
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoelectrocatalytic H2 evolution from integrated photocatalysts adsorbed on NiO†
Nils
Põldme‡
a,
Laura
O'Reilly‡
b,
Ian
Fletcher
c,
Jose
Portoles
c,
Igor V.
Sazanovich
d,
Michael
Towrie
d,
Conor
Long
b,
Johannes G.
Vos
b,
Mary T.
Pryce
*b and
Elizabeth A.
Gibson
 *a
*a
aSchool of Natural and Environmental Science, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: Elizabeth.gibson@ncl.ac.uk
bSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland. E-mail: Mary.pryce@dcu.ie
cNEXUS XPS Laboratory, Newcastle University, Stephenson Building, Newcastle upon Tyne, NE1 7RU, UK. E-mail: nexus@ncl.ac.uk
dCentral Laser Facility, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK. E-mail: Igor.Sazanovich@stfc.ac.uk
First published on 4th October 2018
Abstract
A new approach to increasing the faradaic efficiency of dye-sensitised photocathodes for H2 evolution from water, using integrated photocatalysts, furnished with ester groups on the peripheral ligands, [Ru(decb)2(bpt)PdCl(H2O)](PF6)2 (1) and [Ru(decb)2(2,5-bpp)PtI(CH3CN)](PF6)2 (2), (decb = 4,4′-diethylcarboxy-2,2′-bipyridine, bpp = 2,2':5′,2′′-terpyridine, bpt = 3,5-bis(2-pyridyl)-1,2,4-triazole) is described. Overall, 1|NiO is superior to previously reported photocathodes, producing photocurrent densities of 30–35 μA cm−2 at an applied bias of −0.2 V vs. Ag/AgCl over 1 hour of continuous white light irradiation, resulting in the generation of 0.41 μmol h−1 cm−2 of H2 with faradaic efficiencies of up to 90%. Furthermore, surface analysis of the photocathodes before and after photoelectrocatalysis revealed that the ruthenium bipyridyl chromophore and Pd catalytic centre (1) were photochemically stable, highlighting the benefits of the approach towards robust, hybrid solar-to-fuel devices.
Introduction
In recent years, p-type dye sensitised solar cells (p-DSCs), have attracted considerable attention. This is because of their potential applications in photoelectrocatalytic devices for H2 evolution, CO2 reduction and also in the advancement of tandem DSCs.1,2 Frequently, NiO-based electrodes are used in these devices. The efficiency of conventional DSCs can be increased, in theory, by replacing the Pt counter electrode with a photocathode which captures light transmitted by a TiO2-based photoanode. In addition, these devices can be augmented with catalysts to drive photoelectrocatalytic water splitting in so-called dye sensitised photoelectrochemical cells (DSPEC).DSPECs have many important advantages over other water splitting systems. Firstly, in contrast to homogeneous systems sacrificial electron donors are no longer required. This is because the electrons for H+ reduction are provided by water oxidation at the anode and delivered to the photocathode through the external circuit.3,4 Secondly, by combining the molecular photosensitiser with the molecular or colloidal catalyst on a semiconductor surface, the photon absorption, charge transfer, and catalyst activity can be optimised, leading to higher efficiency and lower processing costs.5 Thirdly, using a molecular photosensitiser provides the opportunity to tune the absorption properties of the system. Combining a dye-sensitised photocathode with a dye-sensitised photoanode, presents an exciting opportunity to use the low energy part of the visible spectrum on one electrode and the high energy part on the other.5 Thus, more of the spectrum is harnessed, resulting in these tandem cells achieving a high photon to H2 efficiency.
Mesoporous NiO has been used in p-DSCs and tandem cells since the late 1990's. It is one of the few stable p-type semiconductors with a wide band-gap and, consequently, the electrode film does not compete with the dye for visible light absorption.6–11 Recently, it has been used in a DSPEC to reduce H+ to H2.5,12–17 Nanostructured NiO films can be produced at reasonably low temperatures in a cost-efficient manner, making them ideal for large-scale applications. NiO is also stable in the mildly acidic aqueous environments required for H2 production. The valence band (VB) is approximately 0.4 V vs. NHE in pH 6.8 phosphate buffer (approx. 0.62 V vs. NHE at pH 3), which lies between the frontier orbitals of typical photosensitisers, such as Ru(bpy)32+.18 Excitation of a photosensitiser adsorbed on NiO can result in extraction of an electron from the NiO (hole injection) reducing the sensitiser and initiating the photocatalytic reaction.18,19 The exact mechanism for H2 evolution, which requires two electrons, is unclear at this stage.17,20–22
Unfortunately, rapid and efficient recombination of the reduced dye and the hole in the NiO valence band reduces photocatalytic efficiency of these systems. Obviously, research into ways to increase the photosensitiser efficiency and reduce the non-productive back reaction will be of enormous benefit.23,24 Understanding the excited state dynamics of the sensitiser is essential, while accurate modelling of the long-lived charge separated state and the localisation of the charge remote from the NiO surface can help in reducing the efficiency of the charge recombination processes.
As mentioned above, polypyridyl ruthenium(II) complexes have been explored as photosensitisers. We have previously studied the H2 generating capability of compound 2 (Fig. 1) in solution (CH3CN/TEA/H2O), and obtained a turnover number (TON) of 650 after 6 h irradiation at 470 nm.25 Analysis of transient absorption (TA) data indicated that the ester ligands on bipyridine lowers the energy of the 3MLCT (metal-to-ligand charge transfer) state in which the unpaired spin is located on a peripheral carboxy-bipy ligand. This excited state is long-lived compared to the unsubstituted bipyridine complex perhaps explaining the high TONs achieved for this complex. In this manuscript, we have used time-resolved infrared (TRIR) spectroscopy and time resolved UV-visible spectroscopy (TA) to characterise the excited states, and the excited state dynamics of complexes 1 and 2 (Fig. 1) in CD3CN and also when immobilised on NiO surfaces (1|NiO and 2|NiO), respectively. H2 production by 1|NiO and 2|NiO in DSPEC's was confirmed under two different applied potentials, Eappl = −0.4 V and −0.2 V vs. Ag/AgCl, thereby negating the need for sacrificial agents. The performance and stability of the photocathodes are discussed below.
 | ||
| Fig. 1 The structures of the dye-catalyst assemblies 1 and 2 (R = CO2Et). | ||
Results
Photocatalyst adsorption
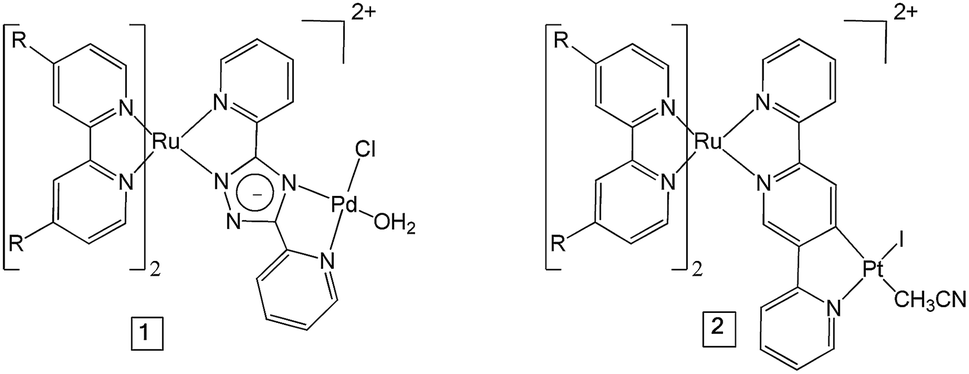
The UV-visible absorption spectra of complexes 1 and 2 dissolved in acetonitrile solution and immobilised on NiO are provided in Fig. 2. The spectra are generally broader and blue-shifted when compared to homogeneous solution (λmax = 490 nm for 1 and 480 nm for 1|NiO, λmax = 480 nm for 2 and ca. 470 nm for 2|NiO) which is consistent with an electronic interaction between the ground state of the dye and the NiO surface. Time-Dependent Density Functional calculations (TD-DFT), described below, suggest that a Ru-to-carboxy-bipy charge-transfer transition is responsible for the low-energy absorption maximum.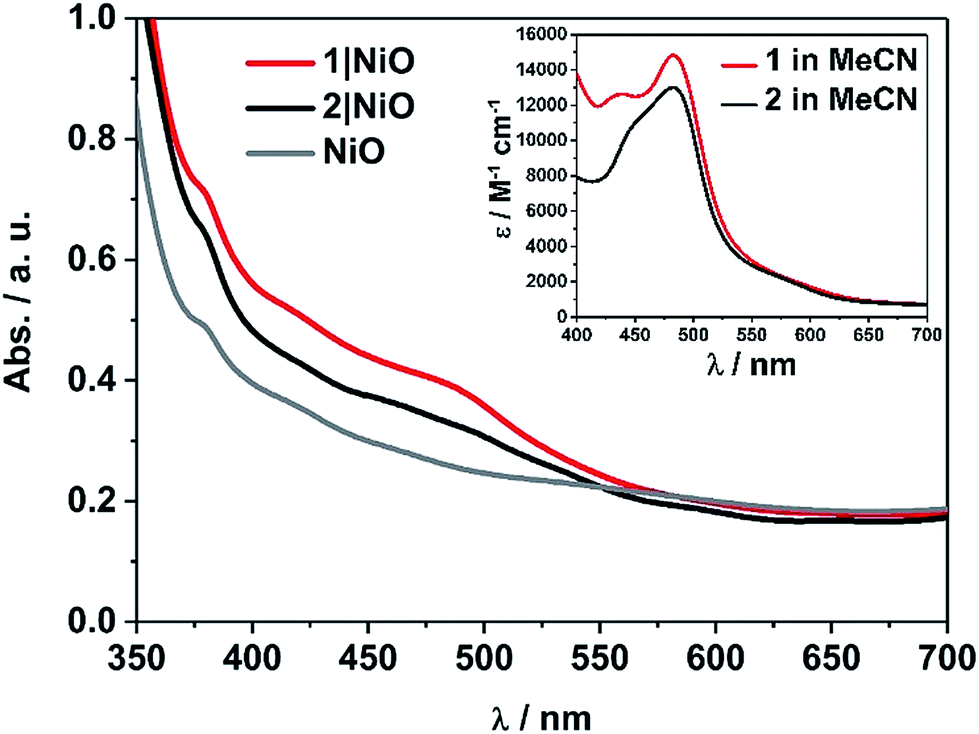 | ||
| Fig. 2 UV-vis absorption spectra of 1 (red) and 2 (black) adsorbed on NiO and bare NiO (gray). Inset: UV-vis absorption spectra of 1 (red) and 2 (black) in MeCN (0.028 mM and 0.032 mM, respectively). | ||
The loading of the photocatalyst onto the NiO surface was quantified from the absorption of 1 and 2 on NiO (Fig. 2).74 For 1, assuming that the absorption coefficient does not change substantially on grafting, the photocatalyst loading was 9 nmol cm−2, and for 2, it was 5.3 nmol cm−2. These values are reasonably similar, consistent with the similar anchoring system, and are a similar order of magnitude to dye-sensitised photocathodes reported elsewhere.26 The FTIR spectra (Fig. S37 and S38†) of complexes 1 and 2 in KBr have a carbonyl band at 1724 and 1726 cm−1, respectively, and when the complexes are immobilised on NiO, the carbonyl bands shift to 1720 cm−1 for both complexes. In both cases there is a marginal shift to lower frequency, which could indicate an interaction between the ester and NiO.
Time resolved IR and TA spectroscopy
The photoelectrochemical process is initiated when a dye absorbed on the NiO surface absorbs light, stimulating hole injection into the NiO material and reduction of the dye.19 To probe the photoexcitation, hole injection and recombination dynamics, time-resolved infrared and transient absorption studies were performed on both complex 1 and 2 in deuterated acetonitrile solution and when immobilised onto NiO films, following excitation at 470 nm. The data are shown in Fig. 3 and 4 below and Fig. S27–S33 in the ESI.†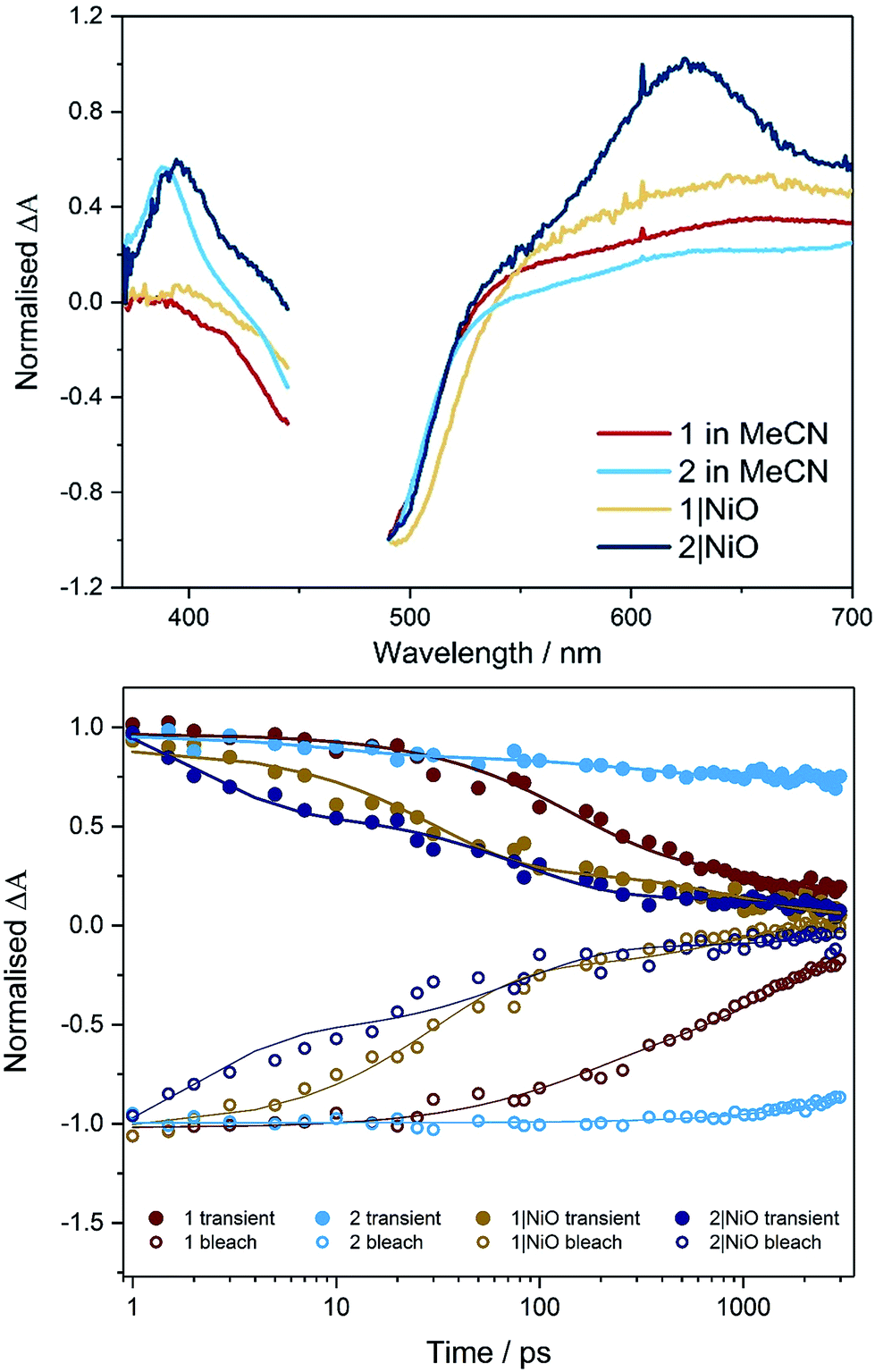 | ||
| Fig. 3 Transient absorption spectra (top, 1 ps) and kinetic traces (bottom) for 1 (red) and 2 (light blue) in CD3CN and 1|NiO (yellow) and 2|NiO (dark blue) following excitation (λ = 470 nm) probed at 490 nm (bleach, open symbols) and 620 nm (transient, filled symbols). On the bottom figure, the rings represent data points and the lines are the exponential fit to the data. | ||
 | ||
| Fig. 4 TRIR Spectra (top, 1 ps) and kinetic traces (bottom) for 1 (red) and 2 (light blue) in CD3CN and 1|NiO (yellow) and 2|NiO (dark blue) following excitation (λ = 470 nm). On the bottom figure, the symbols represent data points (open = bleach, filled = transient absorption) and the lines are the exponential fit to the data. | ||
Transient absorption spectroscopy in solution
The transient absorption spectra obtained following pulsed photolysis (λexc = 470 nm) of complexes 1 and 2, in CD3CN solution are given in Fig. 3 and S27–S29 in the ESI.† In the case of compound 2, a ground state bleach occurs within the pulse, a broad absorption that extends from 500 nm is evident, together with an additional absorption band at ca. 380 nm, which also persists beyond the 3 ns window of the experiment. These transient absorption features (LMCT and π → π* transition on bpy˙−), are typical for ruthenium bipyridyl complexes and are assigned to the 3MLCT excited state species which persist on the ns to μs timescale.25,27 For compound 1, only two features are evident, a ground state bleach which occurs within the pulse, and a broad absorption extending from ca. 560 nm. This broad absorption feature decays over approximately τ = 2 ns, concomitant with recovery of the ground state bleach. On closer inspection using global fitting of the transient absorption, unlike 2, two time constants were extracted for 1 (τ1 = 137, τ2 = 1830 ps), which were associated with a 40 nm red-shift the visible band. Furthermore, TD-DFT calculations on the model complex, [Ru(dmcb)2(bpt)PdCl(H2O)](PF6)2 suggest that the initially formed singlet Ru-to-dmcb charge-transfer excited state rapidly crosses to the triplet surface forming a triplet Ru-to-bpt charge transfer state and not a Ru-to-dmcb charge-transfer state. This explains the absence of absorptions ca. 380 nm in TA spectra for complex 1.Transient absorption on NiO
When compounds 1 and 2 were immobilised onto NiO films (1|NiO, 2|NiO) (Fig. 3, S27, S28, S30 and S31†), the excited species were generated within the laser pulse (200 fs). The initial spectral shape is consistent with the 3MLCT excited species superimposed on a second coexisting species which is assigned to the reduced dye and is consistent with hole injection into the NiO (holes injected into NiO have a broad featureless absorption spectrum extending throughout the red-NIR region18). To extract the dynamics of these processes, global analysis was performed and the fit was evaluated by inspecting the systematic residuals. Fitting a parallel multiexponential model to the data gave the decay associated difference spectra shown in Fig. S30 and S31 in the ESI.† For 1|NiO, two lifetimes were extracted, τ1 ≈ 30 ps, τ2 ≈ 1 ns. The shape of the long-lived species, τ2, is similar to the excited state absorption spectra of 1 in solution. The short-lived species, τ1, absorbs broadly between 540 to 700 nm, with a maximum at 648 nm and does not contain the positive band at 385 nm, which is characteristic of the reduced decb ligand.28 We assign τ1 to the reduced dye, 1−|NiO+, and τ2 to the triplet 3MLCT excited species, 1*|NiO.In the case of 2|NiO, global fitting gave three distinguishable components (Fig. 3), τ1 ≈ 2 ps, τ2 ≈ 80 ps, τ3 ≈ 4 ns. All three contained a positive transient at 370–400 nm, which is associated with the reduced decb ligand, the ground state bleach between 440 to 540 nm (both of these spectral characteristics were evident in the TA experiments carried out in solution as described above), but differed in the region beyond 520 nm. The spectrum associated with the nanosecond component, τ3, is similar in shape to the excited state absorption spectra of 2 in solution. The shortest-lived component, τ1, contained a Gaussian-shaped absorption band at ca. 630 nm. On this timescale (<5 ps), there is approximately 40% recovery of the parent depletion at 500 nm. This species is attributed to hole injection and the formation of the reduced dye (2−|NiO+), and the timebases are similar to that previously reported for bis[2,20-bipyridine][4,4′-dicarboxy-2,20-bipyridine]ruthenium(II) sensitised NiO.29 The component in between, τ2, contains a broad excited state absorption signal above 520 nm which is similar in structure to ligand-to-metal charge transfer (LMCT) transitions associated with the terpyridyl bridging ligand.28 The presence of this component, which is absent in the solution spectra, suggests that the equilibrium between excited states is different when the dye is adsorbed on NiO compared to solution.
Time-resolved IR in solution
TRIR studies in the fingerprint region (Fig. 4, S32 and S33†) were performed in deuterated acetonitrile. Following excitation at 470 nm, a depletion of the carbonyl group was observed at 1730 cm−1 for compound 1 and at 1714 cm−1 for compound 2. A new band to the low energy side of the parent bleach was detected for both complexes and this is assigned to the carbonyl band of the 3MLCT excited state species. For complex 1, this band decays with concomitant recovery of the parent bleach over τ = ca. 2 ns. In the case of compound 2, both the excited state feature and the parent bleach persist for longer than 2 ns. The shorter lifetime for compound 1 containing the triazole bridging ligand, agrees with studies previously observed for ruthenium triazole complexes.30Time-resolved IR on NiO
Time-resolved IR studies were also performed using 1|NiO and 2|NiO (Fig. 4, S32 and S33 in ESI†). As observed in the solution studies, upon excitation, depletion of the carbonyl group occurs within the laser pulse (200 fs) for both compounds, with a new carbonyl band at lower frequency as previously observed in solution studies (discussed above). In the case of 1|NiO, near full recovery of the parent depletion and decay of the band at 1689 cm−1 is observed within 200 ps. Consistent with the solution studies, these spectral changes are assigned to the 3MLCT excited state species. In addition, there is a further species with very weak bands at higher frequency to that of the parent depletion for both 1|NiO and 2|NiO. As these bands are very weak and at the edge of the detection window, it is difficult to get reliable kinetic data. However, similarly to the 3MLCT excited species which forms within the excitation pulse, these weak bands in the range 1750–1810 cm−1, also form within this timeframe. For 2|NiO, similar spectral features are observed to that for complex 1.Computational studies of [Ru(bipy)2(bpt)PdCl](PF6)2, [Ru(dmcb)2(bpt)PdCl](PF6)2, [Ru(dmcb)2(bpt)PdCl(H2O)](PF6)2 and [Ru(dmcb)2(bpt)PdCl](PF6)
Following the satisfactory optimisation of the ground-state singlet structure for the three model complexes at the B3LYP/LanL2DZ level, the TD-DFT method was used to estimate the energy and electronic structure of the fifty low-lying singlet excited states. These calculations were undertaken firstly to verify that the methods used reliably represented the onset absorptions of the complexes when compared to the experimental UV-visible spectra. Secondly, these calculations can be used to construct electron density difference maps for each of the excited states, and these maps are useful in characterising the nature of the excited state in terms of electron drift relative to the ground-state electronic structure. For instance, MLCT transitions can be clearly characterised, as in the case of the lowest energy optically accessible singlet state (S17, 432.6 nm) of [Ru(bipy)2(bpt)PdCl](PF6)2 modelled in acetonitrile (Fig. S21(b)†). Here this excited state can be characterised as a ruthenium to bpt charge-transfer state. In contrast however, the lowest energy optically accessible singlet state of [Ru(dmcb)2(bpt)PdCl](PF6)2 (S12, 479.3 nm) has mainly ruthenium to dmcb charge transfer character (Fig. S26†). Clearly the ester-substituents on the “ancilliary” bipy ligands have a dramatic effect on the nature of the accessible excited states and have a greater role in the catalytic mechanism than as simple binding sites to the NiO surface.As the catalytic processes take place in aqueous environments, the electronic structure of [Ru(dmcb)2(bpt)PdCl(H2O)](PF6)2 in water was modelled both on the singlet and triplet surfaces. The lowest energy optically accessible singlet (Fig. 5) excited state exhibits mainly ruthenium-to-dmcb charge-transfer character, and if it is assumed that this state is efficiently populated, then the closest triplet state in energy terms is the T8 which has predominantly ruthenium/bpt to palladium charge-transfer character. Thus, the intersystem crossing process results in a significant change to the electronic structure, increasing the electron density on the palladium atom and reducing the electron density on the ruthenium and bpt ligand. This explains why the TA experiments on 1 fail to produce features consistent with a Ru-to-bipy charge-transfer excited state at ca. 380 nm. More importantly, the energy of T8 lies 2.64 eV (254 kJ mol−1) above the ground state singlet energy which is sufficient to split water. Finally the electronic structure of the singly reduced [Ru(dmcb)2(bpt)PdCl](PF6) was modelled. The spin distribution in this doublet species was mapped, which indicated the presence of the excess spin on the Pd atom and its coordination sphere.
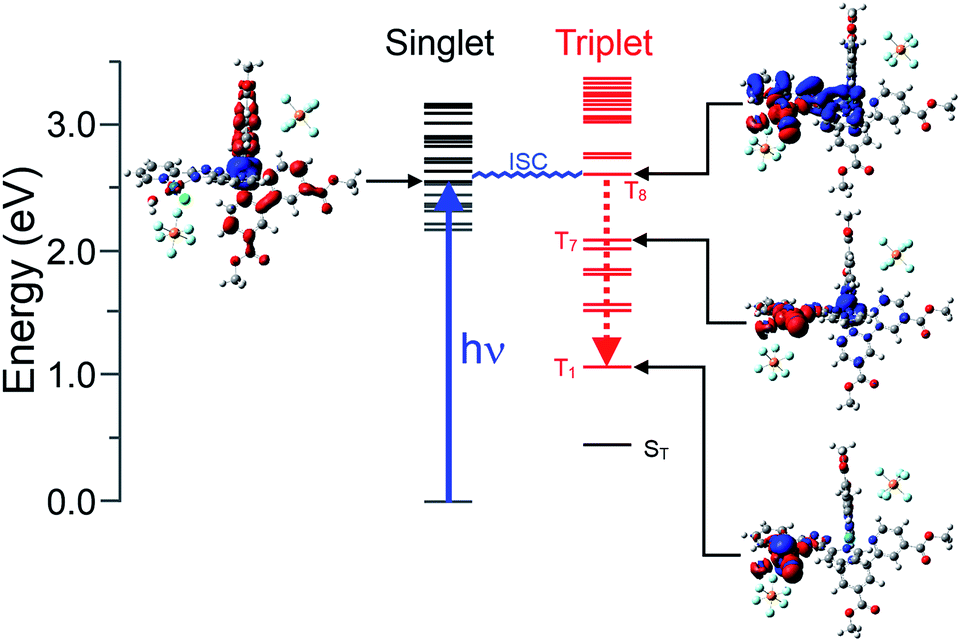 | ||
| Fig. 5 A photophysical model for the excitation of [Ru(dmcb)2(bpt)PdCl(H2O)](PF6)2 into its lowest energy optically accessible singlet excited state (8th excited state, corresponding to a photon energy of 484 nm in water, black lines) followed by inter system crossing (ISC) to the triplet surface (red lines) and internal conversion to the triplet surface; the singlet state energy at the triplet geometry is indicated by ST. Vertical excitation to the lowest energy optically accessible state is indicated as the blue arrow, and the electron density difference maps or selected states are also presented to either side of the energy level. | ||
Photoelectrochemistry and hydrogen evolution
Linear sweep voltammetry (LSV) measurements were carried out on the 1|NiO and 2|NiO electrodes, immersed in aqueous electrolyte solution with 0.1 M KCl. The pH was adjusted with the addition of diluted HCl (pH 1 to pH 7). The potential was swept from Eappl = 0 V to −0.6 V vs. Ag/AgCl for each pH value, under chopped light conditions, during which the samples were irradiated with white light (AM1.5, 100 mW cm−2) over 10 s intervals, under steady-state illumination and in the dark (Fig. S2 in the ESI†). The photocurrent density increased slightly when the pH was raised. This is possibly due to slower charge injection and increased recombination at lower pH.31 The least acidic environment where H2 was detected was pH 3 and this was chosen for further experiments. At pH 3, for both 1|NiO and 2|NiO, the photocurrent increased compared to the dark current until Eappl = −0.4 V vs. Ag/AgCl (Fig. 6). At more negative potentials, the magnitude of both the dark current and photocurrent density increased considerably.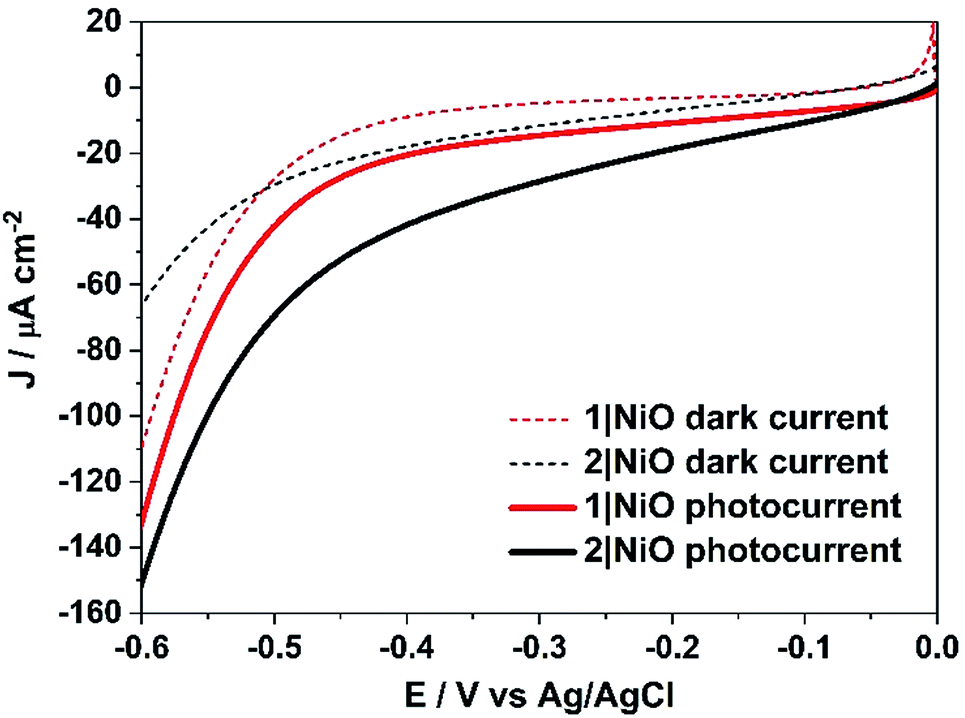 | ||
| Fig. 6 Linear sweep voltammograms of 1|NiO and 2|NiO immersed in aqueous 0.1 M KCl electrolyte (pH 3). The potential was swept from Eappl = 0 to −0.6 V vs. Ag/AgCl (3.0 M NaCl) in the dark and under simulated light illumination (AM1.5, 100 mW cm−2). | ||
Three potentials were chosen at which to monitor H2 evolution, Eappl = −0.40 V vs. Ag/AgCl (ca. −0.01 V vs. RHE), where the photocurrent and dark current were most stable, Eappl = −0.60 V vs. Ag/AgCl (ca. −0.21 V vs. RHE), and Eappl = −0.20 V vs. Ag/AgCl (ca. 0.21 V vs. RHE), all of which are lower than the conduction band edge of TiO2 (i.e. in a tandem PEC device, the bias which will be applied by the photoanode driving the overall water splitting), approximately −0.54 V vs. RHE.12,32
E appl was fixed while the current was measured and Ar was continuously flowed through the electrolyte solution and the exhaust was sampled by in-line GC analysis (see Experimental). Control experiments were carried out on a bare fluorine-doped tin oxide (FTO) substrate and a non-sensitised NiO|FTO electrode under the same conditions (pH 3 aqueous electrolyte with 0.1 M potassium hydrogen phthalate) to distinguish between the activity of the substrate and the sensitised electrode and to check for any electrochemically active impurities (see Fig. S4 in the ESI†). No H2 or photocurrent was detected during the control measurements with FTO and the current density was considerably lower for the bare electrodes compared to the sensitised electrodes measured under same conditions. However, although no H2 was detected, a steady increase in photocurrent was observed for the NiO|FTO sample at Eappl = −0.6 V. The reason for the increase in photocurrent is possibly a reduction of some Ni3+ surface states during illumination with white light.33
The trends observed for the photoelectrodes 1|NiO and 2|NiO during the chronoamperometry experiments under chopped light irradiation (Fig. 7) were consistent with the LSV experiments (Fig. 6), which is representative of the good reproducibility of the system. For 1|NiO, stable cathodic photocurrents were recorded at Eappl = −0.2 V (Jphoto = ca. 37 μA cm−2) and Eappl = −0.4 V (Jphoto = ca. 44 μA cm−2), over 10 dark/light cycles (Fig. 7(a)). The dark current recorded during those measurements was negligible compared to the photocurrent and only increased slightly when the more negative potential was applied. For 2|NiO, a significant increase in photocurrent was observed at the more negative bias, Eappl = −0.4 V (Jphoto = ca. 53 μA cm−2) compared to Eappl = −0.2 V (Jphoto = ca. 31 μA cm−2) (Fig. 7(b)). As for 1|NiO, the dark current was very low compared to the photocurrent. An initial spike in the photocurrent was observed during the chronoamperometry of 2|NiO at Eappl = −0.2 V and −0.4 V. We attribute this rapidly decaying photocurrent to local capacitance effects, the reduction of the dye-catalyst assembly and/or Ni3+ on the surface.17,21 Another explanation could be slow electron transfer from the catalyst to the H+ or slow diffusion of the products from the pores leading to charge recombination.15,17,22 The spike in photocurrent was much less pronounced for 1|NiO which indicates that charge-transfer in the forward direction (e.g. from NiO to the catalyst, to the substrate) is faster than charge-recombination.17,20–22 After these initial spikes, the current was stable and the absence of capacitive features after the first on–off cycle suggests that the current is not diffusion limited. The chopped light illumination was followed by a constant white light illumination for up to a 1 h to test the electrode stability (Fig. S3 ESI†). A small, steady decrease in photocurrent was observed for both samples under almost all Eappl, due to the decrease in active surface area upon bubble formation on the electrode. This behaviour was also observed for a Pt-coated FTO electrode (Fig. S4 in the ESI†) and on shaking the cell, the current was restored. To remove the formed bubbles on the sample surface, the cell was manually shaken in the end of each measurement and additional samples of outlet gas were analysed with GC until no more H2 was detected. Unlike the photocurrent, the dark current at Eappl = −0.2 V and Eappl = −0.4 V did not vary over the duration of the experiment and the magnitude was <10% of the total current recorded. At more negative potentials than Eappl = −0.6 V, more significant changes in current vs. time were observed. A larger dark current (Jdark = 78 μA cm−2) was recorded for 1|NiO (Fig. 7a) and 2|NiO (Fig. 7b), compared to the previous measurements, which decreased over time. In addition, for 2|NiO, the dark current decreased when the light was switched off and the photocurrent increased when light was turned on. This is an indication of changes occurring on the electrode surface during irradiation.
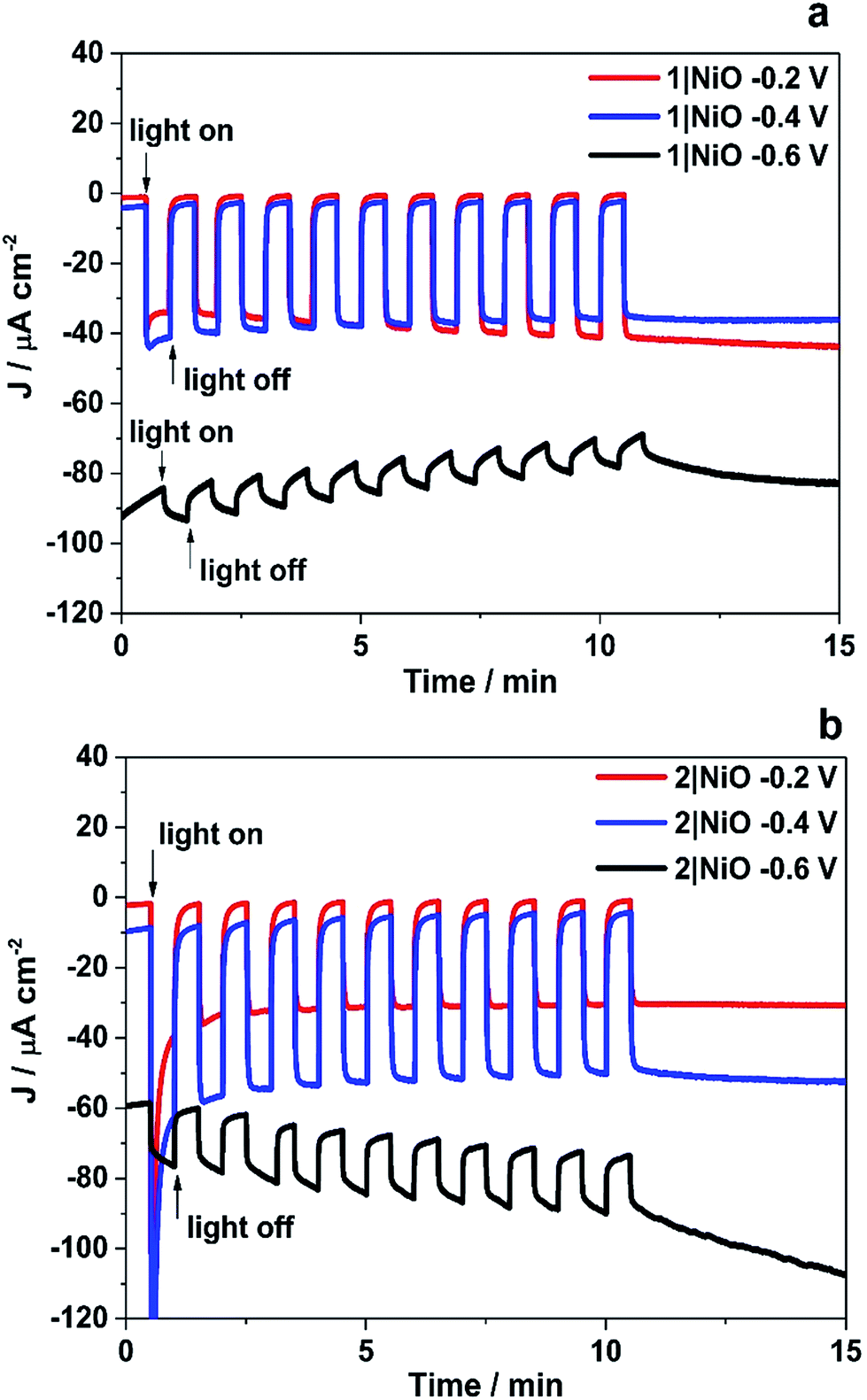 | ||
| Fig. 7 Chronoamperometry measurements of 1|NiO (a) and 2|NiO (b) immersed in pH 3 buffer containing 0.1 M potassium hydrogen phthalate. Eappl = −0.2 V, −0.4 V and −0.6 V vs. Ag/AgCl (3.0 M NaCl). Chopped light illumination was applied with 30 s intervals (10 cycles of dark current/photocurrent). | ||
H2 was detected from both 1|NiO and 2|NiO under white light illumination at Eappl = −0.2 V to −0.6 V and the volume detected vs. time is presented in Fig. 8. No H2 was detected during the control measurements in the dark, except for 2|NiO under Eappl = −0.6 V vs. Ag/AgCl, where the rate of H2 evolution decreased after the light was switched off. The faradaic efficiencies (ηFar) calculated for 1|NiO and 2|NiO are presented in Table 1. The efficiencies of both photocathodes decrease with the increase in applied potential. Reported values of ηFar for H2 evolution elsewhere range from <10% to >100%.2 Our values are estimates as the peaks in the chromatogram corresponding to H2 were small and integrated manually. The photocurrent density and volume of H2 were affected by differences in film thickness and care was taken to ensure these were consistent between samples (1.5 μm). Turnover numbers (TONs) for the immobilised photocatalysts 1|NiO and 2|NiO were estimated from the number of dye molecules adsorbed and the quantity of H2 produced at each Eappl during 1 hour and are included in Table 1. The best TON for 1 resulted from measurement under Eappl = −0.2 V vs. Ag/AgCl, which was 46 and increasing the potential resulted in lower TON. The estimated TONs for 2|NiO were higher than 1|NiO at all Eappl. However, as the photocatalyst was shown to be decomposing to Pt0 during the post-catalysis characterisation of electrodes by XPS (Fig. 10), the TON for 2 is not accurate.
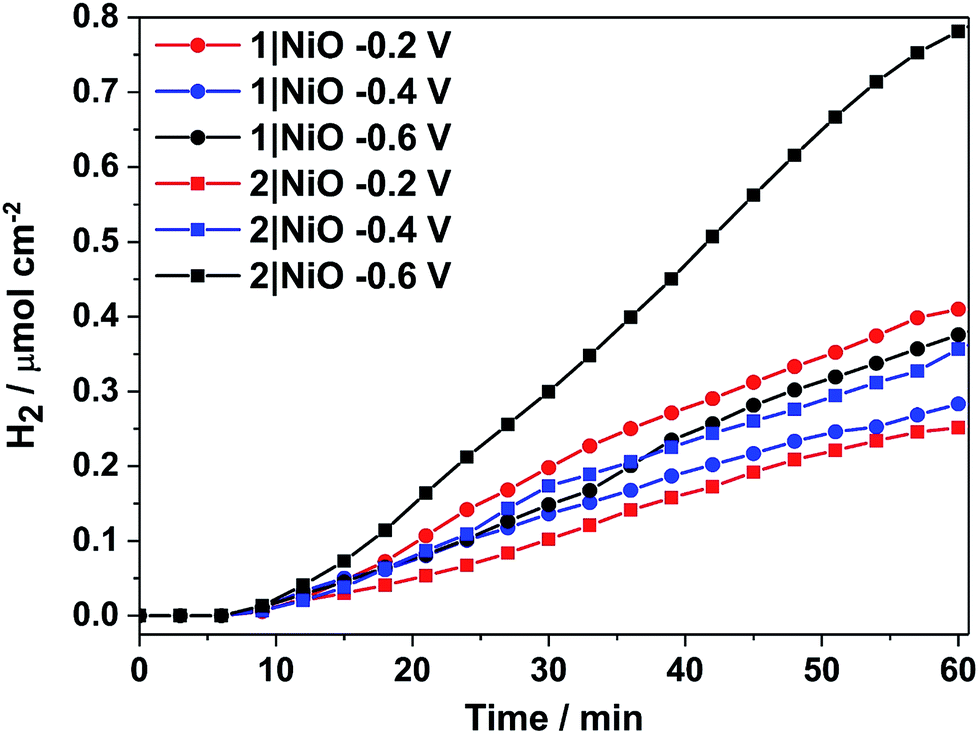 | ||
| Fig. 8 Cumulative photoelectrocatalytic H2 production by 1|NiO and 2|NiO during the chronoamperometry measurements at different Eappl (Fig. 7 and S3 in the ESI†). Chopped light illumination was applied during the first 10 min followed by constant light illumination until the end of the measurement (AM1.5, 100 mW cm−2). | ||
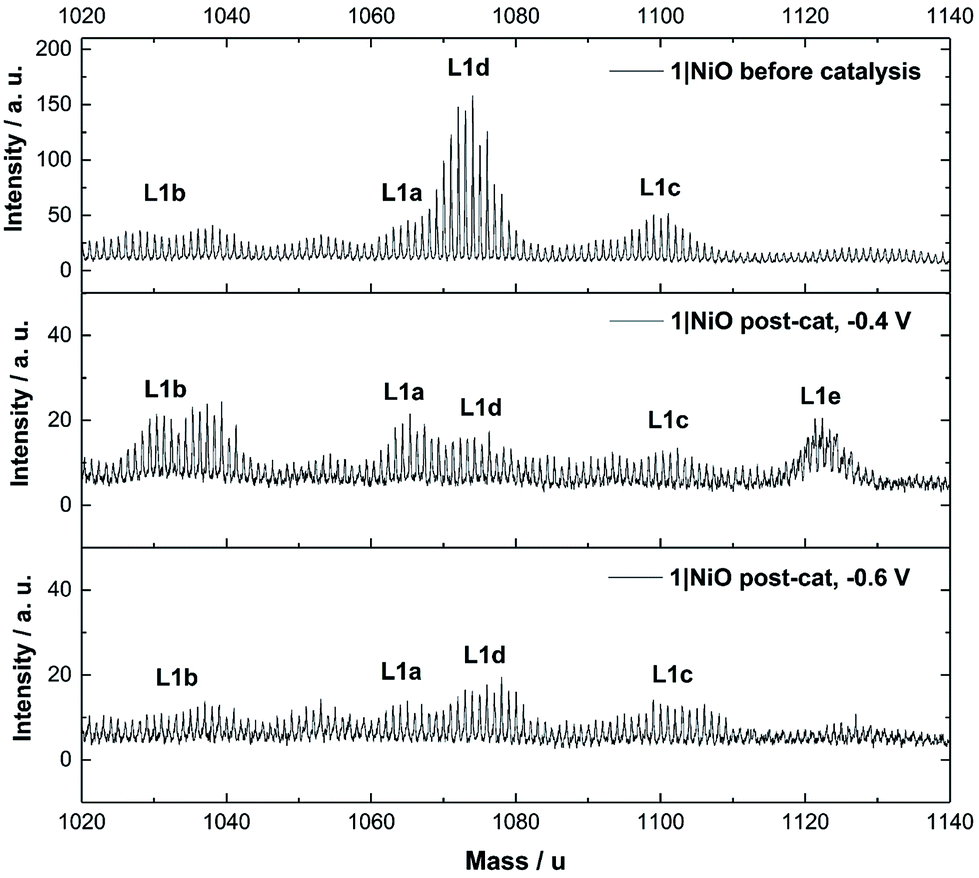 | ||
| Fig. 9 ToF-SIMS positive ion spectra of 1|NiO taken from three different samples: top: – pre-catalysis, middle: post-catalysis at −0.4 V vs. Ag/AgCl and bottom: post-catalysis −0.6 V vs. Ag/AgCl. 1020–1140 mass per unit range. Assignments are as follows: L1a – [Ru(decb)2(bpt)PdCl]2+ calcd. m = 1065, L1b – [Ru(decb)2(bpt)Pd]3+ calcd. m = 1030, L1c – [Ru(decb)2(bpt)PdCl(H2O)2]2+ calcd. m = 1101, L1d – [Ru(decb)2(bpt)Pd(CH3CN)]3+ calcd. m = 1071, L1e – [Ru(decb)2(bpt)PdCl(H2O)3]2+ calcd. m = 1119. | ||
| 1|NiO | 2|NiO | |||||
|---|---|---|---|---|---|---|
| a J photo is the average photocurrent, Jtotal is the photocurrent plus dark current, Pphoto is the percentage of photocurrent of the total current and ηFar is the faradaic efficiency of H2 production. ηFar was calculated using Jphoto in case of Eappl = −0.2 V and Eappl = −0.4 V. *ηFar was calculated from the overall charge passed during the measurement (dark and photocurrent) in case of Eappl = −0.6 V vs. Ag/AgCl. TON calculated from the dye-loading (mol cm−2) and the [H2] over 1 hour. | ||||||
| E appl/V vs. Ag/AgCl | −0.2 | −0.4 | −0.6 | −0.2 | −0.4 | −0.6 |
| J photo/μA cm−2 | 34.8 ± 1.4 | 31.2 ± 8.7 | 12.3 ± 1.9 | 31.8 ± 6.2 | 47.6 ± 7.5 | 43.7 ± 27.1 |
| J total/μA cm−2 | 35.6 ± 1.8 | 34.2 ± 9.4 | 88.2 ± 4.9 | 33.2 ± 7.1 | 52.5 ± 12.0 | 114.7 ± 37.6 |
| P photo/% | 97.8 | 91.2 | 14 | 95.8 | 90.7 | 38.1 |
| η Far/% | 88.6 | 67.5 | 32.3* | 59.1 | 56.9 | 44.4* |
| [H2]/μmol h−1 cm−2 | 0.41 | 0.28 | 0.37 | 0.25 | 0.36 | 0.78 |
| TON | 46 | 31 | 41 | 47 | 68 | 147 |
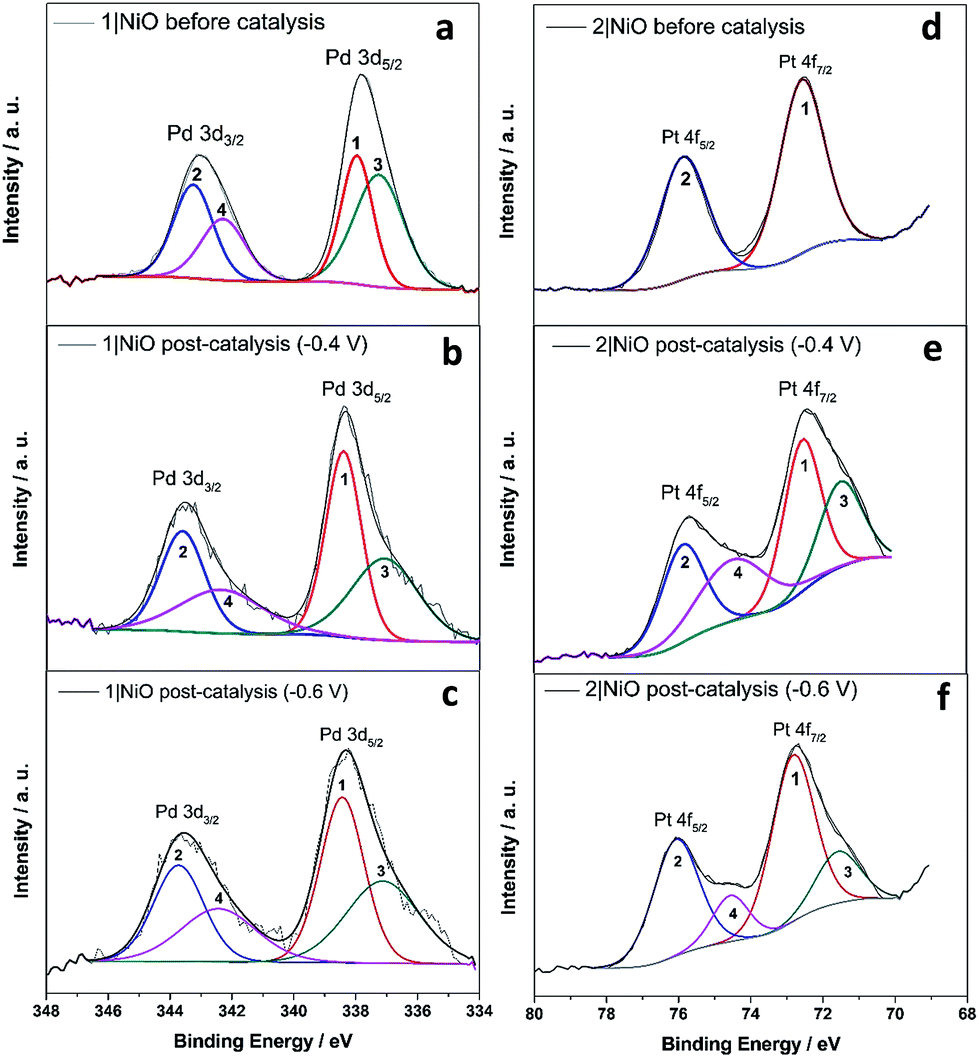 | ||
| Fig. 10 Pd 3d XPS spectrum of 1|NiO (a) before photoelectrocatalysis, after photoelectrocatalysis under (b) Eappl = −0.4 V and (c) Eappl = −0.6 V; Pt 4f XPS spectrum of 2|NiO (d) before photoelectrocatalysis, after photoelectrocatalysis under (e) Eappl = −0.4 V and (f) Eappl = −0.6 V vs. Ag/AgCl. | ||
Electrode characterisation pre- and post-catalysis
The Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) analysis was carried out on 1|NiO and 2|NiO pre- and post-catalysis (Fig. S11–S18 in the ESI†). Higher mass molecular ions corresponding to 1 and 2 were present in the SIMS spectra of as-deposited 1|NiO and 2|NiO (pre-catalysis), respectively. This is consistent with the successful adsorption of photocatalysts 1 and 2 on the surface of the nanostructured NiO electrode. Post-catalysis, there was some evidence for the presence of phthalate buffer residues with the detection of a fragment ion at m/z = 121 due to the PhCO2− species. We did not observe any desorption of the photocatalyst in the presence of the buffer (see above) but the results suggest that non-competitive co-adsorption of some phthalate is possible.The SIMS spectra, 1020–1140 mass per unit, of 1|NiO before and after photoelectrocatalysis are presented in Fig. 9 and contain the higher mass molecular ions corresponding to dye-catalyst assembly 1. This indicates that 1 did not decompose during the experiment and is intact on the surface of the NiO during the photoelectrocatalysis under Eappl = −0.4 V vs. Ag/AgCl. The SIMS spectra for dye-catalyst assembly 2, also contain peaks corresponding to higher mass molecular ions in the post-catalysis samples (Fig. S18 in the ESI†). The intensity of these peaks diminishes going from Eappl = −0.2 V to −0.6 V vs. Ag/AgCl. This indicates that 2 has partially detached or decomposed during photoelectrocatalysis. In Fig. 9, several peaks are present in the range corresponding to dye-catalyst assembly 1. Inspection of the isotope patterns reveals that there is a combination of H2O, Cl− and CH3CN coordinated to the Pd catalytic centre. Possible identities of these species are provided in the figure caption.
X-ray Photoelectron Spectroscopy (XPS) measurements were carried out on 1|NiO and 2|NiO pre- and post-catalysis (Eappl = −0.4 V and Eappl = −0.6 V vs. Ag/AgCl). The results are compared in Fig. S5–S10 in the ESI.† There were no significant differences between the Ni 2p XPS spectra of pre- and post-catalysis samples (Fig. S8 and S9 in the ESI†), which indicates that there are no substantial changes to the NiO electrode during the experiments. In particular, the Ni3+ to Ni2+ ratio was similar and there was no visible Ni0 peak at lower binding energy, which has been reported elsewhere for post- catalysis samples.34 The binding energy for Ni metal 2p3/2 peak on Ni and NiO samples has been reported at 852.6 eV.35,36
For 1|NiO, the C 1s and Ru 3d regions (Fig. S5 and S6†) were largely unchanged, confirming that the dye-catalyst assembly 1 is present on NiO before and after photoelectrocatalysis. In both 1|NiO and 2|NiO samples, the binding energies for the Ru 3d spin–orbit doublet peaks are situated at 281.1 eV and 285.3 eV for Ru 3d5/2 and Ru 3d3/2, respectively, which is consistent with reported values for ruthenium tris(bipyridine) electrocrafted on boron doped diamond electrode (Ru 3d5/2: 281.3 eV, Ru 3d3/2: 285.5 eV).37 There are no additional peaks present in the Ru 3d region to suggest that the Ru has decomposed. This is consistent with the presence of molecular ion fragments corresponding to [Ru(bpy)2(L)]+ in the corresponding TOF-SIMS (Fig. S13 and S17†). Therefore, we conclude that the photosensitiser part of both molecules is stable during the photoelectrocatalysis, even at potentials more negative than Eappl = −0.6 V vs. Ag/AgCl.
The Pd 3d spin–orbit doublet in the spectra for 1|NiO, pre- and post-catalysis, (Fig. 10(a–c)) contain two major components in each band (1, 2, 3 and 4). Component 1 at 338.4 eV under Pd 3d5/2 peak is consistent with spectra reported previously for Pd(II) (PdCl2 (ref. 70) and bis(1,2-ethanediamine-N,N′)dichloropalladium71). The 3d5/2 binding energy for Pd(0) should lie between 334 eV and 336 eV, but component (3) has a maximum at 337.1 eV.72 Therefore, we do not attribute components (3) and (4) to Pd(0). Instead, (3) and (4) could correspond to a Cl-free complex, e.g. [Ru(decb)2(bpt)Pd(CH3CN)]3+ (L1d) identified in the ToF-SIMs spectrum in Fig. 9(a).
In contrast, each doublet peak in the Pt 4f spin–orbit doublet in the 2|NiO, pre-catalysis, spectrum (Fig. 10(d)) is composed of one component, with binding energies for (1) Pt 4f7/2 = 72.6 eV and (2) Pt 4f5/2 = 75.9 eV. These binding energies are consistent with those reported previously for Pt(II) iodo-complexes.35,38 Additional components (3 and 4) are present at lower binding energies in the spectrum for 2|NiO, post-catalysis, (Fig. 10(e) and (f)). The additional component (3) under Pt 4f7/2 doublet peak has the binding energy of 71.6 eV, which is consistent with Pt(0).36,39,73 This indicates that photocatalyst 2 decomposes during the photoelectrocatalysis and metallic Pt is formed on the NiO surface. These findings are consistent with the ToF-SIMS data, which showed the loss of higher mass species on post-catalysis samples of 2|NiO at Eappl = −0.6 V vs. Ag/AgCl (Fig. S18 in the ESI†). From these results, we reason that during the photoelectrochemical experiments, 2 decomposes to give Pt0 on the surface of the electrode and this could drive H2 evolution in this system.
Discussion
The role of the photocathode in a DSPEC is to use the electrons generated by water oxidation at a photocathode to drive reduction of H+ to H2. The dye-sensitised photoelectrochemical devices reported previously used either co-adsorbed dyes and catalysts12 or covalently linked (but electronically de-coupled) metal ion coordination supramolecular dye-catalyst assemblies.16,34 High photocurrents have been reported elsewhere for an organic dye with an H2-evolving catalyst in solution (however, pH 0 was used, which could be problematic when coupled with an oxygen-evolving photoanode).40 Our approach, instead, uses integrated photocatalysts.In solution, the photophysics of the two photocatalysts is distinct, as summarised in Fig. S40.† In photocatalyst 2, the electron transfer is in the direction of the peripheral ligands, whereas in photocatalyst 1, the electron transfer is in the direction of the bridging ligand. In the presence of a sacrificial electron donor, e.g. triethylamine, 2 performs surprisingly well considering its apparently unfavourable structure and this has been attributed to its long-lived excited state (≈μs).28 In contrast, while the structure of 1 favours electron transfer in the direction of the catalytic centre, the excited state decay (≈ns) could compete with diffusion controlled processes such as reductive quenching by triethylamine, and may explain why no hydrogen was observed in the solution studies.
In this work, we have probed the first steps in photocatalysis using TRIR and TA and demonstrated spectroscopically that rapid photoinduced electron transfer from NiO to the adsorbed photocatalyst occurs. The spectra were complex due to overlapping signals for the charge-separated state and residual excited state in both 1|NiO and 2|NiO sets of spectra and multi exponential decay kinetics. This is consistent with studies on model dyes such as, [Ru(4,4′-(CO2H)-bpy)3]2+, [Ru(4,4′-(CO2H)-bpy)(bpy)2]2+ and [Ru(4,4′-(PO4H2)-bpy)(bpy)2]2+, reported previously.24,29,41 To help make sense of the possible pathways through which the excited state could evolve, the possible configurations are shown in Fig. 11, which, to simplify the analysis, assumes only one decb ligand is bound. Absorption of visible light, leads to the population of the 1MLCT, which rapidly relaxes to the 3MLCT, either on the decb ligands (in 2) or the bridge (in 1). According to Bräutigam et al. a competition between the population of bipyridyl ligands pointing away from the surface (injection-favouring) compared to those anchored to the surface (recombination-favouring) takes place, leading to the observation of a transient absorption spectrum corresponding to the charge-separated state or excited state, respectively.29 The TRIR experiments are consistent with this hypothesis, highlighting the loss of symmetry when the dyes are adsorbed onto NiO. In 1|NiO, electron transfer to the bridge is more favourable than to the peripheral decb ligand, so one would expect that injection would be more favourable in this system. Inspection of the amplitudes for the DAS for the immobilised photocatalysts, in Fig. S30 and S31,† reveals that in 1|NiO, about 30% of the bleach corresponds to 1*, for 2|NiO, roughly 50% of the bleach corresponds to 2*. The rapid (<ps) rate of electron transfer from NiO to the photocatalysts, combined with the relative yield compared to residual excited state, implies that this injection process may take place from the 1MLCT and the ligand on which this is localised, may determine whether or not charge injection occurs. The absence of a signal corresponding to the reduced photocatalysts at longer timescales (>100–200 ps), despite the long-lived excited species 1*|NiO (τ ≈ 1 ns) and 2*|NiO (τ ≈ 4 ns), implies that injection does not occur from these 3MLCT states.
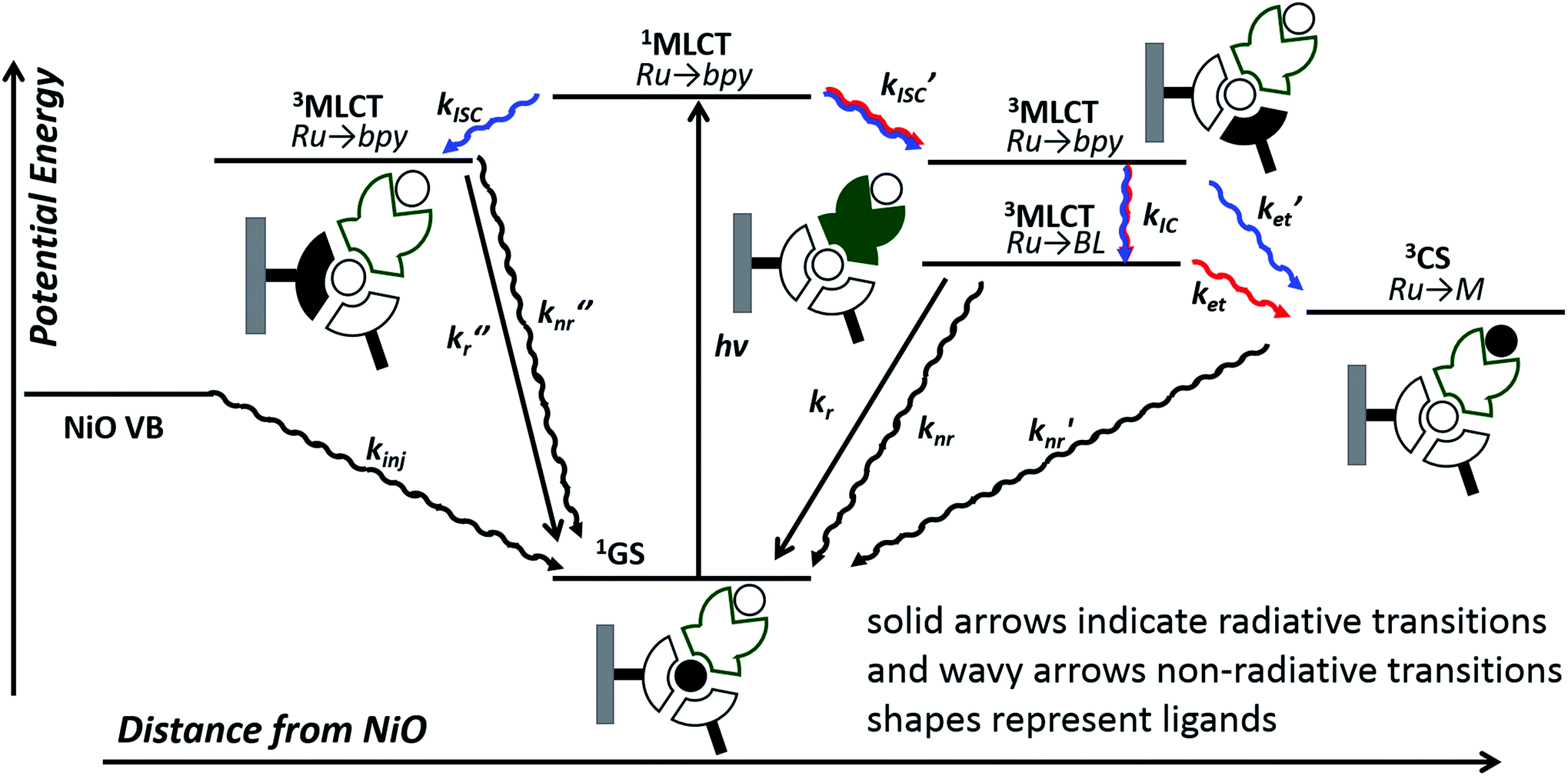 | ||
| Fig. 11 Possible excited state configurations of 1 (red) and 2 (blue) when immobilised on NiO based on the TA and TRIR experiments, DFT and TD-DFT calculations in this work and models proposed in ref. 21, 24, 25 and 28. BL = bridging ligand, bpy = bipyridyl, M = Pt or Pd, CS = charge-separated state. | ||
However, this may not be the whole story under operation of the device. The lifetime of the charge-separated state was found to be extremely short, which is consistent with previous studies of organic dyes on NiO and suggests that recombination between the holes in NiO and the reduced dye dominates.42 This implies that photoelectrocatalysis is unfavourable. However, Dillon et al. demonstrated that changing the bias applied to the photocathode from +0.4 to −0.4 V vs. Ag/AgCl extends the lifetime of a reduced dye, [Ru(4,4′-(PO4H2)-bpy)(bpy)2]+, from ps to μs due to filling intra-bandgap states (see inset Fig. 11 for our interpretation). This could both increase the rate of injection and slow down recombination. We have not applied a bias to the films in the TRIR or TA studies reported for 1|NiO and 2|NiO, (D'Amario et al. estimated a Fermi Level of +250 mV vs. Ag/AgCl for NiO prepared under similar conditions), however, a reduction of the rate of recombination is consistent with the increase in photocurrent as Eappl was increased. The photocurrent onset in our experiments is ca. 0.43 V more negative than the valence band edge of NiO. In the TA experiments, in non-aqueous electrolyte under applied bias, by Dillon et al., ca. 70% filling of the surface states was estimated at this relative applied potential, giving <60% yield of reduced dye, compared to zero yield when no bias was applied. At Eappl > −500 mV (where all intra band gap states should be filled), Dillon et al. observed a change in kinetics, which they attributed to dye desorption or a build-up of cations at the electrode surface which could stabilise the reduced dye.24 This is again consistent with the change in photoelectrochemical behaviour observed in this work for 1|NiO and 2|NiO at extreme negative bias (−0.6 V vs. Ag/AgCl).
The photocurrent densities for 1|NiO and 2|NiO are comparable with the best co-immobilised systems on NiO and ITO. However, 1|NiO is superior to previously reported photocathodes in terms of the faradaic efficiency and stability of the photocurrent.10,43 We attribute the high faradaic efficiency of our photocathodes to the direct coupling of the photosensitiser with the catalyst. In the NiO systems reported previously, the dye and the catalyst have been either separate molecules (either co-deposited on the surface or a catalyst in solution) or tethered through non-conjugated linkers.2 In such systems, the faradaic yields reported are typically less than 70% and in some cases below 10%, possibly due to slow or inefficient charge-transfer to the catalyst (leading to charging and discharging of the electrode), the presence of side reactions (e.g. reduction of O2) or the catalyst acting as a redox shuttle by diffusing from the cathode to the anode. Integrating the sensitiser and catalyst within one molecule permits rapid (i.e. not diffusion-limited) electron transfer to the catalytic centre. While we do not yet know the precise mechanism for hydrogen evolution in the integrated photocatalyst system, the transient spectroscopy confirms that electron transfer from NiO to the catalyst occurs rapidly upon excitation and the high faradaic yield and stable photocurrents are consistent with catalysis out-competing charge-recombination. Nonetheless, the system is far from optimum at this stage, for example, the light harvesting efficiency of both 1|NiO and 2|NiO is low. Thicker and more porous films may improve the performance,16 and, in future, chromophores with higher absorption coefficients will be designed to increase the photocurrent density.
We were encouraged by the stability of the Ru chromophore on the NiO surface as, rather than esters, carboxylic acid or phosphonic acid anchoring groups are generally used to adsorb the dye, which present challenges in synthesis and purification5,10,12,13,16,17 Slight changes in the Pd and Pt catalyst structure during operation were anticipated, e.g. replacement of the halide ligands with solvent. Kaeffer et al., for example, reported halide ligand substitution (Br− for Cl−) in cobalt-diimine–dioxime complexes post-catalysis in their co-immobilised system.26 These results are consistent with reactivity at the catalytic metal centre. The reduction of the Pd(II) centre is expected to be accompanied by dissociation of a chloride anion.44,45
Initial inspection of the photocurrent density and volume of H2 produced, suggested that 2, containing the Pt catalyst, outperformed 1, which contains the Pd catalyst. However, the increase in current observed (particularly at the most negative applied bias) during irradiation indicated that the electrode surface was changing during the experiment. The XPS and ToF-SIMS results confirm that the Pt complex is unstable in water, especially under irradiation at Eappl = −0.6 V vs. Ag/AgCl, and we attribute the increasing current over time to the formation of electrocatalytically active Pt(0) on the NiO surface. It is also possible that the strong transient reductive photocurrents observed for NiO/Ru–Pt (Fig. 7) correspond to the in situ reduction of the Pt(II) complex, generating Pt(0) particles. Du et al. have previously reported the formation of colloidal platinum during photocatalytic H2 evolution using a system containing platinum(II) bi- and terpyridyl chloro complexes, in the presence of a sacrificial electron donor (MeOH or triethanolamine) and an electron relay (TiO2).46 In contrast, the surface analysis experiments showed that 1, which contained the Pd centre, was present on the NiO surface after 1 h PEC at potentials from Eappl = −0.2 to −0.6 V vs. Ag/AgCl. These results highlight the importance of characterising the electrodes after the catalysis. Whilst in situ characterisation of heterogenous catalysts is challenging, hybrid systems containing molecular catalysts furnished with spectroscopic handles present an opportunity to extract mechanistic information.
While the performance of the photocathodes is encouraging, we anticipate that much higher yields are possible with some minor alterations to our experimental setup. Better device engineering is necessary to improve mass transport. The geometry of the custom-built PEC cell (Fig. S1†) is not optimised and bubble formation on the photocathode surface led to a drop in the active area. In this study, no membrane was used to separate the anode and photocathode so the presence of oxygen formed at the Pt anode could provide a recombination pathway. While reasonable photocurrents were observed in pH 7 buffer, pH 3 was optimum for H2 evolution. Raising this will be desirable for tandem devices.
Conclusions
Light driven H2 production from water by two new photocathodes, comprised of integrated photocatalysts 1 and 2 adsorbed on NiO, has been described. Stable photocurrents and sustained H2 production was observed for both photocathodes immersed in pH 3 buffer by PEC experiments with in-line gas chromatography, in the absence of sacrificial agents. The faradaic efficiencies were estimated between 30–90%, depending on Eappl. Surface analysis experiments revealed that the Ru photosensitiser and the Pd catalyst are stable during photoelectrochemical H2 evolution over a range of Eappl, whereas the Pt catalyst decomposed at more negative potentials. These findings were consistent with the observed photocurrent during PEC H2 production, which was most stable for 1|NiO. These results are promising for the development of efficient photoelectrocatalytic devices for storing the energy from sunlight in chemical bonds. Future efforts will be directed towards increasing the absorptivity of the photocathodes and raising the optimum pH through modifications to the ligand structure.Experimental
Materials
3,5-Bis (pyridin-2-yl)-1,2,4-triazole (Hbpt),47 4,4′-di(ethylcarboxy)-2,2′-bipyridine (decb),48cis-[Ru(decb)2Cl2],49cis-[Pt(DMSO)2I2],27cis-[Pd(DMSO)2Cl2], [Ru(decb)2(2,5-bpp)](PF6)·2H2O, and [Ru(decb)2(2,5-bpp)PtI(CH3CN)](PF6)2,27 were prepared using literature methods, solvents obtained were used without further purification.[Ru(decb)2(Hbpt)](PF6)2·2H2O
[Ru(decb)2(Hbpt)](PF6)2·2H2O was prepared using a modified literature method.50 84 mg (0.37 mmol) of 3,5-bis(2-pyridyl)-4-hydro-1,2,4-triazole(Hbpt) were dissolved in 60 ml EtOH/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and heated for 20 minutes. 200 mg (0.26 mmol) of [Ru(decb)2Cl2] in ethanol was added slowly over 30 minutes. The reaction was refluxed for a further 6 h, with a total volume of 100 ml. The ethanol was removed in vacuo. Following this, 40 ml of water was added to the reaction mixture. The red aqueous reaction mixture was filtered and an aqueous solution of NH4PF6 was added in excess to the filtrate and a precipitate formed. The precipitate was washed with diethyl ether and collected by filtration. For further purification, the filtrate was recrystallized with acetone/H2O (3:1), yielding a black/brown solid. Yield: 212 mg (0.17 mmol, 65%). 1H-NMR (400 MHz, DMSO-d6) δ[ppm] = 1.30–1.40 (m, 12H), 4.39–4.49 (m, 8H), 7.39 (dd, 1H), 7.49–7.67 (m, 2H), 7.84 (dd, 1H), 7.93–8.25 (m, 8H), 8.33 (d, 1H), 8.62 (d, 1H), 9.17–9.40 (m, 4H). Elemental analysis for C44H45F12N9O10P2Ru calc.: C 42.2%, H 3.6%, N 10.1%. Found: C 42.12%, H 3.18% and N 10.28%.
:1) and heated for 20 minutes. 200 mg (0.26 mmol) of [Ru(decb)2Cl2] in ethanol was added slowly over 30 minutes. The reaction was refluxed for a further 6 h, with a total volume of 100 ml. The ethanol was removed in vacuo. Following this, 40 ml of water was added to the reaction mixture. The red aqueous reaction mixture was filtered and an aqueous solution of NH4PF6 was added in excess to the filtrate and a precipitate formed. The precipitate was washed with diethyl ether and collected by filtration. For further purification, the filtrate was recrystallized with acetone/H2O (3:1), yielding a black/brown solid. Yield: 212 mg (0.17 mmol, 65%). 1H-NMR (400 MHz, DMSO-d6) δ[ppm] = 1.30–1.40 (m, 12H), 4.39–4.49 (m, 8H), 7.39 (dd, 1H), 7.49–7.67 (m, 2H), 7.84 (dd, 1H), 7.93–8.25 (m, 8H), 8.33 (d, 1H), 8.62 (d, 1H), 9.17–9.40 (m, 4H). Elemental analysis for C44H45F12N9O10P2Ru calc.: C 42.2%, H 3.6%, N 10.1%. Found: C 42.12%, H 3.18% and N 10.28%.
[Ru(decb)2(bpt)PdCl(H2O)](PF6)2·2H2O
28 mg (0.11 mmol) of cis-Pd(DMSO)2Cl2 and 65 mg (0.05 mmol) of [Ru(decb)2(Hbpt)](PF6)2·2H2O were added to 15 ml of hot EtOH and stirred under nitrogen until dissolved. The reaction was brought to reflux temperature for 24 h. A precipitate formed and the solution was allowed to cool to room temperature. The precipitate was collected by vacuum filtration and washed with cold EtOH and diethyl ether. Yield: 58 mg (0.04 mmol, 84%). 1H-NMR (400 MHz, acetonitrile-d3) δ[ppm] = 1.35–1.45 (m, 12H), 4.39–4.49 (m, 8H), 7.29 (t, 1H), 7.50 (d, 2H), 7.65 (t, 1H), 7.70 (d, 1H), 7.75–7.82 (m, 3H), 7.83 (d, 1H), 7.89 (d, 1H), 7.95–8.05 (m, 2H), 8.27 (d, 1H), 8.36 (t, 1H), 8.51 (d, 1H), 8.95–9.08 (m, 3H), 9.18 (d, 1H), 9.60 (d, 1H), 11.12 (d, 1H). Elemental analysis for C44H46F12N9O11P2RuPdCl calc.: C 37.48%, H 3.2%, N 8.94%. Found: C 37.08%, H 2.71% and N 9.03%.Photocathode preparation
Mesoporous NiO photocathodes were prepared by following a reported procedure.51 A NiCl2 precursor solution was prepared by dissolving anhydrous NiCl2 (1 g) and the tri-block co-polymer F108 (poly (ethylene glycol)-block-poly (propylene glycol)-block-poly (ethylene glycol)) (1 g) in ethanol (6 g) and ultrapure Milli-Q water (3 g). The precursor solution described above was spread onto fluorine doped tin oxide (FTO) conducting glass substrates (Pilkington TEC15, sheet resistance 15 Ω per square) using Scotch tape as a spacer (0.79 cm2), followed by sintering in an oven at 450 °C for 30 min. Undyed NiO films were prepared to a thickness of 1.5 μm, measured using a Bruker DektakXT stylus profilometer and averaged over 5 samples. Dye sensitised electrodes 1|NiO and 2|NiO were prepared by soaking the NiO electrodes in acetonitrile solutions of the dye-catalyst assemblies 1 and 2 (0.3 mM) for 16 h at room temperature.Optical and IR spectroscopy
The ultraviolet-visible (UV-vis) absorption spectra of the dye-catalyst assemblies in solution and adsorbed on nanostructured NiO films were recorded using a Shimadzu 1800 UV-vis spectrophotometer. The infrared spectra were measured on a Varian FTS 800 FT-IR spectrometer. Solid samples of sensitised NiO films were removed from the FTO substrate with a spatula and were mixed with potassium bromide (99%, spectroscopic grade, Fisher Scientific) using a pestle and mortar and pressed into disks.Electrochemical and photoelectrochemical experiments
Electrochemical (EC) and photoelectrochemical (PEC) measurements were carried out using an IviumStat potentiostat. Electrochemical measurements were conducted on the dye-sensitised nanostructured NiO photocathodes using a custom-made three-electrode photoelectrochemical cell (Fig. S1 in ESI†). A platinum wire was used as the counter electrode. Ag/AgCl (3.0 M NaCl, EoAg/AgCl = 0.210 V vs. NHE) was used as the reference electrode for the measurements in aqueous solutions, where of 0.1 M potassium hydrogen phthalate and 0.1 M KCl were used as a supporting electrolyte. All potentials are reported vs. Ag/AgCl reference electrode. For conversion to NHE, the following equation was used: ENHE = EAg/AgCl + EoAg/AgCl. For conversion to RHE, Nernst equation was used (ERHE = EAg/AgCl + 0.059 pH + EoAg/AgCl). During the PEC experiments, irradiation was provided by a 300 W Xe lamp (Oriel) fitted with an AM1.5 filter (Newport). This was calibrated using a calibrated reference solar cell (Newport) to give a power density of 100 mW cm−2 (1 sun) at the photocathode surface with the irradiated area of 0.79 cm2. The cell was degassed with Ar for at least 20 minutes prior to each measurement. The pH was measured using a pH mV−1 Benchtop Meter (Hanna instruments). During chronoamperometry measurement chopped light illumination was first applied with 30 s intervals (1 cycle: 30 s light on/30 s light off), which was followed by constant light illumination. Before every PEC measurement, the working electrode (WE) was held 30 s to 1 min under the potential applied during the measurement, Eappl, in the dark to stabilise the background current. The faradaic efficiency, ηFar, was calculated by dividing the amount of H2 produced experimentally (mol) with the theoretical H2 production (mol) according to the charge generated from the photocurrent. The results from a control experiment with Ru(dcbpy)3Cl2 (dcbpy = 2,2′-bipyridyl-4,4′-dicarboxylic acid) Eappl = −0.4 V vs. Ag/AgCl is provided in Fig. S35.†Ultrafast transient absorption spectroscopy
Picosecond Transient Absorption and Time-Resolved Infra-Red (TRIR) spectra were recorded using the ULTRA instrument located in the Central Laser Facility at the Rutherford Appleton Laboratory. Briefly, two Ti: sapphire amplifiers of 10 kHz and 1 kHz were synchronized using a common 65 MHz oscillator. The 1 kHz output was used as a pump and the 10 kHz as probe. The pump laser was tuned to 470 nm by optical parametric amplification (OPA, Light Conversion, TOPAS). For TAS, the probe pulse was provided by a white light continuum (WLC), which was generated by focusing 800 nm into CaF2. The mid-IR probe pulses were generated using OPA with difference frequency mixing. The IR probe beam was split to form reference and probe beams which were passed through spectrographs onto MCT array detectors (IR Associates). High speed data acquisition systems (Quantum Detectors) allowed 10 kHz acquisition and processing of the probe and reference pulses to generate a pump-on pump-off infrared absorption difference signal. Spot sizes in the sample region were ca. 150 and 50 μm for the pump and probe, respectively, with a pump energy of 50 nJ. For all measurements, the pump polarization was set to magic angle relative to the probe.Samples were prepared by adsorbing the dye on a mesoporous NiO film deposited on a CaF2 window (Crystran). The NiO films were prepared by spraying a saturated solution of NiCl2 in acetylacetone onto the surface of the CaF2 window, which was preheated to 450 °C on a hot plate; this was then allowed to cool slowly to room temperature to give a compact film of NiO. The mesoporous layer was then deposited on top of the compact layer using the F108-templated precursor solution described above; the excess was removed by doctor blade. The film was sintered at 450 °C for 30 min, and an additional layer of precursor solution was applied and sintered to increase the film thickness. All spectra were recorded in IR cells (Harrick Scientific Products Inc.) with CaF2 windows. For samples prepared in solution, a 200 μm path length was used. In all experiments, the cell was rastered in the two dimensions orthogonal to the direction of beam propagation to minimize localized sample decomposition.
Gas chromatography
Gas chromatography (GC) measurements were carried out using a Shimadzu chromatograph with the thermal conductivity detector (TCD) operating at 50 °C and fitted with a ShinCarbon ST Micropacked column (Restek) using Ar as a carrier gas. The experimental setup for the in-line, continuous gas sampling and analysis method is described in full by Summers et al.52Briefly, Ar was continuously flowed through the electrolyte solution and into a 6-port, 2-position switch (VICI) at a constant flow (typically 10 cm3 min−1). Flow from the PEC cell was directed through a cold trap to a rheodine switch which injected a gas samples to the GC at three-minute intervals, maintained using a mass flow controller (Bronkhorst, E-Flow series). A 200 μl sample was analysed automatically every 3 min. The calibration of the amount of H2 detected was carried out by varying the flow rate of H2 (mol min−1) into the system using the 6-port, 2-position switch. The H2 peak area varied linearly with the flow rate and the peak area was plotted against the flow rate on a calibration plot. The gradient of the line was used to calculate the H2 production rate (mol min−1) and to get the total amount of H2 produced during an experiment, the production rate was integrated with respect to time.
X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) was carried out on Kratos Axis Nova XPS spectrometer using a monochromatic Al Kα source. The samples were mounted on a clean aluminium platen and immobilised using double sided adhesive tape. The largest analysis area available in this spectrometer (300 μm × 700 μm) was used. All the measurements were repeated on three different analysis positions with non-overlapping analysis areas. Charge compensation was used throughout the measurements. Spectra were analysed using CasaXPS software53 (version 2.3.16). Gaussian (70%) Lorentzian (30%), defined in CasaXPS as GL(30), profiles were used for each component. Spectra have been calibrated to obtain the adventitious C 1s spectral component binding energy of 284.7 eV.Time-of-flight secondary ion mass spectrometry
Static SIMS analyses were carried out using an ION-TOF ‘TOF-SIMS IV – 200’ instrument (ION-TOF GmbH, Münster, Germany) of single-stage reflectron design.54 Positive and negative ion spectra and images of the samples were obtained using a Bi32+ focused liquid metal ion gun at 25 keV energy, incident at 45° to the surface normal and operated in ‘bunched’ mode for high mass resolution. This mode used 20 ns wide ion pulses at 6.7 kHz repetition rate. Charge compensation was effected by low-energy (ca. 20 eV) electrons provided by a flood gun. The total ion dose density was less than 1 × 1016 ions per m2. The topography of the sample surface and the ion gun mode of operation limited the mass resolution in this work to ca. m/Δm = 5000. Positive and negative ion static SIMS spectra and images were recorded from the samples at room temperature. Raw data containing the secondary ions recorded at each pixel was acquired with a 128 × 128 pixel raster and a field of view of 200 μm × 200 μm. The samples for ToF-SIMS analysis were mounted directly onto a sample holder using small pieces of silicone-free double-sided tape (3M grade 665).Quantum chemical methods
Quantum chemical methods were used to model the electronic structure of the ground state singlet and lowest energy triplet states of three model systems [Ru(bipy)2(bpt)PdCl](PF6)2 [Ru(dmcb)2(bpt)PdCl](PF6)2, and [Ru(dmcb)2(bpt)PdCl(H2O)](PF6)2. Density functional theory (DFT) modelled the ground-state structures (singlet and triplet) and time-dependent density functional (TD-DFT) methods were used to characterise the low-lying electronic excited states. Either the hybrid B3LYP55–57 or its Coulomb adjusted variant cam-B3LYP58 was used with the double-zeta quality LanL2DZ basis set.59–62 However, the B3LYP functional performed better than the cam-B3LYP in modelling the energies of the low-lying singlet excited states and thus the simulated UV/vis spectra were a closer fit to the experimental. For this reason, the results obtained using the B3LYP are reported here.The same general approach was used for all complexes. Initial structures were generated by molecular mechanics methods and these were optimised at the B3LYP/LanL2DZ model chemistry. The stability of the wavefunction was then tested followed by calculation of the vibrational frequencies. Absence of imaginary frequencies confirmed that all optimised structures were located at a minimum on their respective potential energy hypersurface. Because of difficulties with optimising structure containing ethylester functional groups, the model systems chosen contained either unsubstituted bipy ligand or the dimethylcarbonylate substituted ligands (dmcb). These allowed the effect that ester functional groups have on the electronic structures of the excited states of these systems to be estimated. Calculations were performed in the presence of either acetonitrile or water as indicated using the PCM (Polarisable Continuum Method) approach.63–65
All calculations were performed using the Gaussian 16, Revision A.03 (ref. 66) programme suite. Molecular structures and electron-density difference maps were visualised using GaussView 03.67 Orbital contributions for excited states and simulations of UV/vis. Spectra were obtained using AOMix version 6.88.68,69 Calculations were performed on the Fionn system at the Irish Centre for High End Computing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leverhulme Trust for a project grant RGS108374, The North East Centre for Energy Materials EP/R021503/1, STFC for access to the CLF ULTRA facility for transient spectroscopy, EU access grant 16140001, NEXUS national EPSRC XPS facility for conducting the XPS measurements and surface characterisation by ToF-SIMS measurements. The authors wish to acknowledge the SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. NP thanks Newcastle University for a PhD studentship, and LOR thanks the Irish Research Council for financial support. Data supporting this publication is openly available under an ‘Open Data Commons Open Database License’. Additional metadata are available at: http://dx.doi.org/10.17634/172828-2.Notes and references
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 4, 2551–2564 Search PubMed.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- L. Francàs, C. Richmond, P. Garrido-Barros, N. Planas, S. Roeser, J. Benet-Buchholz, L. Escriche, X. Sala and A. Llobet, Chem.–Eur. J., 2016, 22, 5261–5268 CrossRef.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- J. He, H. Lindström, A. Hagfeldt and S. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS.
- C. Y. Lee, H. S. Park, J. C. Fontecilla-Camps and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 5971–5974 CrossRef CAS PubMed.
- D. Mersch, C. Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS.
- B. Shan, A. K. Das, S. Marquard, B. H. Farnum, D. Wang, R. M. Bullock and T. J. Meyer, Energy Environ. Sci., 2016, 9, 3693–3697 RSC.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 56, 510–514 Search PubMed.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- L. Tong, A. Iwase, A. Nattestad, U. Bach, M. Weidelener, G. Götz, A. Mishra, P. Bäuerle, R. Amal, G. G. Wallace and A. J. Mozer, Energy Environ. Sci., 2012, 5, 9472 RSC.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS.
- P. B. Pati, L. Zhang, B. Philippe, R. Fernández-Terán, S. Ahmadi, L. Tian, H. Rensmo, L. Hammarström and H. Tian, ChemSusChem, 2017, 10, 2480–2495 CrossRef CAS.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 242–247 RSC.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Chem. Sci., 2017, 8, 541–549 RSC.
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef CAS.
- A. M. Brown, L. J. Antila, M. Mirmohades, S. Pullen, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 8060–8063 CrossRef CAS.
- C. E. Castillo, M. Gennari, T. Stoll, J. Fortage, A. Deronzier, M.-N. Collomb, M. Sandroni, F. Légalité, E. Blart, Y. Pellegrin, C. Delacote, M. Boujtita, F. Odobel, P. Rannou and S. Sadki, J. Phys. Chem. C, 2015, 119, 5806–5818 CrossRef CAS.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- D. W. Kim, S. C. Riha, E. J. DeMarco, A. B. F. Martinson, O. K. Farha and J. T. Hupp, ACS Nano, 2014, 8, 12199–12207 CrossRef CAS.
- A. L. Smeigh, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2012, 48, 678–680 RSC.
- R. J. Dillon, L. Alibabaei, T. J. Meyer and J. M. Papanikolas, ACS Appl. Mater. Interfaces, 2017, 9, 26786–26796 CrossRef CAS.
- T. Kowacs, Q. Pan, P. Lang, L. O'Reilly, S. Rau, W. R. Browne, M. T. Pryce, A. Huijser and J. G. Vos, Faraday Discuss., 2015, 185, 143–170 RSC.
- N. Kaeffer, C. D. Windle, R. Brisse, C. Gablin, D. Leonard, B. Jousselme, M. Chavarot-Kerlidou and V. Artero, Chem. Sci., 2018, 9, 6721–6738 RSC.
- T. Kowacs, L. O'Reilly, Q. Pan, A. Huijser, P. Lang, S. Rau, W. R. Browne, M. T. Pryce and J. G. Vos, Inorg. Chem., 2016, 55, 2685–2690 CrossRef CAS.
- Q. Pan, L. Freitag, T. Kowacs, J. C. Falgenhauer, J. P. Korterik, D. Schlettwein, W. R. Browne, M. T. Pryce, S. Rau, L. González, J. G. Vos and A. Huijser, Chem. Commun., 2016, 52, 9371–9374 RSC.
- M. Bräutigam, J. Kübel, M. Schulz, J. G. Vos and B. Dietzek, Phys. Chem. Chem. Phys., 2015, 17, 7823–7830 RSC.
- S. Fanni, T. E. Keyes, C. M. O'Connor, H. Hughes, R. Wang and J. G. Vos, Coord. Chem. Rev., 2000, 208, 77–86 CrossRef CAS.
- K. A. Click, B. M. Schockman, J. T. Dilenschneider, W. D. McCulloch, B. R. Garrett, Y. Yu, M. He, A. E. Curtze and Y. Wu, J. Phys. Chem. C, 2017, 121, 8787–8795 CrossRef CAS.
- G. Boschloo and D. Fitzmaurice, J. Phys. Chem. B, 1999, 103, 2228–2231 CrossRef CAS.
- L. D'Amario, R. Jiang, U. B. Cappel, E. A. Gibson, G. Boschloo, H. Rensmo, L. Sun, L. Hammarström and H. Tian, ACS Appl. Mater. Interfaces, 2017, 9, 33470–33477 CrossRef.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- C. Agnès, J.-C. Arnault, F. Omnès, B. Jousselme, M. Billon, G. Bidan and P. Mailley, Phys. Chem. Chem. Phys., 2009, 11, 11647 RSC.
- D. T. Clark, D. B. Adams and D. Briggs, J. Chem. Soc., Chem. Commun., 1971, 603, 602–604 RSC.
- J. F. Moulder, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin-Elmer Corporation, 1992 Search PubMed.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS.
- J. C. Freys, J. M. Gardner, L. D'Amario, A. M. Brown and L. Hammarström, Dalton Trans., 2012, 41, 13105–13111 RSC.
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45, 6215–6218 CrossRef CAS.
- M. Karnahl, C. Kuhnt, F. Ma, A. Yartsev, M. Schmitt, B. Dietzek, S. Rau and J. Popp, ChemPhysChem, 2011, 12, 2101–2109 CrossRef CAS.
- P. Du, J. Schneider, F. Li, W. Zhao, U. Patel, F. N. Castellano and R. Eisenberg, J. Am. Chem. Soc., 2008, 130, 5056–5058 CrossRef CAS PubMed.
- J. F. Geldard and F. Lions, J. Org. Chem., 1965, 30, 318–319 CrossRef CAS.
- A. R. Oki and R. J. Morgan, Synth. Commun., 1995, 25, 4093–4097 CrossRef CAS.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- R. Hage, A. H. J. Dijkhuis, J. G. Haasnoot, R. Prins, J. Reedijk, B. E. Buchanan and J. G. Vos, Inorg. Chem., 1988, 27, 2185–2189 CrossRef CAS.
- S. Sumikura, S. Mori, S. Shimizu, H. Usami and E. Suzuki, J. Photochem. Photobiol., A, 2008, 199, 1–7 CrossRef CAS.
- P. A. Summers, J. Dawson, F. Ghiotto, M. W. D. Hanson-Heine, K. Q. Vuong, E. Stephen Davies, X. Z. Sun, N. A. Besley, J. McMaster, M. W. George and M. Schroder, Inorg. Chem., 2014, 53, 4430–4439 CrossRef CAS.
- N. Fairley, http://www.casaxps.com, © Casa software Ltd, 2005.
- J. Schwieters, H. G. Cramer, T. Heller, U. Jürgens, E. Niehuis, J. Zehnpfenning, A. Benninghoven and U. Jurgens, J. Vac. Sci. Technol., A, 1991, 9, 2864–2871 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- C. Lee and R. G. Parr, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 193–200 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- T. H. Dunning Jr and P. J. Hay, ed. H. F. Schaefer III, Modern Theoretical Chemistry, Plenum Press, New York, 1997 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 94107 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas and J. B. Foresman, D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- GaussView 3.0, Gaussian Inc., Pittsburgh, PA, USA: 2003 Search PubMed.
- S. I. Gorelsky, AOMix program, http://www.sg-chem.net/version6.88, University of Ottawa, Ottawa, 2013 Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- C. Sleigh, A. Pijpers, A. Jaspers, B. Coussens and R. J. Meier, J. Electron Spectrosc. Relat. Phenom., 1996, 77, 41–57 CrossRef CAS.
- T. H. Fleisch and G. J. Mains, J. Phys. Chem., 1986, 90, 5317 CrossRef CAS.
- P. Brant, L. S. Benner and A. L. Balch, Inorg. Chem., 1979, 18, 3422 CrossRef CAS.
- J. Thiele, N. T. Barrett, R. Belkhou, C. Guillot and H. Koundi, J. Phys.: Condens. Matter, 1994, 6, 5025 CrossRef CAS.
- L. A. Gallagher, S. A. Serron, X. Wen, B. J. Hornstein, D. M. Dattelbaum, J. R. Schoonover and T. J. Meyer, Inorg. Chem., 2005, 40, 2089–2097 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Electrochemical and photoelectrochemical characterisation, surface analysis, TA and TRIR spectra. See DOI: 10.1039/c8sc02575d |
| ‡ LO'R and NP contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |