
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDalmanol biosyntheses require coupling of two separate polyketide gene clusters†
Zhen Zhen
Zhou
a,
Hong Jie
Zhu
a,
Li Ping
Lin
bc,
Xuan
Zhang
a,
Hui Ming
Ge
 a,
Rui Hua
Jiao
a and
Ren Xiang
Tan
*ab
a,
Rui Hua
Jiao
a and
Ren Xiang
Tan
*ab
aState Key Laboratory of Pharmaceutical Biotechnology, Institute of Functional Biomolecules, Nanjing University, Nanjing 210023, China. E-mail: rxtan@nju.edu.cn
bState Key Laboratory Cultivation Base for TCM Quality and Efficacy, Nanjing University of Chinese Medicine, Nanjing 210023, China
cState Key Laboratory Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 27th November 2018
Abstract
Polyketide–polyketide hybrids are unique natural products with promising bioactivity, but the hybridization processes remain poorly understood. Herein, we present that the biosynthetic pathways of two immunosuppressants, dalmanol A and acetodalmanol A, result from an unspecific monooxygenase triggered hybridization of two distinct polyketide (naphthalene and chromane) biosynthetic gene clusters. The orchestration of the functional dimorphism of the polyketide synthase (ChrA) ketoreductase (KR) domain (shortened as ChrA KR) with that of the KR partner (ChrB) in the bioassembly line increases the polyketide diversity and allows the fungal generation of plant chromanes (e.g., noreugenin) and phloroglucinols (e.g., 2,4,6-trihydroxyacetophenone). The simultaneous fungal biosynthesis of 1,3,6,8- and 2-acetyl-1,3,6,8-tetrahydroxynaphthalenes was addressed as well. Collectively, the work may symbolize a movement in understanding the multiple-gene-cluster involved natural product biosynthesis, and highlights the possible fungal generations of some chromane- and phloroglucinol-based phytochemicals.
Introduction
All organisms, particularly single-celled microbes, produce arrays of structurally unforeseeable small molecules (or natural products), which are scientifically termed as secondary metabolites (SMs) owing to their indirect involvement in central metabolism.1 Attention to SMs has led to the discovery of renowned antibiotics (e.g., penicillin and lovastatin), agrochemicals (e.g., beauvericin and gibberellin), and other useful chemicals.2,3 However, the hit rate of finding such sorts of valuable SMs from nature declines substantially owing to: (i) the reisolation of known compounds4 and (ii) the lack of sufficient amounts of SM samples for an in-depth usability evaluation.5 But in theory, the microbial SMs identified so far occupy only a tiny portion of the chemical space that outnumbers 1060 predictable molecules with the molecular weight smaller than 500 Da.6 The question raised in this regard is how to access and expand the skeletal diversity and productivity of natural products.7 Of particular significance is the knowledge about the logic and mechanisms involved in regulating microbial SM biosynthesis at a gene cluster level.8,9In plants, the genes responsible for the phytochemical biosynthesis are usually scattered in different parts to facilitate the efficient organ-based responses to biotic and abiotic stresses,10 except for a few gene clusters encoding the specialized metabolic pathways.11 In fungi and bacteria, the genes that govern the assembly of a particular secondary metabolite are usually arranged in a cluster.12 As an expansion to the “dogma”, the biosynthetic gene cluster of polyketides interacts with those of terpenoids to produce meroterpenoids (e.g., austinol and dehydroaustinol) composed typically of polyketide- and isoprenoid-featured substructures.13 Inspired by such hybridized forms of SMs,14 we were curious about the possible interaction between different polyketide biosynthetic gene clusters, and if existing such inter-cluster couplings may multiply remarkably the skeletal diversity of microbial polyketides.15
Dalmanol A (1) and acetodalmanol A (2), sharing a unique 6/6/6/6/6 ring system, were obtained as immunosuppressive agents from mantis-associated fungus Daldinia eschscholzii (Fig. 1A).16 Our previous16 and present attention to its SM profile confirms that the fungus produces, in addition to 1 and 2, two groups of pentaketides with one and two benzene nuclei. The pentaketides with a benzene ring include 1-(2,6-dihydroxyphenyl)butane-1,3-dione (DPB) derived hemiketal (3), 1-(2,6-dihydroxyphenyl)but-2-en-1-one (PBEO, 4), 1-(2,6-dihydroxyphenyl)butan-1-one (5), 5-hydroxy-2-methyl-4H-chromen-4-one (6), and 2,3-dihydro-5-hydroxy-2-methylchromen-4-one (7) (Fig. 1B). The polyketides with two aromatic nuclei are exemplified by 1,3,6,8-tetrahydroxynaphthalene (4HN) and acetyl-1,3,6,8-tetrahydroxynaphthalene (A4HN).16,17 This observation encouraged us to hypothesize that 1 and 2 could be biosynthesized through the hybridization of chromane-related precursor(s) with 4HN and A4HN, which were also presumed to be involved in the construction of dalmanol B and acetodalmanol B.16 Since no more polyketide synthase (PKS) gene is included in the gene clusters controlling the assembly of 4HN17 and A4HN,18 we hypothesized that 1 and 2 could be among the compounds that may showcase the mechanism underlying the coupling of two separate polyketide biosynthetic gene clusters. Here, the intra- and intercellular couplings between the two distinct (naphthalene- and chromane-encoding) polyketide gene clusters is addressed to play a decisive role in the biosynthesis of 1 and 2 with the cross-cluster hybridization most likely triggered by an unspecific monooxygenase found in fungi. In addition to ascertaining the simultaneous fungal biosynthesis of 4HN and A4HN, the functionally dimorphic orchestration of the ketoreductase (KR) domain and KR partner in the chromane bioassembly line is demonstrated to allow the fungal production of the plant chromones and phloroglucinols.
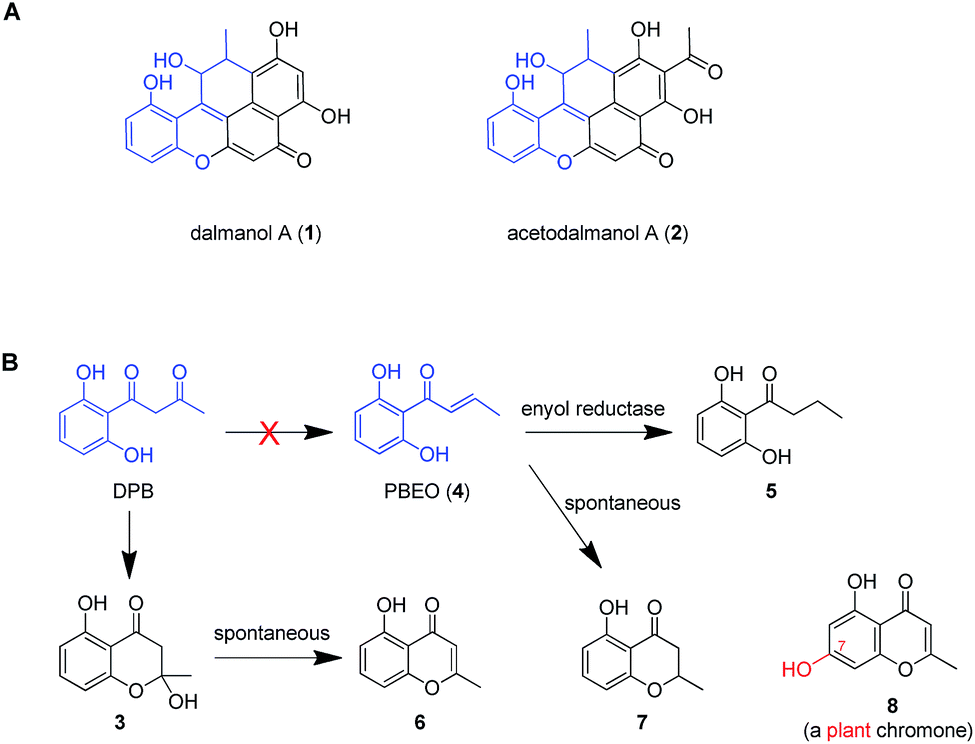 | ||
| Fig. 1 Representative frameworks of chromane-related polyketides. (A) Dalmanol A (1) and acetodalmanol A (2) signify hybridizations between the chromane and naphthalene biosynthetic pathways. (B) The biosynthetic network of fungal pentaketides 5–7 that are skeletally identical with noreugenin (8) from plant (Rheum palmatum).20 DPB, 1-(2,6-dihydroxyphenyl)butane-1,3-dione. PBEO (4), 1-(2,6-dihydroxyphenyl)but-2-en-1-one. | ||
Results
Identification of individual gene clusters encoding chromane- and naphthalene-based polyketides
The investigation was facilitated by our whole-genome sequencing of the D. eschscholzii genome while addressing the biosynthetic pathways of dalesconols A–C.17 As supposed, co-present in the fungal genome are the genes encoding both partially reducing (PR) and non-reducing (NR) PKSs. In view of the attribution of the NR-PKS to the 4HN generation,17 the PR-PKS might be simultaneously involved in the production of 1, 2, and 5–7 (Fig. 1), which are abundant enough to be analyzed by the liquid chromatography hyphened with high-resolution mass spectrometry (LC-HR/MS).16 Both type III and type I PKSs (including NR-PKS and PR-PKS) have been found to involve in the aromatic polyketide biosynthesis in fungi and plants.19 In particular, a type III PKS catalyzes in plants (e.g., Rheum palmatum) the biosynthesis of noreugenin (8),20 a 7-hydroxylated derivative of 6 (Fig. 1B). We were prompted to delete first the only type III PKS gene (GME9039) in the D. eschscholzii genome. However, such a mutation procedure exerted no effect on the production of 1, 2, and 5–7, as indicated by the LC-HR/MS screening of the EtOAc extract of the mutant culture. Therefore, the type I PKS is required for the assembly of these polyketides. Thus, the gene expression level of all NR- and PR-PKSs (Table S2†) in the fungal genome was assessed using quantitative reverse transcription PCR (qRT-PCR). Highly expressed were an NR-PKS (encoded by GME5781) and a PR-PKS (encoded by GME8183) (Fig. S1†). The NR-PKS is pksTL which was demonstrated to catalyze the biosynthesis of naphthalene-based dalesconols A–C.17 The PR-PKS encoded by GME8183, hereafter referred to as ChrA, might catalyze the architectural construction of the chromane-based pentaketides such as 5–7. The chromane-encoding gene cluster seems to be widespread in the fungal kingdom since these pentaketides and/or their congeners have been characterized from taxonomically distant fungi such as Daldinia loculata,21Cryptosporiopsis sp.,22Phialophora gregata,23 and Nodulisporium sp.24 To confirm the postulation, the ChrA gene was deleted from the fungal genome to obtain the ΔChrA mutant, which was shown to produce none of 1 and 2 as evidenced from the LC-HR/MS screening of the extract derived from its culture (Fig. 2B). Bioinformatic analysis showed that ChrA encodes a type I PR-PKS (ChrA) with 2089 amino acid residues, containing the domains of β-ketoacyl synthase (KS), acyltransferase (AT), dehydratase (DH), C-methyltransferase (MT), ketoreductase (KR), and acyl carrier protein (ACP) (Table S3†). Since 1 and 2 are non-methylated, the MT domain of ChrA was assumed to be deficient in spite of its possession of the residues (such as D/ExGxGxG) required for S-adenosyl-L-methionine (SAM) binding and methylation (Fig. S2†).25 Such a methyltransferase deficiency was noticed earlier in some eukaryotic cells.26 Adjacent to the ChrA gene is ChrB, which was found to encode a KR partner. Deletion of ChrB abolished the production of 1 and 2. As expected, the naphthol trimers, dalesconols A and B, became more abundant in the ΔChrA and ΔChrB mutant cultures (Fig. S15†). Next, deletion of the pksTL gene from the fungal genome as detailed17 gave the ΔpksTL mutant which was unable to generate 1 and 2 as well as dalesconols A and B (Fig. 2B and S17†). Similarly, more accumulation of chromane-based pentaketides (5–7, 9, and 10) was observed in the ΔpksTL mutant (Fig. S16†). Moreover, the formation of 1 was restored by supplementing 4HN in the ΔpksTL mutant culture (Fig. S22†). Accordingly, the fungal construction of 1 and 2 requires the co-participation of the naphthalene- and chromane-biosynthesizing gene clusters. These two clusters were found to reside in scaffolds 20 and 36 respectively, and are at least 493 kb apart even if the two scaffolds anchored most closely on the same chromosome (Fig. 2A and S4†).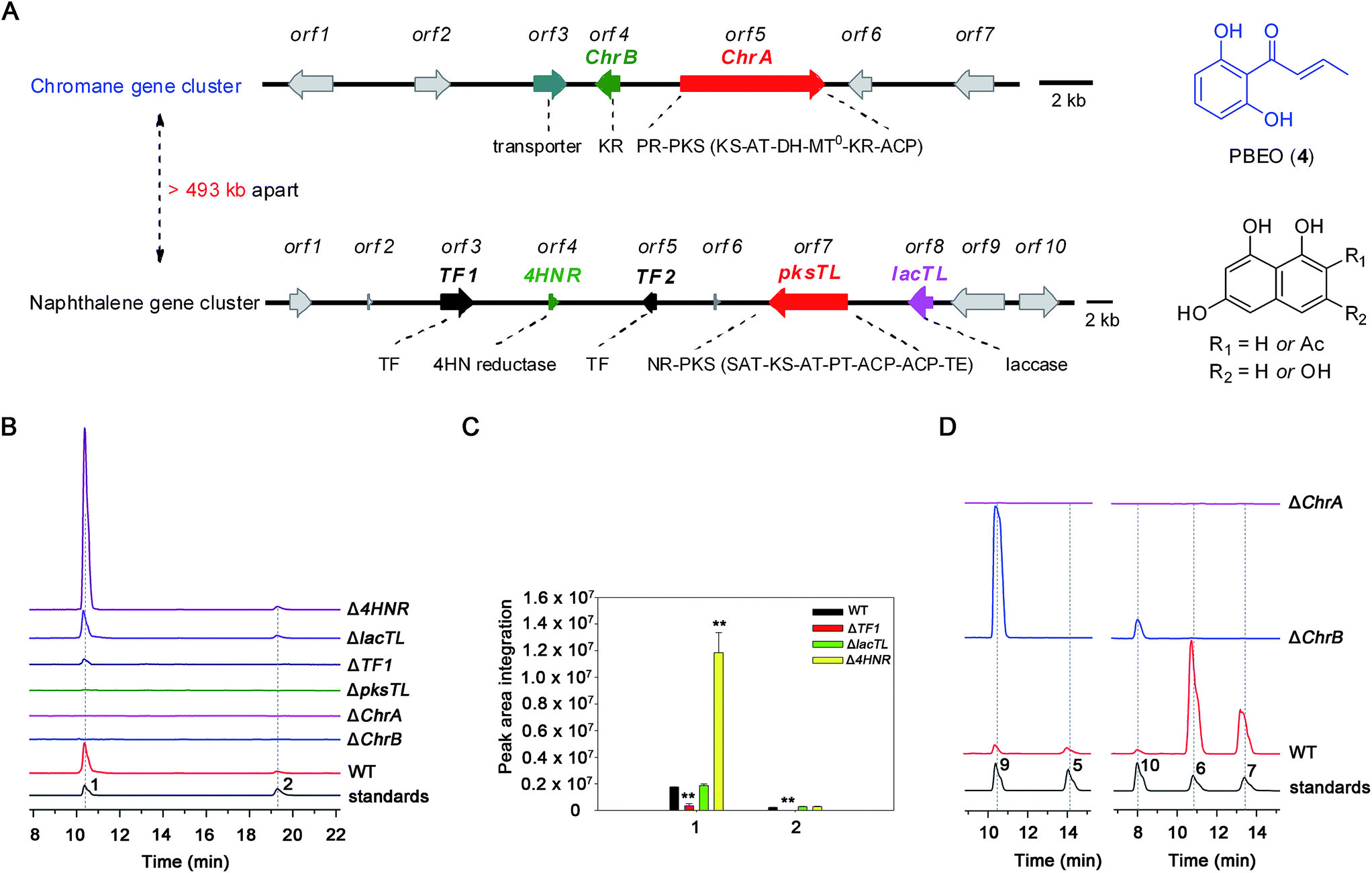 | ||
| Fig. 2 Polyketide productivity comparison among the wild-type (WT) and mutants of D. eschscholzii. (A) The two gene clusters encoding naphthalene and chromane. The WT strain and mutants (Δ4HNR, ΔlacTL, ΔTF1, ΔpksTL, ΔChrA, and ΔChrB) were compared through the LC-HR/MS profiling for the production of: (B) dalmanol A (1) and acetodalmanol A (2) (peak areas integrated (C)) and (D) 5–7, 9 and 10, with authentic samples co-analyzed as references. Data shown as mean ± SD (n = 3). ** indicated P < 0.01, by Student's t test. | ||
Simultaneous biosynthesis of 1,3,6,8- (4HN) and 2-acetyl-1,3,6,8-tetrahydroxynaphthalenes (A4HN)
The above observation prompted us to hypothesize that the abundance of 1 and 2 could be influenced by the fluctuation in the 4HN and A4HN availability. To address the assumption, the 4HN reductase (4HNR) encoding gene (4HNR) was knocked out to give the Δ4HNR mutant, which displayed an escalated abundance of 1, but with a negligible change in the production of 2 (Fig. 2B and C). Meanwhile, the chromane-based pentaketides such as 6 and 7 was discerned to be less abundant in the Δ4HNR mutant (Fig. S16†). Furthermore, deletion of the laccase encoding gene (lacTL) exerted no effect on the abundance of 1 and 2 (Fig. 2B and C). Such mutation results signalled the co-involvement of the pksTL, ChrA, and ChrB genes in the generation of 1 and 2, while confirming the participation of 4HNR in the biosynthesis of melanin18 and dalesconols A–C (Table S4†).17 In particular, the 4HNR deletion from the fungal genome results exclusively in the 4HN boost, which in turn increased the production of 1, but not 2. This observation raised our curiosity about the fungal generation of 4HN and A4HN. Previously, 4HN was reported to result largely from the A4HN deacetylation through different mechanisms.18,27 For example, in Wangiella dermatitidis, 4HN is produced by deacetylating A4HN under the catalysis of WdYg1p, a serine hydrolase homolog of Ayg1p.18 In Colletotrichum lagenarium, the deacetylation of A4HN into 4HN is catalyzed by the bifunctional thioesterase (TE) domain of the PKS, which is simultaneously involved in the cyclization of the enzyme-bound monocyclic hexaketide intermediate.27 The TE domain of pksTL in D. eschscholzii bears 46% and 81% sequence identity with those of W. dermatitidis (WdPKS1)18 and C. lagenarium (PKS1)27 (Fig. S14†).Moreover, some fungal PKS TE domain could be substrate-promiscuous as evidenced both from the simultaneous production of the skeletally alike penta- and hexaketides,16,27 and from the earlier observation that TE domain is not a determinant of the chain-length specificity of polyketides.28 We intuited that there might exist a ‘parallel production mechanism’ for 4HN and A4HN, namely, the TE domain of pksTL in D. eschscholzii might be able to catalyze in parallel the constructions of 4HN and A4HN from the penta- and hexa-ketide precursors, respectively. Herein, this assumption was reinforced by taking the advantage of the involvement of 4HN and A4HN in the biosynthesis of 1 and 2, respectively (Fig. 3). The rationalization seems obvious. The 4HNR deletion quenches the reduction of 4HN into 1,3,8-trihydroxynaphthalene (3HN)17 to generate a ‘4HN boost’. Such a sharp escalation of the 4HN level should lead to a clearly increased A4HN accumulation if 4HN was formed via the A4HN deacetylation process as reported.18,27 However, our deletion of the 4HNR gene enriched 1 only, without substantial effect on the production of 2, suggesting that the A4HN availability was negligibly dependent, or independent, of the 4HN boost resulting from the 4HNR deletion (Fig. 2B and C). This observation could be more convincingly reasoned by the parallel generation of 4HN and A4HN in D. eschscholzii (Fig. 3), although that the pksTL-catalyzed generation of 4HN as in case of PKS1 in C. lagenarium27 (Fig. 3) could not be definitely ignored.
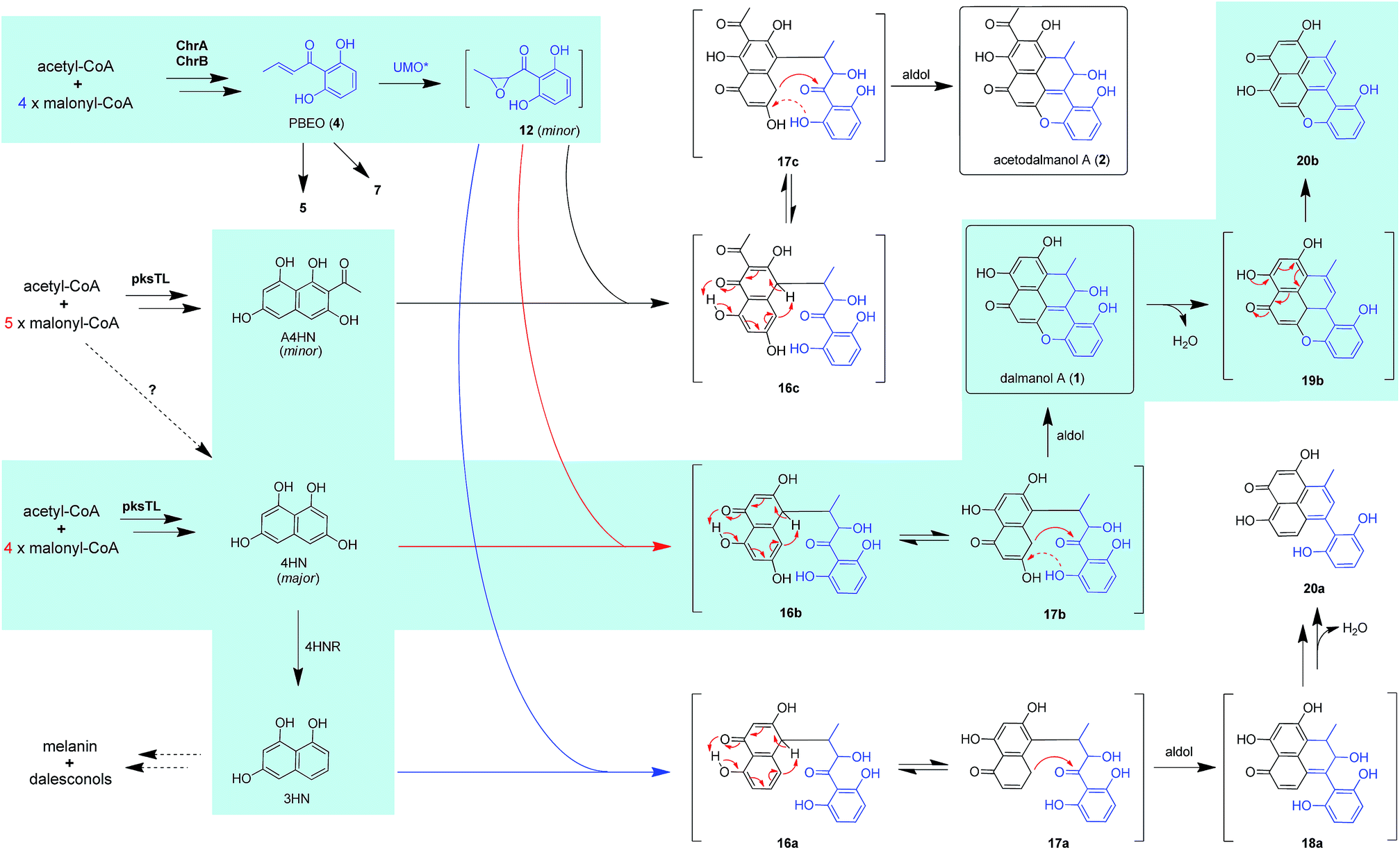 | ||
| Fig. 3 Proposed biosynthetic pathways for dalmanol A (1) and acetodalmanol A (2). The dalmanol carbon skeleton formation requires the naphthalene and chromane biosynthetic gene clusters, which interact intra- and intercellularly with the presumable participation of an unspecific fungal monooxygenase expressible in D. eschscholzii and A. oryzae. UMO*, unspecific monooxygenase. Dotted and solid red arrows in 17b and 17c indicate the electron transfer in the intramolecular addition and aldol reactions, respectively. | ||
Functional analysis of transcription factors in naphthalene biosynthetic gene cluster
Bioinformatic analysis indicated the presence of two transcription factor genes (TF1 and TF2) in the naphthalene-encoding gene cluster (Fig. 2A and Table S4†). Deletion of TF1 from the fungal genome significantly reduced the abundance of 1 and completely abolished the production of 2. More abundant chromane-based pentaketides (5–7, 9, and 10) were produced by the ΔTF1 mutant (Fig. S16†). Overexpression of TF1 (TF1 OE) under the control of strong α-amylase (amyB) promoter greatly increased the amount of 1, but decreased that of 2 (Fig. S18†). This observation underpinned that TF1 was an upregulator specific for the naphthalene biosynthetic gene cluster (Fig. 2B and C), and that 4HN and A4HN might have to compete for the same outside-cluster molecular partner to form 1 and 2, respectively. To ascertain whether other naphthols such as 3HN are involved in the outside-cluster molecule, a large-scale fermentation of the TF1 OE strain was carried out to lead to the characterization of a new minor shunt compound 20a, which could be reasoned to be a hybrid of 3HN with the outside-cluster pentaketide intermediate (12). Co-existed with 20a was 20b (a dehydrated product of 1, Fig. 3), reinforcing the proposed competition among naphthols for the intermediate 12 (Fig. S56–S65†). The upregulation of naphthalene-encoding gene cluster by TF1 was confirmed by the negligible production of other naphthol-condensed biomolecules including melanin and dalesconols A and B16,17 by the ΔTF1 strain of D. eschscholzii (Fig S17†). Compared with that of wild type, more abundant chromane-related pentaketides (5–7, 9, and 10) were observed in the ΔpksTL or ΔTF1 mutants (Fig S16†). However, the abundance of pentaketides 6 and 7 was reduced upon deleting 4HNR, because of the increased availability of 4HN in the Δ4HNR mutant. Unfortunately, our repeated deletion of TF2 in the gene cluster was unsuccessful.Heterologous expression of individual gene clusters in Aspergillus oryzae
To better understand the inter-cluster coupling, we were motivated to identify/confirm the individual gene function in the naphthalene- and chromane-encoding gene clusters. However, pksTL and ChrA were unable to be expressed in Escherichia coli. The frustration was overcome by using as an expression host Aspergillus oryzae NSAR1, a quadruple auxotrophic mutant strain (niaD−, sC−, ΔargB, adeA−) as outlined elsewhere.33 As expected, expression of the pksTL gene in A. oryzae yielded 4HN as a major product (Fig. S19†). Co-expression of pksTL and 4HNR in A. oryzae afforded 3HN, thus confirming the 4HNR catalyzed 4HN reduction (Fig. S19†). Next, the ChrA gene was expressed in A. oryzae to produce 9 and 10, both accumulating in the ΔChrB mutant of D. eschscholzii (Fig. 2D and S20†). The ChrB gene was expressed alone in A. oryzae, which was unable to form 5 and 7 upon supplementing 9 and 10, respectively. Moreover, none of 5–7 was detectable in the co-culture of the transformants of ChrA-AO and ChrB-AO (Fig. S20, S26, and S27†). These data confirmed ChrA as a pentaketide synthase and ChrB as a KR partner capable of directing, at early-stage, the generation of 7-deoxygenated pentaketides 5–7 (Fig. 4). With this ascertained, such heterologous expressions were optimized to characterize the nascent intermediates 3 and 4 (Fig. S31–S41†). Furthermore, the co-transformant of ChrA and ChrB (ChrA/ChrB-AO) produced 3–7, 9, and 10. This observation suggested the production of 9 and 10via a shunt path which was in parallel or competition with the ChrB catalyzed production of 3–7 (Fig. 4). Furthermore, the conversion of 3 into 6, and 4 into 7, was shown to be non-enzymatic since such processes were accomplishable in sterile ME medium (Fig. 4, S24 and S25†). The biosynthesis of 10 is catalyzed by ChrA, a type I PKS, which is distinct from the type III PKS involved in the biosynthesis of 8 in the plant R. palmatum (Fig. S13†).20 However, we were frustrated by the formation of 5 and 9 because no enoyl reductase-encoding gene could be found in the chromane biosynthetic gene cluster. This could only be explained by assuming the presence of an unspecific enoyl reductase outside the cluster,28 since 5 and 9 were detected in the cultures of D. eschscholzii and A. oryzae (a heterologous expression host used in the study). Such an unspecific enoyl reductase might be present in some, if not all, fungi since 5 was re-isolated from the cultures of other fungi such as Cryptosporiopsis sp.,22Phialophora gregata23 and Nodulisporium sp.24 Moreover, the ChrA KR domain might function as a bifunctional ‘gate-keeper’ that allowed the generation of 3 (when inactive) or 4 (if active), in collaboration with ChrB as a KR partner (Fig. 4). This set of heterologous (co-)expressions pinpointed the metabolic network about pentaketides 3–7 and 9–10 (Fig. 4; pathways II, III, and IV).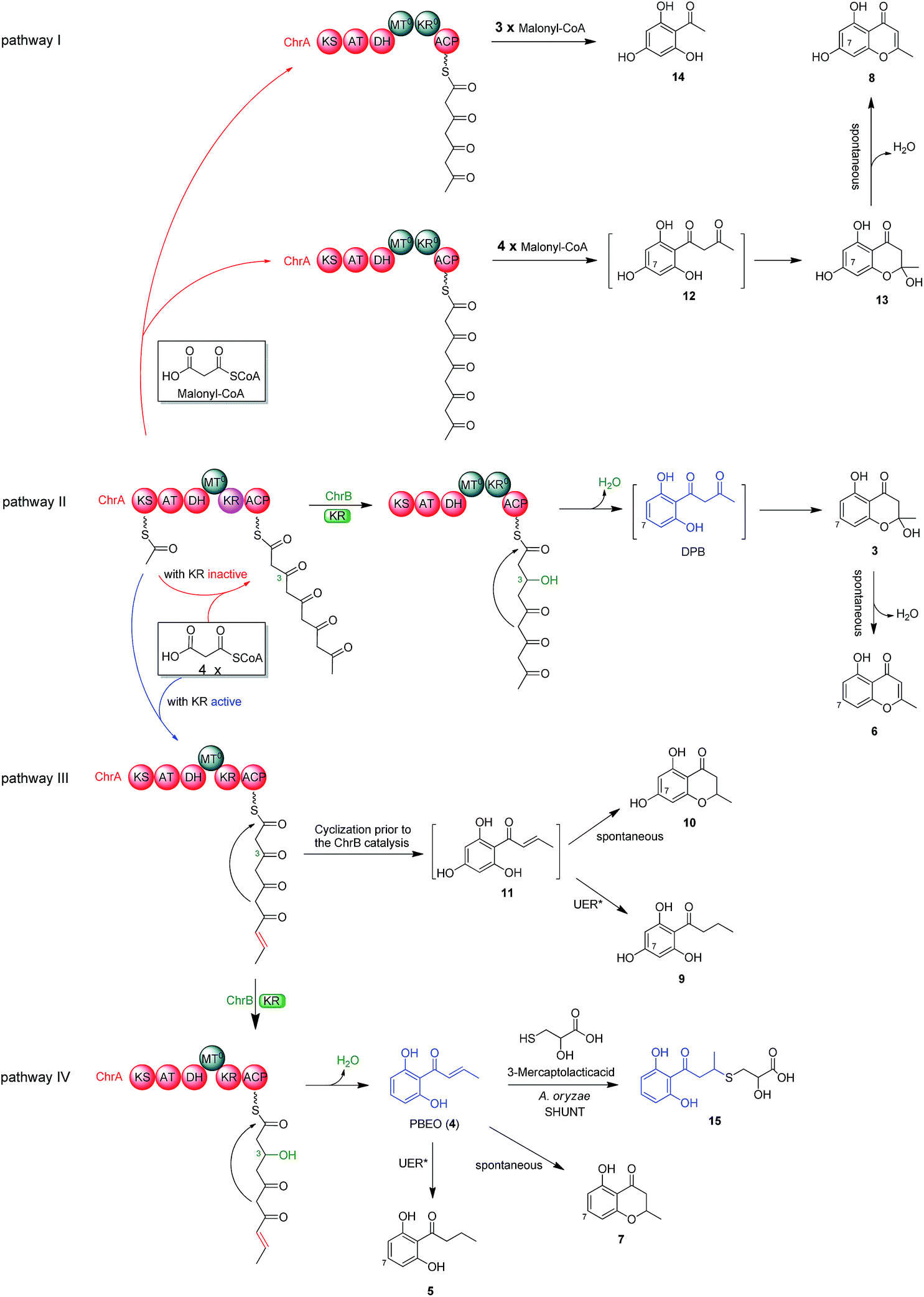 | ||
| Fig. 4 Metabolic network of polyketides encoded by the chromane gene cluster in D. eschscholzii. ChrA is a partially reducing polyketide synthase (PR-PKS) with the domains of β-ketoacylsynthase (KS), acyltransferase (AT), dehydratase (DH), C-methyltransferase (MT0, inactive), ketoreductase (KR, active/inactive) and acyl carrier protein (ACP). ChrB is a ketoreductase partner that catalyzes the selective 3-ketone reduction of acyclic pentaketide with acetonyl or allyl side-chain terminals. UER*, unspecific enoyl reductase expressible in D. eschscholzii and A. oryzae. | ||
Heterologous expression of KR domain mutated ChrA in A. oryzae
The aforementioned heterologous expression of ChrA in A. oryzae afforded a new pentaketide 13, which is spontaneously transformed to 8 (Fig. S21†), a plant chromone.20 To understand the catalytic feature of the KR domain of ChrA, three conserved catalytic residues (Lys, Ser, and Tyr) were sequentially mutated as outlined (Fig. S3†).29 Analogously, the catalytic residues (Lys, Ser, Tyr, and Asn) in ChrA was identified as described by Yi Tang and co-workers.29 The KR-mutated ChrA genes (ChrA-M1 for K1829D, ChrA-M2 for S1853A, and ChrA-M3 for Y1866A) were individually expressed in A. oryzae to produce, in addition to 10 and 13, the fungal tetraketide 14 (viz., 2,4,6-trihydroxyacetophenone).16 In comparison to 13, an acetyl unit reduction in 14 was noticed, suggesting that such KR mutations could reduce the extension cycle specificity of ChrA. This phenomenon was discerned when addressing terrein biosynthesis in Aspergillus terreus.28 This observation was substantiated by the production of 14 upon co-expressions of ChrB with original and KR-mutated ChrA genes (Fig. 5, traces vi–ix). However, such co-expressions generated four additional peaks, three of which were shown to arise from 3, 6, and 7 (Fig. 5, traces vii–ix). The fourth peak was identified to be 15, which is an adduct of 4 with 3-mercaptolactic acid (a described fungal metabolite) formed via an outlined pathway (Fig. 4, pathway IV).30 The above observations could only be explained by the functional dimorphism (active and silent states) of ChrA KR domain that serves as a gate-keeper to diversify the chromane-based polyketides (Fig. 4). It is noteworthy that the KR mutations mitigated, but not abolished as discerned,29 the function of the fungal ChrA.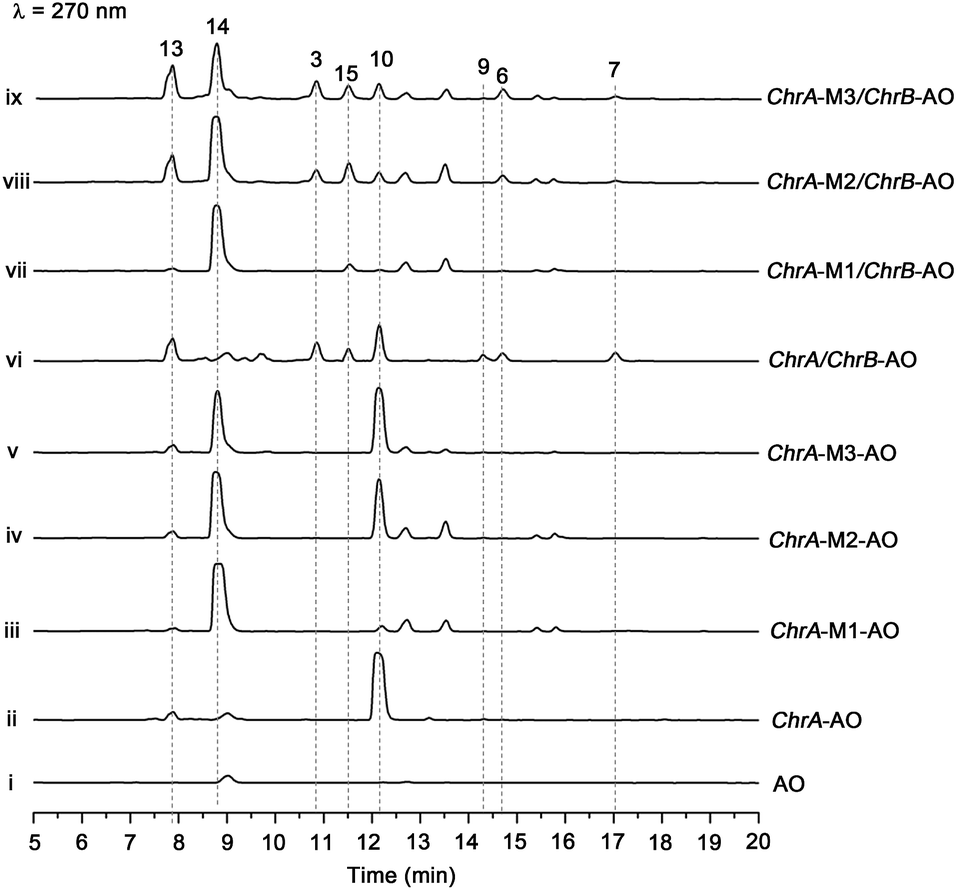 | ||
| Fig. 5 Co-expression of KR-mutated ChrA and ChrB in A. oryzae (AO). HPLC traces for metabolites produced by AO ((i), as a blank reference), the transformants [with ChrA (ii), ChrA-M1 for K1829D (iii), ChrA-M2 for S1853A (iv), and ChrA-M3 for Y1866A (v)], and the co-transformants with ChrA and ChrB (vi), ChrA-M1 and ChrB (vii), ChrA-M2 and ChrB (viii), and ChrA-M3 and ChrB (ix). | ||
Coupling between chromane- and naphthalene-encoding gene clusters
Scrutiny of the structural feature of 1 and 2 prompted us to hypothesize that the PBEO (4) epoxidation might be required for the coupling between the naphthalene and chromane biosynthetic gene clusters. The stability of 4 was examined indicating that it is relatively stable in acetone and unable to be oxidized with air exposure, and tends to form 7 in MeOH, DMSO, and the culture medium (as trapped by xenobiotic indoles31) (Fig. S25†). With the rationalization, 4 was likely epoxidized into 12 under a monooxygenase catalysis, followed by independent couplings with 4HN and A4HN to give 1 and 2, respectively (Fig. 3). To address the assumption, ChrA and 4HNR were co-deleted from the D. eschscholzii genome to afford the ΔChrA/Δ4HNR mutant, which was reasoned to boost a 4HN accumulation without producing 3 and 4 (see above). As expected, the generation of 1 by the ΔChrA/Δ4HNR mutant was restored by addition of 4, but unrestorable by re-supplementing 3, suggesting that 4 and 4HN were precursors of 1 (Fig. S23†). The production of 2 failed to be restored by the experiment because A4HN is much less abundant and nearly independent of the 4HN boost (see above). By getting the two on-pathway intermediates in hand, 4 and 4HN were incubated together in ME medium used for culturing D. eschscholzii to see if spontaneous coupling could take place. However, 4 rapidly converted to 7 at 5 min, and completely converted to 7 at 30 min after co-incubation, but no coupling product was detected (Fig. S28†). These results showed that 4 and 4HN could not spontaneously coupled to generate 1. Furthermore, no monooxygenase-encoding gene was found within the chromane- and naphthalene-encoding gene clusters. The enzyme catalyzing the PBEO (4) epoxidation could be outside the clusters in the light of the alkene epoxidation catalyzed by cytochrome P450 monooxygenase32,33 or flavin-containing monooxygenase (FMO).34,35 Owing to its analogy to styrene, 4 could be epoxidized under the catalysis of styrene monooxygenase, an FMO found in bacteria.36 However, our bioinformatic analysis showed the absence of any bacterial styrene monooxygenase in the fungus (Fig. S29†). Previously, a fungal P450 monooxygenase was evidenced to catalyze the styrene metabolism initialized with the epoxidation of vinyl side-chain.37 In the D. eschscholzii genome,17 a total of 119 cytochrome P450s and 79 FMOs have been annotated (Table S10†). Since it is too tedious to identify the epoxidation-catalyzing enzyme of the fungus, we alternatively ascertained the epoxidase type via a gene-implied enzyme inhibition strategy.38 Thus, an FMO (methimazole) and two P450 inhibitors (phenylbutazone and proadifen) were supplemented separately in the fungal cultures. As a result, the fungal exposure to the P450 inhibitors substantially reduced the production of 1 and 2 whereas methimazole exerted negligible effect (Fig. S30†). Accordingly, the P450, but not FMO, monooxygenase(s) most likely catalyzed the epoxidation of 4 in the fungus.To confirm the assumption above, the two naphthalene and chromane biosynthetic gene clusters were co-expressed in A. oryzae. Gratifyingly, 1 and 2 were produced by the co-transformant with the pksTL, ChrA, and ChrB genes (pksTL/ChrA/ChrB-AO) (Fig. 6). These results indicated that the monooxygenase catalyzing the formation of 12 from 4 could be unspecific since expressible in both A. oryzae and D. eschscholzii.28 Accordingly, 1 and 2 result likely from the monooxygenase-triggered coupling between the naphthalene and chromane biosynthetic gene clusters. Moreover, the involvement of the unspecific enzyme in the inter-cluster coupling strengthened our curiosity about whether the naphthalene and chromane biosynthetic gene clusters would interact if located in different fungal cells. To address the hypothesis, the pksTL-AO transformant was co-cultured with the co-transformant with ChrA/ChrB-AO. As expected, 1 and 2 were formed upon the co-cultivation of these (co-)transformants (Fig. 6). This experimentation highlighted that the two polyketide gene clusters can interact intercellularly, too. The finding may be of considerable value in the rational design of the microbial co-culture approach which has been accepted as a promising access to the improved secondary metabolite chemodiversity for drug discovery.39
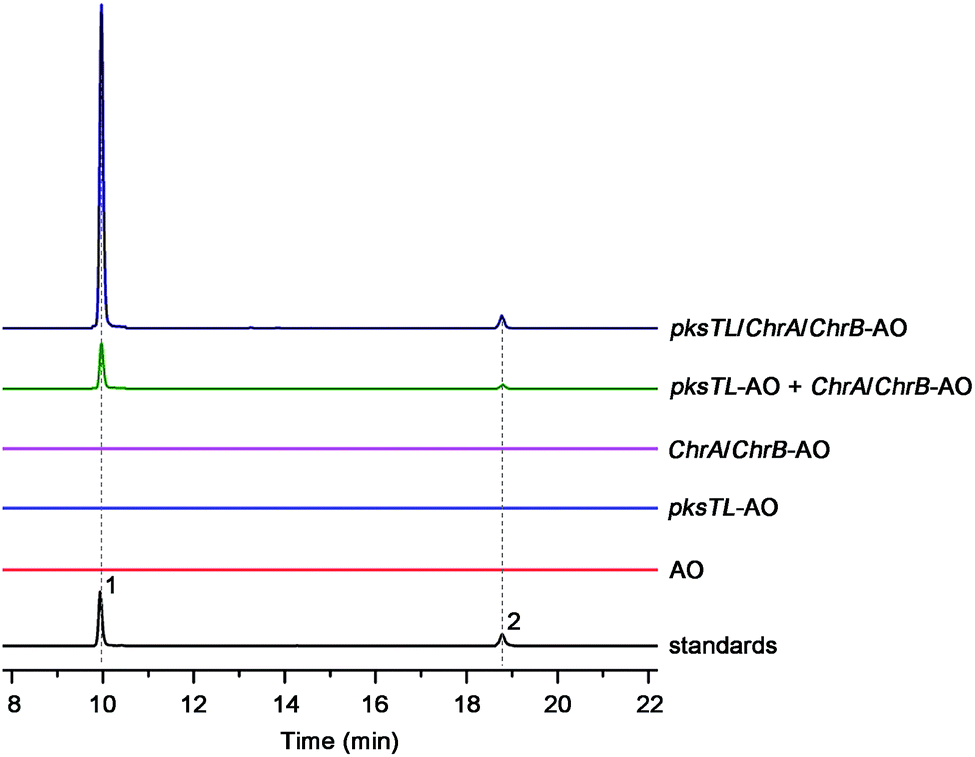 | ||
| Fig. 6 Production of 1 and 2via heterologous co-expression and co-cultivation. Co-expression of pksTL, ChrA, and ChrB in A. oryzae gave a co-transformant (pksTL/ChrA/ChrB-AO) capable of producing 1 and 2, which were afforded as well by co-culturing the transformant (pksTL-AO) with the co-transformant (ChrA/ChrB-AO). However, none of 1 and 2 was generated if culturing separately the pksTL-AO transformant and the ChrA/ChrB-AO co-transformant. | ||
Discussion
As versatile biosynthetic precursors of natural products16,40 and virulence factors such as melanin,18 4HN and/or A4HN are widespread polyketides in fungi (this work and many others) with the former producible in bacteria.41,42 Chromane-based polyketides have been characterized as the SMs from diverse fungi16,21–24 and plants.20,43 Also found are the natural hybrids of chromone with piperidine43 and of 4HN with chromane16,24 or isoprenoid motifs.40 Therefore, the fresh portion of small molecule chemical space could be occupied by SMs biosynthesizable through the ‘collaboration’ of multiple gene clusters. This rationalization has been reinforced herein by the elucidated biosynthesis of dalmanol A (1) and acetodalmanol A (2), which reveals the intra- and intercellular hetero-coupling between the naphthalene- and chromane-encoding gene clusters. The cross-cluster coupling manner underlying the formation of 1 and 2 is different from the meroterpenoid bioassembly (co-supported by the polyketide and terpenoid biosynthetic gene clusters),13 and within-cluster PKS pairings.44,45The cross-cluster coupling could be either spontaneous or enzymatic. Thus, PBEO (4) and 4HN were incubated together in media used for the fungal culture. To our surprise, 4 became 7 rather than reacted with 4HN (Fig. S28†). Thus, such a unique coupling could only be triggered by an unspecific monooxygenase found in some (if not all) fungi such as D. eschscholzii and A. oryzae.28 Such a monooxygenase is outside the two polyketide biosynthetic gene clusters and catalyzes the conversion of pentaketide precursor PBEO (4) to the epoxide intermediate 12 which is too labile or less abundant to be identified in the work. The difficulty in characterizing 12 might have been intensified by the competitive conversion of 4 to 7 and 5via the non-enzymatic intramolecular addition and the reduction catalyzed by an unspecific enoyl reductase expressible in D. eschscholzii and A. oryzae (Fig. 4, pathway IV). Similarly, an unspecific ketoreductase was found to involve in the terrein biosynthesis.28 This rationalization agrees with the oxygenation pattern of 1 and 2 and the monooxygenase-catalyzed 2,3-epoxidation of some indole motifs (a mimic of the polarized double bond of 4, Fig. 3) in the fungal culture.46 The inter-cluster coupling gains reinforcements from the production of 1 and 2via the heterologous co-expression of pksTL, ChrA and ChrB in A. oryzae, and through the co-culture of the two (co-)transformants, pksTL-AO and ChrA/ChrB-AO. The finding seems to be different from the post-biosynthesis coupling between small-molecule phenols via a non-enzymatic nucleophilic addition reaction47 and the phenolic radical couplings catalyzed by laccase,17 multicopper oxidase,48 and the within-cluster P450 enzymes.49,50 Interestingly, the addressed coupling between the naphthalene and chromane biosynthetic gene clusters enabled us to identify the simultaneous fungal biosynthesis of 4HN and A4HN. Along with the transformant co-culture protocol, the driving force for the cross-cluster coupling may help to rationally design and efficiently accomplish the generation of new SMs via microbial co-cultivation approaches.
The chromane-based polyketides are found in fungi (this work and others) and plants such as Aloe arborescens (family Aloaceae),51Dysoxylum acutangulum (Meliaceae),43Eugenia caryophyllata (Myrtaceae),52Crossosoma bigelovii (Crossosomataceae),53Knoxia corymbosa (Rubiaceae),54 and Schumanniophyton magnificum (Rubiaceae).55 Many chromane derivatives have promising biological activities.56 This work highlights the biosynthetic relationship of this family of SMs by genetic manipulation. Interestingly, both ChrA ketoreductase domain (ChrA KR) and ChrB as a KR partner in the same cluster are functionally dimorphic; namely, they exert or lose their catalytic activity during the assembly of chromane polyketides (Fig. 4). Such a KR dimorphism orchestration of ChrA KR and ChrB allows the generation of diversified chromane polyketides (Fig. 4). Previously, the KR domain dimorphism of polyketide synthases was believed to likely arise from a couple of mechanisms such as the module skipping57 and chain-length control.58,59 Our mutation confirmed that the KR domain affects some polyketide chain-length. In particular, the replacement of Lys1829 with Asp residue in ChrA KR afforded an remarkably escalated level of 2′,4′,6′-trihydroxyacetophenone (14), an anti-obesity phytochemical60 that serves as a (bio)synthetic starter motif for arrays of bioactive phloroglucinols.61,62 More interestingly, when ChrA KR and ChrB both lose the catalytic activity, the fungal chromane biosynthetic pathway constructs 13 that tends to form noreugenin (8), a plant chromone used as a common starting material for the synthesis of bioactive chromone natural products such as ptaeroxylin and eranthin.63 These findings suggest that the fungal chassis with such a useful and operable chromane biosynthetic gene cluster could be a proper host for the synthetic biology approaches to chromane- and phloroglucinol-based bioactive phytochemicals and/or precursors thereof.
Conclusions
We have addressed the two-gene-cluster based biosynthetic pathways of dalmanol A (1) and acetodalmanol A (2). Their unique carbon skeletons form via the monooxygenase triggered cascade reactions between a pair of polyketides constructed by two distinct polyketide (naphthalene and chromane) biosynthetic gene clusters. Such a cross-cluster interaction allowed the identification of the simultaneous fungal biosynthesis of 4HN and A4HN. A combination of transformant (co-)cultivations with gene deletions and heterologous (co-)expressions disclosed the metabolic network about pentaketides 3–7, 9, and 10, which are produced via the competitive shunt pathways switched likely by the functional dimorphism of ChrA KR and/or the nascent intermediate aromatization before the ChrB catalyzed cyclization (Fig. 4). In the chassis of D. eschscholzii and probably other chromane-synthesizing fungi,21–24 the ChrA with an inactivated KR domain seems to be of significance in establishing the synthetic biology approaches to the chromane- and phloroglucinol-based phytochemicals. Collectively, the work sheds light on the understanding of the multiple-gene-cluster based natural product biosynthesis, which is valuable for generating skeletally related phytochemicals and other small molecules with new chemical and biological space.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was co-financed by the NSFC (81530089, 21672101 and 21702099) and MOST grants (2018ZX09711001-007-004). We thank Professor Katsuhiko Kitamoto (University of Tokyo) for providing fungal heterologous expression host (A. oryzae NSAR1) and plasmids (pTAex3, pUSA and pAdeA).References
- S. W. Drew and A. L. Demain, Annu. Rev. Microbiol., 1977, 31, 343–356 CrossRef CAS PubMed.
- N. P. Keller, Nat. Chem. Biol., 2015, 11, 671–677 CrossRef CAS PubMed.
- T. Hautbergue, E. L. Jamin, L. Debrauwer, O. Puel and I. P. Oswald, Nat. Prod. Rep., 2018, 35, 147–173 RSC.
- J. Hubert, J. M. Nuzillard and J. H. Renault, Phytochem. Rev., 2017, 16, 55–95 CrossRef CAS.
- M. C. Leal, C. Sheridan, R. Osinga, G. Dionisio, R. J. M. Rocha, B. Silva, R. Rosa and R. Calado, Mar. Drugs, 2014, 12, 3929–3952 CrossRef CAS PubMed.
- A. Boufridi and R. J. Quinn, Annu. Rev. Pharmacol., 2018, 58, 451–470 CrossRef CAS PubMed.
- M. J. Smanski, H. Zhou, J. Claesen, B. Shen, M. A. Fischbach and C. A. Voigt, Nat. Rev. Microbiol., 2016, 14, 135–149 CrossRef CAS PubMed.
- X. N. Liu, W. T. Ding and H. F. Jiang, Microb. Cell Fact., 2017, 16, 125 CrossRef PubMed.
- P. Chettri, P. Y. Dupont and R. E. Bradshaw, Mol. Microbiol., 2018, 107, 508–522 CrossRef CAS PubMed.
- J. K. Weng, New Phytol., 2014, 201, 1141–1149 CrossRef PubMed.
- J. K. Weng, R. N. Philippe and J. P. Noel, Science, 2012, 336, 1667–1670 CrossRef CAS PubMed.
- Y. F. Li, K. J. S. Tsai, C. J. B. Harvey, J. J. Li, B. E. Ary, E. E. Berlew, B. L. Boehman, D. M. Findley, A. G. Friant, C. A. Gardner, M. P. Gould, J. H. Ha, B. K. Lilley, E. L. McKinstry, S. Nawal, R. C. Parry, K. W. Rothchild, S. D. Silbert, M. D. Tentilucci, A. M. Thurston, R. B. Wai, Y. J. Yoon, R. S. Aiyar, M. H. Medema, M. E. Hillenmeyer and L. K. Charkoudian, Fungal Genet. Biol., 2016, 89, 18–28 CrossRef CAS PubMed.
- H. C. Lo, R. Entwistle, C. J. Guo, M. Ahuja, E. Szewczyk, J. H. Hung, Y. M. Chiang, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 4709–4720 CrossRef CAS PubMed.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- W. Cai and W. Zhang, Curr. Opin. Biotechnol., 2017, 50, 32–38 CrossRef PubMed.
- Y. L. Zhang, J. Zhang, N. Jiang, Y. H. Lu, L. Wang, S. H. Xu, W. Wang, G. F. Zhang, Q. Xu, H. M. Ge, J. Ma, Y. C. Song and R. X. Tan, J. Am. Chem. Soc., 2011, 133, 5931–5940 CrossRef CAS PubMed.
- W. Fang, S. Ji, N. Jiang, W. Wang, G. Y. Zhao, S. Zhang, H. M. Ge, Q. Xu, A. H. Zhang, Y. L. Zhang, Y. C. Song, J. Zhang and R. X. Tan, Nat. Commun., 2012, 3, 1039 CrossRef PubMed.
- M. H. Wheeler, D. Abramczyk, L. S. Puckhaber, M. Naruse, Y. Ebizuka, I. Fujii and P. J. Szaniszlo, Eukaryotic Cell, 2008, 7, 1699–1711 CrossRef CAS PubMed.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- I. Abe, Y. Utsumi, S. Oguro, H. Morita, Y. Sano and H. Noguchi, J. Am. Chem. Soc., 2005, 127, 1362–1363 CrossRef CAS PubMed.
- A. K. Nadeau and J. L. Sorensen, Tetrahedron Lett., 2011, 52, 1697–1699 CrossRef CAS.
- M. K. Zilla, M. Qadri, A. S. Pathania, G. A. Strobel, Y. Nalli, S. Kumar, S. K. Guru, S. Bhushan, S. K. Singh, R. A. Vishwakarma, S. Riyaz-Ul-Hassan and A. Ali, Phytochemistry, 2013, 95, 291–297 CrossRef CAS PubMed.
- L. E. Gray, H. W. Gardner, D. Weisleder and M. Leib, Phytochemistry, 1999, 50, 1337–1340 CrossRef CAS.
- J. Q. Dai, K. Krohn, S. Draeger and B. Schulz, Eur. J. Org. Chem., 2009, 1564–1569 CrossRef CAS.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- A. Dong, J. A. Yoder, X. Zhang, L. Zhou, T. H. Bestor and X. Cheng, Nucleic Acids Res., 2001, 29, 439–448 CrossRef CAS PubMed.
- A. L. Vagstad, E. A. Hill, J. W. Labonte and C. A. Townsend, Chem. Biol., 2012, 19, 1525–1534 CrossRef CAS PubMed.
- C. Zaehle, M. Gressler, E. Shelest, E. Geib, C. Hertweck and M. Brock, Chem. Biol., 2014, 21, 719–731 CrossRef CAS PubMed.
- H. Zhou, Z. Z. Gao, K. J. Qiao, J. J. Wang, J. C. Vederas and Y. Tang, Nat. Chem. Biol., 2012, 8, 331–333 CrossRef CAS PubMed.
- E. Adelin, M. T. Martin, M. F. Bricot, S. Cortial, P. Retailleau and J. Ouazzani, Phytochemistry, 2012, 84, 135–140 CrossRef CAS PubMed.
- L. P. Lin, P. Yuan, N. Jiang, Y. N. Mei, W. J. Zhang, H. M. Wu, A. H. Zhang, J. M. Cao, Z. X. Xiong, Y. Lu and R. X. Tan, Org. Lett., 2015, 17, 2610–2613 CrossRef CAS PubMed.
- M. Schrewe, M. K. Julsing, B. Buhler and A. Schmid, Chem. Soc. Rev., 2013, 42, 6346–6377 RSC.
- F. H. Jin, J. Maruyama, P. R. Juvvadi, M. Akioka and K. Kitamoto, FEMS Microbiol. Lett., 2004, 239, 79–85 CrossRef CAS PubMed.
- A. Minami, M. Shimaya, G. Suzuki, A. Migita, S. S. Shinde, K. Sato, K. Watanabe, T. Tamura, H. Oguri and H. Oikawa, J. Am. Chem. Soc., 2012, 134, 7246–7249 CrossRef CAS PubMed.
- K. C. Van de Bittner, M. J. Nicholson, L. Y. Bustamante, S. A. Kessans, A. Ram, C. J. van Dolleweerd, B. Scott and E. J. Parker, J. Am. Chem. Soc., 2018, 140, 582–585 CrossRef CAS PubMed.
- T. Heine, A. Scholtissek, A. H. Westphal, W. J. H. van Berkel and D. Tischler, Biochim. Biophys. Acta, 2017, 1865, 1770–1780 CrossRef CAS PubMed.
- H. H. Cox, B. W. Faber, W. N. Van Heiningen, H. Radhoe, H. J. Doddema and W. Harder, Appl. Environ. Microbiol., 1996, 62, 1471–1474 CAS.
- W. Yan, H. M. Ge, G. Wang, N. Jiang, Y. N. Mei, R. Jiang, S. J. Li, C. J. Chen, R. H. Jiao, Q. Xu, S. W. Ng and R. X. Tan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18138–18143 CrossRef CAS PubMed.
- S. Bertrand, N. Bohni, S. Schnee, O. Schumpp, K. Gindro and J. L. Wolfender, Biotechnol. Adv., 2014, 32, 1180–1204 CrossRef CAS PubMed.
- L. Kaysser, P. Bernhardt, S. J. Nam, S. Loesgen, J. G. Ruby, P. Skewes-Cox, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2012, 134, 11988–11991 CrossRef CAS PubMed.
- N. Funa, Y. Ohnishi, I. Fujii, M. Shibuya, Y. Ebizuka and S. Horinouchi, Nature, 1999, 400, 897–899 CrossRef CAS PubMed.
- Y. Sone, S. Nakamura, M. Sasaki, F. Hasebe, S. Y. Kim and N. Funa, Appl. Environ. Microbiol., 2018, 84, e00258-18 CrossRef PubMed.
- P. Klausmeyer, Q. Zhou, D. A. Scudiero, B. Uranchimeg, G. Melillo, J. H. Cardellina, R. H. Shoemaker, C. J. Chang and T. G. McCloud, J. Nat. Prod., 2009, 72, 1879–1883 CrossRef PubMed.
- Y. H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS PubMed.
- M. Sato, J. E. Dander, C. Sato, Y. S. Hung, S. S. Gao, M. C. Tang, L. Hang, J. M. Winter, N. K. Garg, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2017, 139, 5317–5320 CrossRef CAS PubMed.
- L. P. Lin and R. X. Tan, Chin. J. Chem., 2018, 36, 749–753 CrossRef CAS.
- C. Huang, C. Yang, W. Zhang, L. Zhang, B. C. De, Y. Zhu, X. Jiang, C. Fang, Q. Zhang, C. S. Yuan, H. W. Liu and C. Zhang, Nat. Commun., 2018, 9, 2088 CrossRef PubMed.
- M. Kawaguchi, T. Ohshiro, M. Toyoda, S. Ohte, J. Inokoshi, I. Fujii and H. Tomoda, Angew. Chem., Int. Ed., 2018, 57, 5115–5119 CrossRef CAS PubMed.
- S. S. Gao, T. Zhang, M. Garcia-Borras, Y. S. Hung, J. M. Billingsley, K. N. Houk, Y. Hu and Y. Tang, J. Am. Chem. Soc., 2018, 140, 6991–6997 CrossRef CAS PubMed.
- L. S. Mazzaferro, W. Huttel, A. Fries and M. Muller, J. Am. Chem. Soc., 2015, 137, 12289–12295 CrossRef CAS PubMed.
- Y. Mizuuchi, S. P. Shi, K. Wanibuchi, A. Kojima, H. Morita, H. Noguchi and I. Abe, FEBS J., 2009, 276, 2391–2401 CrossRef CAS PubMed.
- Y. W. Zhang and Y. W. Chen, Phytochemistry, 1997, 45, 401–403 CrossRef CAS.
- P. Klausmeyer, Q. Zhou, D. A. Scudiero, B. Uranchimeg, G. Melillo, J. H. Cardellina, R. H. Shoemaker, C. J. Chang and T. G. McCloud, J. Nat. Prod., 2009, 72, 805–812 CrossRef CAS PubMed.
- Y. B. Wang, R. Huang, H. B. Zhang and L. Li, J. Asian Nat. Prod. Res., 2006, 8, 663–670 CrossRef CAS PubMed.
- P. Tane, J. F. Ayafor, B. L. Sondengam and J. D. Connolly, Phytochemistry, 1990, 29, 1004–1007 CrossRef CAS PubMed.
- F. M. Awadallah, T. A. El-Waei, M. M. Hanna, S. E. Abbas, M. Ceruso, B. E. Oz, O. O. Guler and C. T. Supuran, Eur. J. Med. Chem., 2015, 96, 425–435 CrossRef CAS PubMed.
- S. C. Wenzel, B. Kunze, G. Hofle, B. Silakowski, M. Scharfe, H. Blocker and R. Muller, ChemBioChem, 2005, 6, 375–385 CrossRef CAS PubMed.
- X. L. Chen, Z. F. Guo, P. M. Lai, K. H. Sze and Z. H. Guo, Angew. Chem., Int. Ed., 2010, 49, 7926–7928 CrossRef CAS PubMed.
- I. Abe, T. Watanabe, W. Lou and H. Noguchi, FEBS J., 2006, 273, 208–218 CrossRef CAS PubMed.
- E. A. Ferreira, E. F. Gris, J. M. Rebello, J. F. G. Correia, L. F. S. de Oliveira, D. Wilhelm and R. C. Pedrosa, Planta Med., 2011, 77, 1569–1574 CrossRef CAS PubMed.
- S. B. Wu, C. Long and E. J. Kennelly, Nat. Prod. Rep., 2014, 31, 1158–1174 RSC.
- C. Blanchard, D. Pouchain, P. Vanderkam, M. C. Perault-Pochat, R. Boussageon and H. Vaillant-Roussel, Eur. J. Clin. Pharmacol., 2018, 74, 541–548 CrossRef CAS PubMed.
- M. Bruder, P. L. Haseler, M. Muscarella, W. Lewis and C. J. Moody, J. Org. Chem., 2010, 75, 353–358 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03697g |
| This journal is © The Royal Society of Chemistry 2019 |