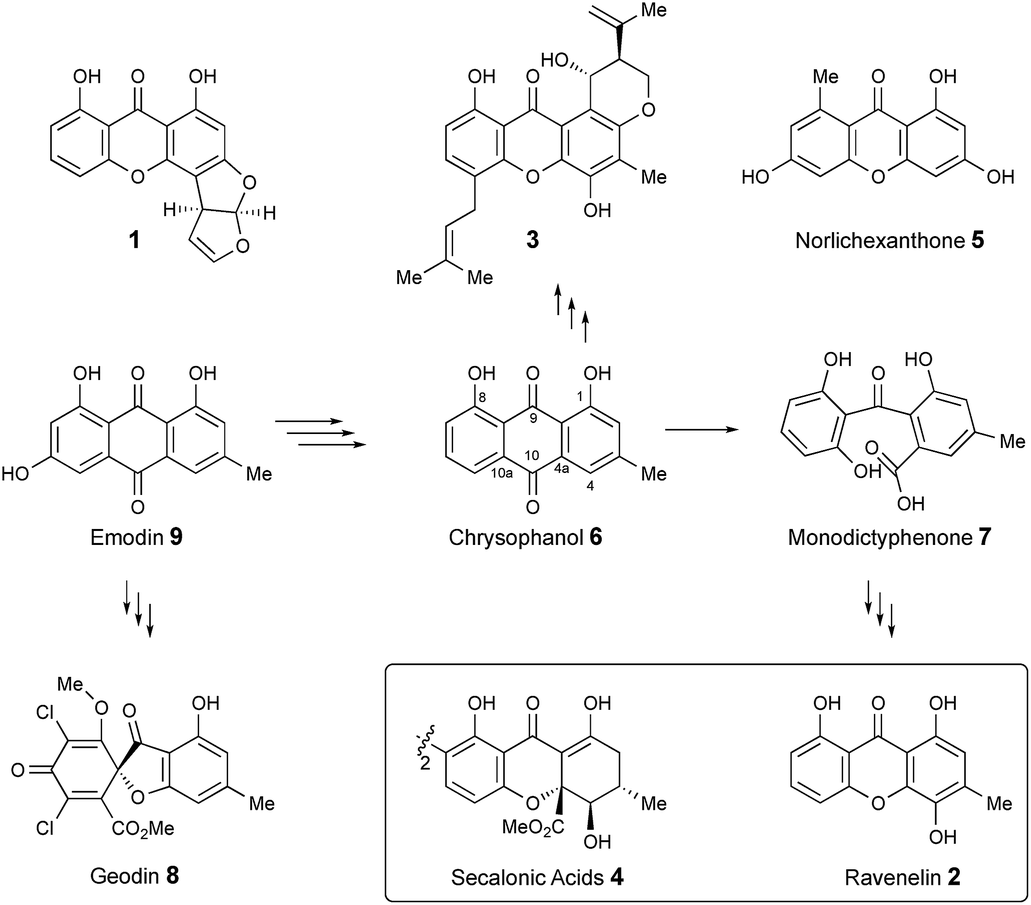
Xanthones and related benzophenones are produced by a variety of filamentous fungi.1 Examples (Scheme 1) include desmethyl-sterigmatocystin 1,2 a key intermediate to the aflatoxin group of mycotoxins produced by Aspergillus flavus, ravenelin 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 from Drechslera ravenelii and prenylated xanthones, e.g. shamixanthone 3 from Aspergillus variecolor.4 More complex derivatives include the dimeric xanthone ergochromes (secalonic acids) e.g.4 from inter alia Claviceps purpurea5 and Penicillium oxalicum.6
3 from Drechslera ravenelii and prenylated xanthones, e.g. shamixanthone 3 from Aspergillus variecolor.4 More complex derivatives include the dimeric xanthone ergochromes (secalonic acids) e.g.4 from inter alia Claviceps purpurea5 and Penicillium oxalicum.6
 | ||
| Scheme 1 Pathways emanating from emodin 9 and related compounds. | ||
While some fungal xanthones are produced via simple folding of a polyketide chain, e.g. norlichexanthone 5 from Lecanora straminea,7 most are produced via Baeyer–Villiger-type oxidation of anthraquinones such as chrysophanol 6 to give benzophenones such as monodictyphenone 7 which are subsequently cyclized to xanthones.8 Recent interest in these pathways has been stimulated by the results of genome sequencing and bioinformatic analysis9 which has allowed identification of the gene clusters in, e.g. Aspergillus nidulans for biosynthesis of shamixanthone 3.10,11 Other related compounds include geodin 812 which is derived from emodin 9, the precursor of chrysophanol 6,13 while cladofulvin from the tomato pathogen Cladosporium fulvum is a dimeric naphthoquinone also derived from 9.14 Despite these advances key questions remain about the precise genes, proteins and chemical steps involved.
In the course of studies on the biosynthesis of maleidrides,15 we examined extracts of the cornexistin producing fungus Paecilomyces variotii.16 These extracts contained mainly the benzophenone monodictyphenone 7 and other unknown aromatic metabolites. Here we determine the structures of the unknowns, the associated biosynthetic gene cluster and reveal key new redox steps on the fungal pathway to chrysophanol 6.
Isolation of metabolites
Extracts of P. variotii K5103 contained several major aromatic metabolites (Fig. 1) which were isolated by semi-preparative HPLC. The material eluting at 11.4 min (typical yield of 20 mg L−1 after 15 days fermentation) had 1H and 13C NMR (see ESI†) and UV spectra which were identical to those for monodictyphenone 7.17 Closer inspection of the 1H NMR spectrum revealed the presence (ca. 25%) of a co-eluting isomer whose NMR spectra (see ESI†) matched those of the previously reported cephalone F 10 from Graphiopsis chlorocephala.18 Monodictyphenone 7 is believed to be formed by a Baeyer–Villiger monooxygenase (BVMO) cleavage of the C-10/C-10a bond of chrysophanol 6 and the formation of cephalone F 10 can be rationalized by alternative cleavage at the C-4a/C-10 bond. The reported 1H NMR spectrum of cephalone F 10 contained signals due to a minor component (ca. 5%) clearly corresponding to those of monodictyphenone 7, suggesting that the putative BVMO in G. chlorocephala has a complementary regioselectivity to that in P. variotii.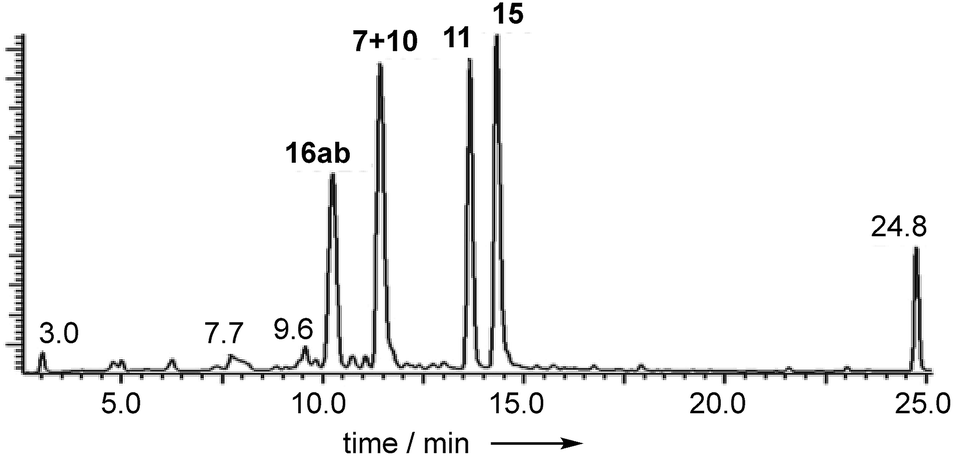 | ||
| Fig. 1 Typical DAD chromatogram of wild type P. variotii K5103 culture extract. Bold, compound numbers; other numbers retention times. | ||
The peak eluting at 13.6 minutes (typical yields of 15 mg L−1) had the same molecular formula as 7, C15H12O6 (calc. 311.0532, measured 311.0536 for [M + Na]+), but whereas the main low resolution mass fragment ion in monodictyphenone 7 occurred at M-44 (–CO2), indicating that a carboxylic acid was present, the main fragment ion now occurred at M-62 (–CO2, –H2O). The NMR spectra also had similarities to monodictyphenone 7, e.g. a methyl group (δH 2.05), a highly conjugated ketone (δC 181.7) and three adjacent aromatic hydrogens (δH 6.91, 7.55 and 6.75) confirmed by their characteristic ortho couplings and COSY correlations.
The methyl signal showed long range couplings and COSY correlations to signals at δH 6.08 and 4.86, the latter being further coupled to a doublet (8.5 Hz) at 4.07 ppm. COSY and HMBC correlations (Fig. 2), were consistent with the dihydroxanthone structure 11. Slow crystallization from methanol gave crystals which allowed a high quality X-ray crystal structure (see ESI†) to be obtained (Flack parameter −0.01(4)) which confirmed structure 11, here named agnestin A, and the (1R,2R) absolute stereochemistry.19 It is relatively stable due to the gauche relationship of the carboxyl and hydroxyl substituents although slow dehydration does occur to give the known monodictyxanthone 1220 whose presence was also detected in older (>25 days) cultures. The related dehydro-compound 13 (Fig. 3) is known from fungal endophytes of Picea glauca,21 and the similar tetrahydro-xanthone analogue, α-diversonolic ester 14 is known from Penicillium diversum.22
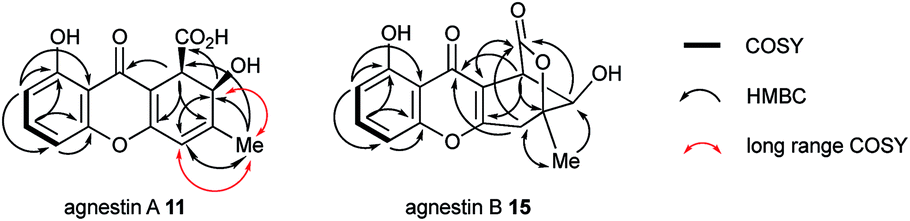 | ||
| Fig. 2 2D correlations (H to C and H to H) observed for agnestin A and agnestin B. | ||
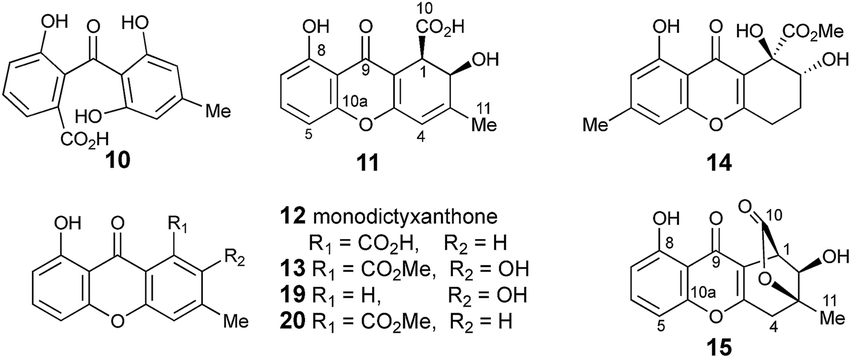 | ||
| Fig. 3 Cephalone F 10 and xanthones discussed in the text. | ||
The peak eluting at 14.3 minutes (typical yields of 5–10 mg L−1, Fig. 1) was a further structural isomer with the molecular formula C15H12O6 (HRMS calc. 311.0532, measured 311.0521 for [M + Na]+). The 1H NMR spectrum indicted a close resemblance to agnestin A 11: three adjacent mutually coupled aromatic hydrogens were clearly present at δH 6.92, 7.58 and 6.78 ppm. The main difference was the presence of two methine singlets (δH 3.87 and 4.11 ppm) and a closely coupled methylene AB signal centred at 3.15 ppm. The two singlets showed long-range COSY correlations to a methyl group (1.53 ppm), and the C-3 resonance at δC 158.1 ppm in agnestin A 11 was replaced by one at 86.8 ppm. These observations, combined with HMBC correlations (Fig. 2) were consistent with the bicyclic lactone-containing structure 15, which we have named agnestin B.
Consistent with this, samples of agnestin A 11 were observed (HPLC) to slowly convert to agnestin B 15 and give colourless crystals on slow evaporation from acetonitrile. X-ray analysis (see ESI†) confirmed the structure of agnestin B 15 and also explained the lack of observable coupling between H-1 and H-2 due to the observed dihedral angle of 77° (see ESI†). The 1R,2R absolute configuration of agnestin B 15 was assigned on the basis of its formation from rearrangement of agnestin A 11.
To examine if either agnestin A 11 or agnestin B 15 was merely an extraction-artefact formed by acid-catalysed rearrangement of the other, a comparison of extraction conditions was made. One culture was worked-up under the normal acidified conditions, while another was extracted under neutral conditions. In each case both agnestins were observed, suggesting that both are true in vivo products.
Extracts of younger cultures of P. variotii (4–5 days) show the presence of a major peak at 10.2 minutes with a molecular formula of C15H14O7 (calc. 305.0667, measured 305.0660 for [M − H]−) corresponding to formal addition of H2O to the agnestins. However, on attempted isolation, the compound rearranged to give mainly agnestin A 11 with some agnestin B 15 (see ESI†). Initial NMR studies of a freeze-dried HPLC-purified fraction showed the presence of two major components, one of which appeared to be a benzophenone due to the presence of the characteristic ortho coupled one-proton triplet and two proton doublet associated with the “symmetrical” 2,6-dihydroxyphenolic ring. The other component has the triplet, doublet, doublet pattern more typical of constraining this ring into a xanthone derivative. Other signals are present at δH 5.8, 4.7, 3.8, 3.2 and 2.1 ppm. Similar signals were observed in a spectrum obtained directly from an HPLC-purified fraction (WET 1H NMR with solvent suppression)23 using a 600 MHz cryo-probed spectrometer. Monitoring the 1H NMR spectrum over time showed that the mixture gradually converted to mainly agnestin A 11 after 15 hours (see ESI†). Simple biosynthetic rationale requires the 2–OH to be introduced by an oxidation at C-2 of monodictyphenone 7, and the molecular formula requires a further reduction to give the dihydroxanthone core.
We propose that this intermediate, agnestin C, is an equilibrating ca. 50:50 mixture of structures 16a and 16b which could be formed via epoxidation of the dihydro-derivative 17 of monodictyphenone 7 to give epoxide 18 followed by rearrangement to give the allylic alcohols 16a/b. In the absence of evidence to the contrary it is of course possible that epoxidation could precede reduction. Elimination of water could give agnestin A 11 or agnestin B 15 by either of the mechanisms shown in Scheme 2.
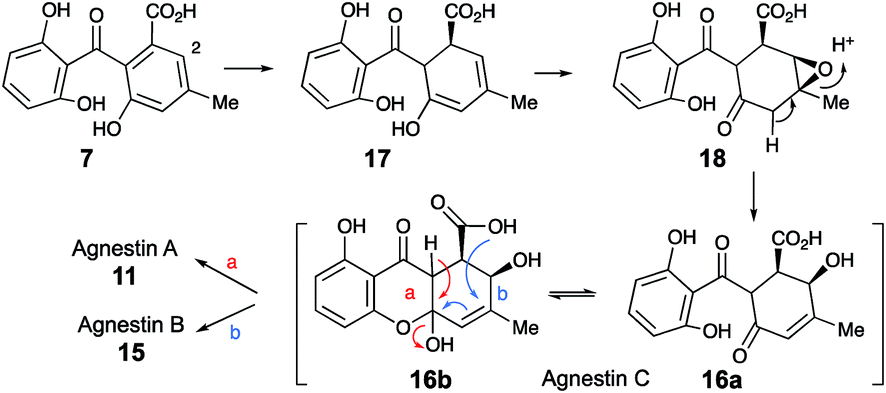 | ||
| Scheme 2 Postulated formation of agnestins A 11 and B 15via agnestin C 16ab. | ||
In addition to monodictyxanthone 12, older (ca. 25 days) cultures contained two further minor xanthone derivatives. These were readily identified by comparison of their spectroscopic characteristics with a compound 19 reported24 by Tarcz et al. and a natural product 20 isolated by Li et al. from co-cultures of two South China coast fungal strains (Fig. 3).25 The former is presumably formed from agnestin A by decarboxylation and oxidation, whereas the latter corresponds to the methyl ester of monodictyxanthone 12. Interestingly, these extracts also contained detectable amounts of the anthraquinones emodin 9 and chrysophanol 6.
Genetic analysis
A draft genome sequence of P. variotii K5103 was previously obtained.16 Identification of a putative biosynthetic gene cluster (BGC) responsible for the biosynthesis of the agnestins (agn) was aided by comparison with the known mdp7 and ged8 clusters from A. nidulans (Table 1).9,10|
|
|||||
|---|---|---|---|---|---|
| Gene | Putative function | Swissprot homolog (% id.) | Mdp homolog (% id.) | Ged homolog (% id.) | Ref. |
| agnL12 | MFS transporter | lepC, B8NJG7 (39) | — | — | 27 |
| agnL11 | Transcription factor | lepB, B8NJG5 (31) | — | — | — |
| agnL10 | Transcription factor | MdpE, Q5BH32 (33) | MdpE, (33) | GedR, (34) | 28 |
| agnL9 | Regulation | MdpA, (43) | MdpA, (43) | GedD, (40) | |
| agnL8 | Dehydratase | SCD1, Q00455 (63) | MdpB, (57) | — | 29 |
| agnL7 | Hydrolase | MdpF, Q5BH31 (69) | MdpF, (69) | GedB, (65) | 30 |
| agnL6 | Reductase | CPUR_05429, M1W270 (74) | MpdC, (72) | — | 31 |
| agnL5 | Unknown | PtaG, A0A067XNI6 (43) | — | — | — |
| agnL4 | Oxidoreductase | TpcG, Q4WQZ1 (59) | MdpK, (59) | GedF, (61) | 32 |
| agnL3 | BVMO | CPUR_05427, M1WG92 (51) | MdpL, (42) | GedK, (43) | 33 |
| agnL2 | Anthrone oxidase | GedH, P0DOB2 (46) | MdpH2, (43) | GedH, (46) | 34 |
| agnL1 | Decarboxylase | TpcK, Q4WQY7 (72) | MdpH1, (59) | GedI, (67) | |
| agnPKS | nr-PKS | MdpG, Q5BH30 (66) | MdpG, (66) | GedC, (65) | 35 |
| agnR1 | Oxidoreductase | — | — | — | — |
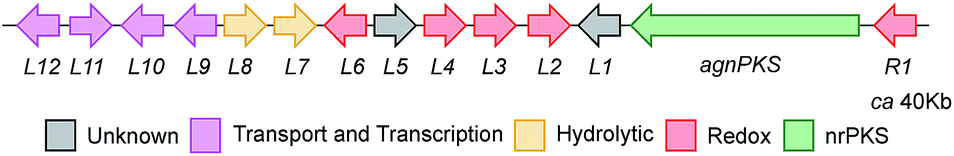
The proposed agn BGC consists of agnPKS, encoding a fungal non-reducing polyketide synthase 66% identical to the MdpG PKS, flanked downstream by agnL1 to agnL12 and upstream by agnR1. Nine genes are common in the mdp, ged and agn clusters (translated protein identities >42%, Table 1). In addition agnL10 encodes a transcription factor with 33% identity to the transcription factor MdpE. Four putative ORFs that are specific to the P. variotii agn cluster were also identified (agnR1, agnL5, agnL11 and agn L12, Table 1).26
To confirm that we had identified the correct cluster, the agnPKS was knocked-out using a bipartite strategy.36 LCMS analysis of the ΔagnPKS strain, showed complete loss of monodictyphenone 7, agnestins and all related compounds (see ESI†). Knockout of agnL3, which shows 42% identity with mdpL, encoding a Baeyer–Villiger oxidase, also caused total loss of monodictyphenone 7 and agnestin biosynthesis. The mutant accumulated emodin 9 and chrysophanol 6 (677 and 791 mg L−1 respectively, see ESI†), consistent with the predicted role for AgnL3 in anthraquinone ring cleavage.
AgnL4 shows high homology (59%) with MdpK which had been assigned10 a role in rearrangement of an epoxide intermediate in the proposed conversion of emodin 9 to monodictyphenone 7 by analogy to that proposed for AflX during the conversion of versicolorin A 24 to desmethylsterigmatocystin 1.2 However, the appropriateness of this analogy has been questioned by Simpson and it has been suggested instead that MdpK may act as a thiolester reductase during the conversion of the Baeyer–Villiger lactone product of MdpL oxidation of chrysophanol 6 to an aldehyde equivalent of monodictyphenone.37
Our results are not consistent with either of these possibilities. Knock-out of agnL4 results in accumulation of emodin 9, but only traces of chrysophanol 6 (see ESI†). Müller has shown38 that the initial “ketoreduction” step during the conversion of 9 to 6in vitro requires prior chemical reduction of 9 to the hydroquinone (shown by NMR to exist in the tautomeric forms 21a and 21b) before MdpC (=AgnL6) mediated reduction to hydroxyketone 22 (and subsequent MdpB [=AgnL8] mediated dehydration to give chrysophanol 6) can occur in vitro (Scheme 3). In a subsequent study with Townsend,39 it was shown that AflM (67% amino acid identity to MdpC, = AgnL6), will also reduce emodin 9 and versicolorin A 24 to their corresponding hydroxyketones 22 and 26, but again only after prior chemical (dithionite) reduction to the corresponding dihydroquinones 25 and 21 respectively (Scheme 3). Thus the most likely in vivo role of AgnL4, and by extension MdpK, is to reduce emodin 9 to its hydroquinone 21, which is the true substrate for AgnL6/MdpC. Likewise, we propose that AflX (43% identity to AgnL4) reduces versicolorin A 24 to its corresponding dihydroquinone 25 before the subsequent AflM-mediated conversion to 6-deoxy-versicolorin A 28 in the sterigmatocystin pathway.
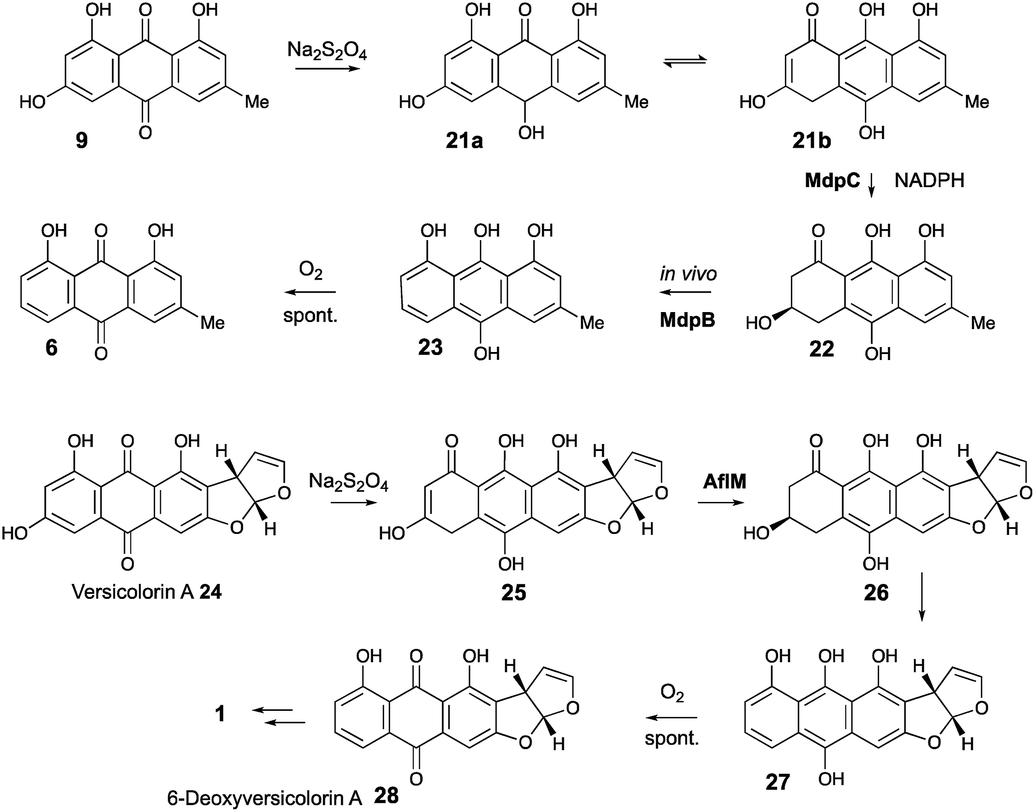 | ||
| Scheme 3 Prior reduction of anthraquinones 9 and 24 is required before phenol reduction. | ||
Based on these observations we now propose a unifying pathway to monodictyphenone and the agnestins (Scheme 4) which is consistent with all available evidence. AgnPKS (=MdpG) assembles and cyclises an octaketide bound to the PKS as a thiolester. This is then hydrolysed by AgnL7 (=MdpF) to give atochrysone carboxylic acid 30 as the first enzyme-free intermediate. AgnL1 then catalyses the concerted decarboxylation-elimination required to convert atochrysone carboxylic acid 30 to emodin anthrone 31 which is then oxidized to emodin 9 by AgnL2 (=MdpH1). Emodin 9 then undergoes reduction catalysed by AgnL4 (=MdpK) to give the dihydroquinone tautomer 21b. This is the substrate for AgnL6 (=MdpC) reduction to 22, followed by AgnL8 (=MdpB) dehydration and likely spontaneous autoxidation to chrysophanol 6. Baeyer–Villiger oxidation by AgnL3 (=MdpL) gives monodictyphenone 7 along with some cephalone F 10. Formation of 7 presumably occurs via hydrolysis of the lactone 32 which has not been previously reported as a natural product. Close examination of the NMR spectra of isolated samples of monodictyxanthone 12 (see ESI†), show the presence of a co-eluting structural isomer. The chemical shifts are fully consistent with values calculated for 32 which we name monodictylactone.
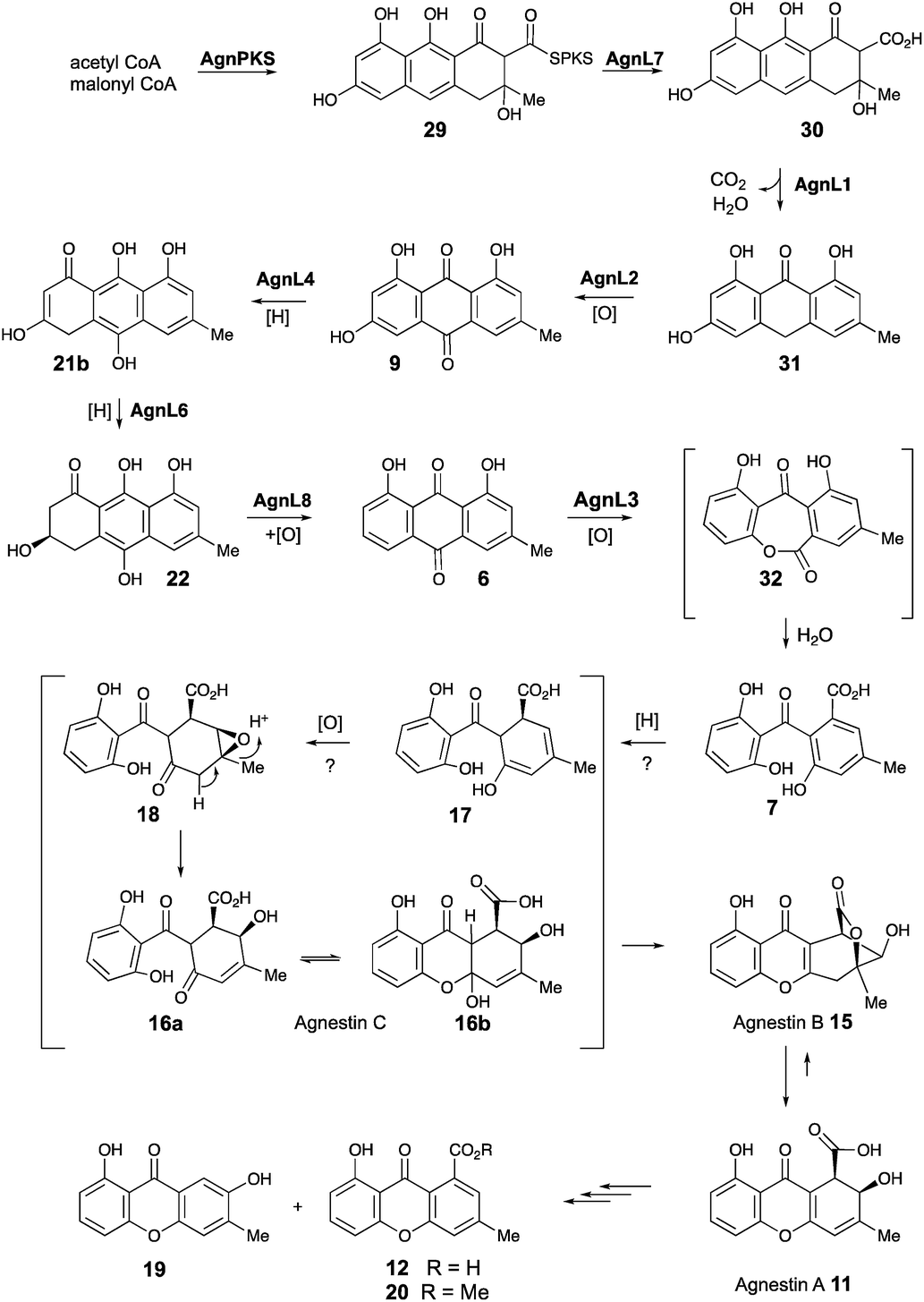 | ||
| Scheme 4 Biosynthesis of agnestins and related phenolic metabolites in P. variotii. | ||
Further conversion to agnestins A 11 and B 15 (Scheme 4), requires reduction to dihydro-monodictyphenone 17, oxidation to agnestin C 16ab probably via18, and rearrangement to either agnestin A 11 or agnestin B 15 directly, although we have demonstrated that 11 and 15 also interconvert. Examination of the agn cluster, reveals agnR1 as the only unassigned oxidoreductase encoding gene present which could be involved in this conversion. KO of agnR1, however, revealed it is not involved in the pathway (see ESI†), and thus genes involved in the proposed oxidation/reduction may be located elsewhere on the genome of P. variotii. Such split BGCs have been observed before in related systems, for example prenyl transferases involved during the biosynthesis of the shamixanthones in A. nidulans are not encoded within the mdp BGC itself.11 The remaining metabolites 12, 19 and 20 are probably formed by spontaneous decarboxylations, dehydrations and methanolysis reactions (Scheme 4).
Thus we have identified the protein responsible for the first essential reductive step in the aromatic deoxygenation of anthraquinones, e.g. emodin 9 to chrysophanol 6. The chemical requirement for this had been elegantly demonstrated by Müller and co-workers using an initial chemical reduction step. We have now firmly established the genetic and biochemical basis for this important process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank BBSRC (BB/J006289/1 and Bristol Centre for Synthetic Biology BB/L01386X/1) and Syngenta for funding. LCMS were provided by EPSRC (EP/F066104/1) and DFG (INST 187/621). 500 MHz NMR (EP/L011999/1) was provided by EPSRC. P. variotii was a gift from Syngenta and was sequenced at The Genome Analysis Centre (Norwich, UK) under contract to Syngenta through Genome Enterprises Ltd.Notes and references
- K.-S. Masters and S. Brase, Chem. Rev., 2012, 112, 3717–3776 CrossRef CAS PubMed
; T. Wezeman, S. Brase and K. S. Masters, Nat. Prod. Rep., 2015, 32, 6–28 RSC
- K. M. Henry and C. A. Townsend, J. Am. Chem. Soc., 2005, 127, 3724–3733 CrossRef CAS PubMed
- A. J. Birch, J. Baldas, J. R. Hlubucek, T. J. Simpson and P. W. Westerman, J. Chem. Soc., Perkin Trans. 1, 1976, 898–904 RSC
- S. A. Ahmed, E. Bardshiri, C. R. McIntyre and T. J. Simpson, Aust. J. Chem., 1992, 45, 249–274 CrossRef CAS
- B. V. Franck and F. H. Münster, Fortschr. Chem. Org. Naturst., 1973, 30, 151–206 CAS
- P. S. Steyn, Tetrahedron, 1970, 26, 51–57 CrossRef CAS PubMed
- A. Rhodes, G. A. Somerfield and M. P. McGonagle, Biochem. J., 1963, 88, 49–357 CrossRef
- I. Fujii, Y. Ebizuka and U. Sankawa, J. Biochem., 1988, 103, 878–883 CrossRef CAS PubMed
- M. L. Nielsen, J. B. Nielsen, C. Rank, M. L. Klejnstrup, D. M. K. Holm, K. H. Broggard, B. J. Hansen, J. C. Frisvad, T. O. Larsen and U. H. Mortensen, FEMS Microbiol. Lett., 2011, 321, 157–166 CrossRef CAS PubMed
- Y.-M. Chiang, E. Szewczyk, A. D. Davidson, R. Entwhistle, N. P. Keller, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2011, 76, 2067–2074 CrossRef PubMed
- J. F. Sanchez, R. Entwistle, J.-H. Hung, J. Yaegashi, S. Jain, Y.-M. Chiang, C. C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS PubMed
- M. T. Nielsen, J. B. Nielsen, D. C. Anyaogu, D. K. Holm, K. F. Nielsen, T. O. Larsen and U. H. Mortensen, PLoS One, 2013, 8, e72871 CrossRef CAS PubMed
- R. Schor and R. J. Cox, Nat. Prod. Rep., 2018, 35, 230–256 RSC
- S. Griffiths, C. Mesarich, B. Saccomanno, A. Vaisberg, P. Wit, R. Cox and J. Collemare, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6851–6856 CrossRef CAS
- K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2016, 55, 6784–6788 CrossRef CAS PubMed
- A. J. Wozniak, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 15, 17088–17091 Search PubMed
- A. Krick, S. Kehraus, C. Gerhäuser, K. Klimo, M. Nieger, A. Maier, H.-H. Fiebig, I. Atodiresei, G. Raabe, J. Fleischhauer and G. M. König, J. Nat. Prod., 2007, 70, 353–360 CrossRef CAS PubMed
- T. Asai, S. Otsuki, H. Sakurai, K. Yamashita, T. Ozeki and Y. Oshima, Org. Lett., 2013, 15, 2058–2061 CrossRef CAS PubMed
- Crystallographic coordinates for 11 and 15 have been deposited with the CCDC with accession numbers 1839028 and 1839029 respectively..
- A. Krick, S. Kehraus, C. Gerhäuser, K. Klimo, M. Nieger, A. Maier, H.-H. Fiebig, I. Atodiresei, G. Raabe, J. Fleischhauer and G. König, J. Nat. Prod., 2007, 70, 353–360 CrossRef CAS PubMed
- M. W. Sumarah, E. Puniani, B. A. Blackwell and J. D. Miller, J. Nat. Prod., 2008, 71, 1393–1398 CrossRef CAS PubMed
- J. S. E. Holker, E. O Brien and T. J. Simpson, J. Chem. Soc., Perkin Trans. 1, 1983, 1365–1368 RSC
- R. J. Ogg, P. B. Kingsley and J. S. Taylor, J. Magn. Reson., Ser. B, 1994, 104, 1–10 CrossRef CAS
- S. Tarcz, X. Xie and S.-M. Li, RSC Adv., 2014, 4, 17986–17992 RSC
- C. Li, J. Zhang, C. Shao, W. Ding, Z. She and Y. Lin, Chem. Nat. Compd., 2011, 47, 382–384 CrossRef CAS
- K. Throckmorton, P. Wiemann and N. Keller, Toxins, 2015, 7, 3572–3607 CrossRef CAS PubMed
- P. Reihl and J. Stolz, J. Biol. Chem., 2005, 280, 39809–39817 CrossRef CAS PubMed
- C. P. Woloshuk, K. R. Foutz, J. F. Brewer, D. Bhatnagar, T. E. Cleveland and G. A. Payne, Appl. Environ. Microbiol., 1994, 60, 2408–2414 CAS
- Y. Kubo, Y. Takano, N. Endo, N. Yasuda, S. Tajima and I. Furusawa, Appl. Environ. Microbiol., 1996, 62, 4340–4344 CAS
- V. Tiranti, C. Viscomi, T. Hildebrandt, I. Di Meo, R. Mineri, C. Tiveron, M. D. Levitt, A. Prelle, G. Fagiolari, M. Rimoldi and M. Zeviani, Nat. Med., 2009, 15, 200–205 CrossRef CAS PubMed
- N. P. Keller, N. J. Kantz and T. H. Adams, Appl. Environ. Microbiol., 1994, 60, 1444–1450 CAS
- J. W. Cary, K. C. Ehrlich, J. M. Bland and B. G. Montalbano, Appl. Environ. Microbiol., 2006, 72, 1096–1101 CrossRef CAS PubMed
- K. C. Ehrlich, B. Montalbano, S. M. Boue and D. Bhatnagar, Appl. Environ. Microbiol., 2005, 71, 8963–8965 CrossRef CAS PubMed
- K. C. Ehrlich, P. Li, L. Scharfenstein and P.-K. Chang, Appl. Environ. Microbiol., 2010, 76, 3374–3377 CrossRef CAS PubMed
- A. Watanabe, I. Fujii, U. Sankawa, M. E. Mayorga, W. E. Timberlake and Y. Ebizuka, Tetrahedron Lett., 1999, 40, 91–94 CrossRef CAS
- M. L. Nielsen, L. Albertsen, G. Lettier, J. B. Nielsen and U. H. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed
- T. J. Simpson, ChemBioChem, 2012, 13, 1680–1688 CrossRef CAS PubMed
- M. A. Schätzle, S. M. Husain, S. Ferlaino and M. Müller, J. Am. Chem. Soc., 2012, 134, 14742 CrossRef PubMed
- D. Conradt, M. A. Schätzle, J. Haas, C. A. Townsend and M. Müller, J. Am. Chem. Soc., 2015, 137, 10867–10869 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: All experimental and characterisation details. CCDC 1839028 and 1839029. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03778g |
| This journal is © The Royal Society of Chemistry 2019 |